

Abstract
Colorectal cancer arises as a consequence of the accumulation of genetic alterations (gene mutations, gene amplification, and so on) and epigenetic alterations (aberrant DNA methylation, chromatin modifications, and so on) that transform colonic epithelial cells into colon adenocarcinoma cells. The loss of genomic stability and resulting gene alterations are key molecular pathogenic steps that occur early in tumorigenesis; they permit the acquisition of a sufficient number of alterations in tumor suppressor genes and oncogenes that transform cells and promote tumor progression. Two predominant forms of genomic instability that have been identified in colon cancer are microsatellite instability and chromosome instability. Substantial progress has been made to identify causes of chromosomal instability in colorectal cells and to determine the effects of the different forms of genomic instability on the biological and clinical behavior of colon tumors. In addition to genomic instability, epigenetic instability results in the aberrant methylation of tumor suppressor genes. Determining the causes and roles of genomic and epigenomic instability in colon tumor formation has the potential to yield more effective prevention strategies and therapeutics for patients with colorectal cancer.
Colorectal cancer results from the progressive accumulation of genetic and epigenetic alterations that lead to the transformation of normal colonic epithelium to colon adenocarcinoma, which is called the polyp → cancer progression sequence (Figure 1). The sequential process of gene mutations and epigenetic alterations is widely believed to drive the initiation and progression of benign adenomas to malignant adenocarcinomas because these mutations affect signaling pathways that regulate hallmark behaviors of cancer.1,2 These mutations create a clonal growth advantage that leads to the outgrowth of progressively more malignant cells, which ultimately manifests itself as invasive adenocarcinoma.
Figure 1.

Depiction of colorectal tumor progression in sporadic and high-risk genetic syndromes. The general paradigm is that a tumor is initiated from a normal colonocyte stem cell that has sustained genetic damage over time due to the local environment and any germline genetic mutation that has been inherited. The damaged DNA provides a growth advantage that drives tumor progression as successive clonal outgrowths are generated, ultimately forming carcinoma. In FAP, tumor initiation is accelerated with the inheritance of a germline APC mutation; in Lynch syndrome, tumor intitiation might be normal to slightly accelerated, but tumor progression is greatly accelerated due to the hypermutable phenotype that occurs with loss of DNA MMR. Photomicrographs depict, in order, normal colon, tubular adenoma, high-grade dysplasia, and cancer.
Importantly, as suggested by Theodor Boveri almost 100 years ago, the acquisition of these mutations is facilitated by the loss of genomic stability, which appears to be a key molecular step in cancer formation.3–5 Indeed, the causative role of genomic instability in the pathogenesis of sporadic colon cancer formation is illustrated by the increased risk of colon cancer observed in the Lynch syndrome and mut Y homologue (MYH)-associated polyposis (MAP), which result, respectively, from germline mutations in genes that encode for DNA repair proteins: the mismatch repair (MMR) system and base excision repair (BER) system.6–8 Further evidence supporting the idea that genomic instability is central to cancer formation comes from recent data showing that germline mutations in genes that encode for other DNA repair proteins are associated with an increased risk of colon cancer, including CHEK2, BRCA1, BRCA2, and BLM, which all are involved in the regulation of double-strand DNA breaks.9–13
It is notable that approximately 30% of the genes in the human genome encode for proteins that regulate DNA fidelity.14 This implies that not only is the maintenance of the integrity of genomic DNA important in eukaryotic cells but also that there are likely a variety of different mechanisms that can cause loss of genomic DNA stability. However, despite advances in our knowledge of the proteins that regulate DNA fidelity, the mechanism responsible for aneuploidy, a hallmark of chromosomal instability (CIN), in the majority of cancers is not known. This has led some investigators to propose that genomic instability can arise in the absence of genetic mutations and is self-propagating in cancer.9 However, in at least a subset of cancers, a specific genetic etiology has been found, which argues that specific, identifiable mechanisms that normally regulate DNA fidelity and are inactivated in cancer mediate the loss of genomic stability in colon cancer. In fact, in 15%–20% of colon cancers, inactivation of the MMR system either through the aberrant CpG island methylation of MLH1 or point mutations in MLH1, MSH2, or other members of the MMR family leads to microsatellite instability (MSI). Furthermore, in some CIN colon cancers, mutations in the mitotic checkpoint regulators have been found.15,16 Other potential mechanisms that may play a role in genomic instability include telomere shortening/telomerase overexpression, impaired DNA replication checkpoint regulation, APC gene mutations, and abnormal centrosome number regulation.4 Recently, the ploidy status in yeast was shown to be primarily regulated by proteins that mediate homologous recombination, sister chromatid cohesion, and mitotic spindle function.17 The mechanisms responsible for causing genomic instability and the specific role that genomic instability appears to play in colon cancer and in driving different molecular pathways of colon cancer formation is discussed in more detail in the following text.
Timing of Genomic Instability in the Adenoma-Carcinoma Sequence
The evolution of normal epithelial cells to adenocarcinoma usually follows a predictable progression of histologic changes and concurrent genetic and epigenetic changes. These alterations provide a growth advantage and lead to clonal expansion of the altered cells. In a manner reminiscent of Darwinian evolution, subsequent alterations with waves of clonal expansion then occur as a consequence of progressive events that provide other growth advantages to the cells such as loss of cell contact inhibition. Predictably, the progressive accumulation of genetic alterations can be identified in more histologically advanced colon neoplasm (Figure 1).18,19 An unresolved issue related to genomic instability, which has been the subject of recent reviews, is whether genomic instability commonly initiates the adenoma-carcinoma sequence or whether it arises during this process and facilitates the formation of colon cancer.9,20,21
The earliest identifiable lesion in colon cancer formation is the aberrant crypt focus (ACF). The true neoplastic potential of this lesion is still unclear, but it does appear that some of these lesions harbor mutations in KRAS or APC and can progress to frank adenocarcinoma. In particular, dysplastic ACF frequently carry mutations in APC and appear to have the highest potential for progressing to colon cancer. The increased likelihood of ACF with mutant APC progressing to cancer may not only result from overactivation of the Wingless/Wnt signaling pathway but also from the induction of chromosome instability. APC has multiple protein-binding domains, including an EB1-binding domain in its C-terminal end. EB1 associates with the growing ends of cytoplasmic and spindle microtubules as well as centrosomes, and the yeast homologue of EB1 is required for a microtubule-dependent cytokinesis checkpoint.22 Fodde et al showed in mouse ES cells homozygous for the mutant ApcMIN or Apc1638N alleles CIN with whole-genome increment changes resulting in polyploidy. This CIN is a consequence of abnormal mitotic spindles that form in these cells because of an abundance of microtubules that inefficiently connect with kinetochores.23 However, although these provocative findings suggest that truncating mutations in APC induce the genomic instability observed in colon adenomas and colon cancers, the significance of these observations is still unclear. The CIN seen in Apc mutant cells is polyploidy rather than gains or losses of individual chromosomes, which is what is seen in colon cancer.24 A unifying model for these discrepancies is that APC inactivation may create a permissive state that allows the evolving tumor clone to tolerate the development of true aneuploidy.9,21,24
Regardless of the role of mutant APC in the regulation of genomic stability in early colon neoplasms, further support for an active role of genomic instability in initiation of colon cancer comes from the observation that both CIN and MSI can be observed in adenomas.25–27 Stoler et al have demonstrated the presence of approximately 11,000 genomic alterations in colon adenomas using inter-(simple sequence repeat) polymerase chain reaction.28 Thus, the timing of loss of genomic stability, either CIN or MSI, appears to be during initiation of adenoma but before progression to frank malignancy. Based on the observation of loss of heterozygosity events in adenomas and using mathematical modeling methods, it can be shown that genomic instability occurs very early in the colon tumorigenesis process and may even precede APC mutations.29 –31 In addition to mathematical modeling showing that it is possible, if not likely, that genomic instability initiates tumor formation in the colon, Bartkova et al provided experimental evidence that suppression of DNA damage and subsequent genomic instability is one of the earliest antitumor mechanisms present in tissues. They have shown that an early event in the formation of several cancers, including bladder, lung, breast, and colon, is the activation of mechanisms involved with DNA damage regulation. In colon adenomas, phosphorylated Chk2 (pT68) can be detected, indicating the activation of double-strand DNA break repair in the tumors.32 Phosphorylated Chk2 is the activated state of Chk2, an effector kinase in the DNA damage network that is activated by ATM (ataxia telangiectasia mutated) when DNA double-strand damage occurs. In addition to phosphorylated Chk2, phosphorylated ATM (Ser1981) and γ-H2AX (Ser 139) can also be detected in colon adenomas, providing further evidence of activation of mechanisms involved in DNA damage regulation and the maintenance of genomic stability. These data suggest that early events in the tumorigenesis process activate mechanisms to maintain DNA stability via the ATR (ataxia telangiectasia mutated and Rad3 related)-ATM checkpoint and that deregulation of this DNA damage response is one of the first events that permit tumor progression.32
MSI
The cause for MSI (sometimes referred to as MIN) was discovered with the observation that its presence is associated with loss of function of the DNA MMR system. The correlation between MSI and inactivation of the MMR system was initially recognized and studied in bacteria and yeast.6,33 With the recognition that MSI occurs in 15%–20% of sporadic human colorectal tumors and in >95% of colon cancers arising in patients with Lynch syndrome, a cancer family syndrome causing an increased risk of colon and extracolonic cancers, it was quickly shown that inactivation of members of the human DNA MMR gene family was the cause of microsatellite unstable colorectal cancer. In contrast, for most colorectal cancers, the mechanism(s) responsible for causing CIN and CpG island methylator phenotype (CIMP) remain to be identified.
Thus, MSI is a marker for loss of DNA MMR activity. In the analysis of tumor DNA, it has become clear that in some tumors, genetic sequences with short repetitive nucleotide sequences such as An or CAn (termed microsatellites, where n is the number of repeats) can become shorter or longer in length when compared with nontumor DNA from the same individual. A microsatellite may lengthen in a daughter cell if there is nucleotide-pairing slippage (looping) along the newly synthesized strand during DNA synthesis or may shorten if the template strand microsatellite has slippage during DNA replication. This alteration in microsatellite length in genomic DNA defines MSI. In the laboratory, MSI is identified by the electrophoretic resolution of amplified microsatellite DNA sequences and the determination of >30% of markers examined having length changes compared with normal DNA from the same individual.33 Although the DNA MMR system recognizes and directs repair of single nucleotide mispairs (see the following text), this aspect is not usually tested when examining tumors for MSI. Multiple point mutations as well as MSI are observed in these tumors, providing the basis for the term “hypermutable” phenotype, and this increase in the spontaneous mutation rate appears to be the mechanism for the apparent rapid neoplastic progression in Lynch syndrome and sporadic tumors that exhibit MSI34,35 (see Figure 1 for Lynch syndrome). If the mismatch occurs in the coding region for a particular gene, the newly introduced point mutation may affect the expression and/or function of that particular gene.36,37 The process of DNA mutations is initially random in MSI cancers affecting any susceptible microsatellite repeat; however, ultimately clones that acquire mutations in key regulatory genes gain a growth advantage that promotes the formation of the carcinoma (see the following text).
The DNA MMR System
The human DNA MMR system is an evolutionarily conserved system that involves a number of proteins that recognize and direct repair of nucleotide mismatches and is responsible for maintaining replicative fidelity of DNA.38,39 The DNA sequences repaired by the MMR system are residual errors that have apparently escaped the normal editing function of DNA polymerase. Base mispairs, if not corrected by the MMR system, may cause nucleotide transitions or transversions, allowing a novel base to alter the authentic genetic sequence.38 Such point mutations in genes that regulate cell growth can accumulate in cells with defective MMR and may promote neoplastic growth. The MMR system replaces the mispair on the newly synthesized daughter strand because these proteins can recognize the methylation pattern (in bacteria) and the nicks present (in higher organisms) in the newly synthesized strand.40,41 The proteins that comprise the MMR system bind to nucleotide base-base mismatches of double-stranded DNA or loops of inaccurately replicated repetitive sequences (microsatellites) and target the DNA area for excision, resynthesis, and ligation.42 Most knowledge of the biochemical function and recognition fidelity of the DNA MMR system comes from studies in Escherichia coli and Saccharomyces cerevisiae. In fact, the names of the human proteins are derived from bacteria and yeast. Mutated S and mutated L proteins are found in bacteria that display MSI; in yeast, the proteins have been named mutS and mutL, and in humans, the proteins are named hMutL and hMutS.
Defects in the MMR genes hMSH2 and hMSH6 (human Mut S homologues), hMLH1 (human Mut L homologue), and hPMS2 (human postmitotic segregation, a Mut L homologue) in humans have been linked to Lynch syndrome (formerly known as hereditary nonpolyposis colorectal cancer), an autosomal dominant condition in which one of the DNA MMR genes is mutated in germline cells43–50 (Table 1). No germline mutation in hMSH3 has been identified in families with this syndrome, perhaps due to redundancy of function of the hMutS homologues.51 In contrast to the germline inactivation of the MMR genes seen in Lynch syndrome, the 15%–20% of sporadic colorectal tumors with MSI inactivate the MMR system by a somatic epigenetic mechanism: the aberrant methylation of the 5′ untranslated region of the hMLH1 gene.52–54 The hypermethylation silences gene transcription of hMLH1, thus inactivating DNA MMR and allowing the phenotype of MSI.
Table 1.
Genes Involved in DNA MMR
| MMR gene | Chromosomal location | MMR partners | MMR fidelity | Lynch syndrome germline mutation (%) |
|---|---|---|---|---|
| hMSH2 | 2p15 | hMSH6, hMSH3 | Complete loss | 31–45 |
| hMSH6 | 2p15 | hMSH2 | Single base pairs, single insertion/deletion loops | 5–8 |
| hMSH3 | 5q11-q13 | hMSH2 | Larger insertion/deletion loops | 0 |
| hMLH1 | 3p21 | hPMS2 (principally), hMLH3, hPMS1 | Complete loss | 32–49 |
| hPMS1 | 2p32 | hMLH1 | Unclear | 0 |
| hPMS2 | 7p22 | hMLH1 | Complete loss? | 0–2 |
| hMLH3 | 7p22 | hMLH1 | Unclear | 0 |
One wild-type allele of an MMR gene is generally sufficient to maintain normal MMR function. For colon cancer to develop in patients with Lynch syndrome, a second somatic event (in addition to the vertically transmitted mutant allele) must occur in the wild-type allele of a colonocyte. This makes both copies of an MMR gene completely inactive55 and causes the hypermutable phenotype seen in tumors in patients with Lynch syndrome. Similarly, both alleles in sporadic colorectal tumor with MSI are inactivated by biallelic hypermethylation of hMLH1.52–54 When a physical deformation remains in the newly replicated DNA double helix (caused by the mispairing of nucleotides or due to slippage and looping at microsatellite loci), a complex termed hMutSα (a heterodimer of hMSH2 and hMSH6 proteins) or hMutSβ (a heterodimer of hMSH2 and hMSH3) identifies the error and binds the DNA at this site.56–59 In particular, based on studies of the bacterial and yeast MMR proteins, hMutSα appears to recognize and bind single–base pair mismatches and loops of DNA containing <2 looped nucleotides, while hMutSβ recognizes loops of DNA containing >2 or more nucleotides60,61 and may repair DNA loops up to 16 nucleotides in length,62 with larger loops repaired by a non-MMR mechanism.63 Subsequently, hMutSα or hMutSβ recruit the hMutLα complex, a heterodimer of hMLH1 and hPMS2 proteins, which targets the newly synthesized, daughter DNA strand for “long patch” excision repair (Figure 2). In vitro nicks in the DNA direct the strand specificity of repair, which can proceed in either the 5′ → 3′ or 3′ → 5′ direction, depending on the location of the nick relative to the mispair.40,41,62,64 The MMR proteins hydrolyze adenosine triphosphate (ATP) during the recognition and repair process.64 The hydrolysis of ATP is believed to act as a “molecular switch” in that the MMR proteins are turned “on” when in the adenosine diphosphate (ADP)-bound form and bound to the DNA double strand and “off” when in the ATP-bound form as a mode to dissociate from the strand.65 Initially based on biochemical studies and later corroborated by the crystallographic structure of the bacterial “S” MMR protein, Gradia et al proposed a “sliding” clamp model in which hMutSα slides along the DNA double strand for finite distances until it exchanges ADP for ATP or encounters a mismatched nucleotide.64 Loss of any of the components of the MMR system will inactivate or attenuate repair, and the heterodimers help stabilize the individual partnered proteins. For instance, when hMSH2 is missing, hMSH6 and hMSH3 become unstable as individual proteins.51 When either hMSH6 or hMSH3 is absent, the other protein increases its expression for interaction with hMSH2.51 It remains unclear how hPMS1 (binds to hMLH1 to form hMutLβ) or hMLH3 (binds to hMLH1 to form hMutLγ) interacts within the entire complex to effect repair, although hMutLγ may play a repair role during meiosis.66,67
Figure 2.

Schematic of DNA MMR. The MMR heterodimer hMSH2-hMSH6 (hMutSα) recognizes single nucleotide mispairs and will bind to DNA as a sliding clamp. The heterodimer hMLH1-hPMS2 (hMutLα) then binds to hMutSα to eventually guide an exonuclease to remove several bases from the newly synthesized DNA strand, with eventual resynthesis of DNA with the correct base pairing. At microsatellite sequences, insertion-deletion loops of one nucleotide are typically recognized by hMutSβ (upper panel), but larger insertion-deletion lops are recognized by the heterodimer hMSH2-hMSH3 (hMutSβ) (lower panel), followed by binding by hMutLα, excision, and resynthesis as described previously. The roles of hMLH1-hPMS1 and hMLH1-hMLH3 in human MMR function are not entirely clear. When the MutS complexes bind DNA, they exchange ADP for ATP. For the MMR proteins to be released from DNA, ATP is hydrolyzed to ADP.
DNA MMR requires the presence of a number of proteins that aid in its function. In E coli, in addition to the MMR proteins MutS and MutL, repair involves MutH (aids in strand discrimination), UvrD (a DNA helicase), ExoI, ExoVII, ExoX or RecJ (involved in DNA excision), Pol III holoenzyme (for DNA resynthesis), SSB (single-stranded DNA binding and protection), and DNA ligase (for nick ligation).66 Similarly, the human DNA MMR system has a number of components in addition to the core MMR proteins that effect repair. These proteins have been primarily identified by homology to E coli, although there is no currently identified human homologue for MutH or UvrD. Exonuclease 1, a 5′ → 3′ exonuclease, is involved in both 5′ and 3′ directed MMR and excises mismatched DNA.68–70 Human polδ directs DNA resynthesis, and proliferating cellular nuclear antigen interacts with hMSH2 and hMLH1 (as well as hMSH6 and hMSH3) to help localize hMutSα and hMutSβ to newly replicated DNA for initiation of MMR and DNA resynthesis.71–76 hMutLα possesses a proliferating cellular nuclear antigen/replication factor C–dependent endonuclease activity that plays a key role in 3′ nick-directed MMR that involves exonuclease 1.77 The single-strand DNA binding replication protein A (RPA) is involved in all stages of MMR. RPA binds to nicked heteroduplex DNA before hMutSα and hMutSβ, stimulates mismatched-provoked excision, protects single-strand DNA gapped regions generated during excision, and facilitates DNA resynthesis.78 RPA also becomes phosphorylated after polδ becomes active to close DNA gaps, which reduces the affinity of RPA for DNA.79 The high mobility group box 1 protein can interact with hMSH2 and hMSH6, binds to mismatches, and has DNA unwinding activity to provoke mismatched excision.80 Human DNA ligase 1 directs nicked DNA ligation.73 Overall, the MMR process is highly conserved from E coli to humans to include the components of MMR, substrate specificity, nick-directed strand specificity, and the bidirectionality for repair.
Tumorigenesis With Loss of DNA MMR
It is believed that certain key tumor suppressors drive the pathogenesis of MSI tumors and that these genes include a unique complement of genes that are commonly mutated in MSI tumors but not in CIN tumors. Rarely, microsatellite sequences are present in the coding region (exons) of critical growth regulatory genes, and because of the tumor-promoting effects of their mutational inactivation, these genes are the ones selected for when mutation in MSI tumors. Mutation of the coding microsatellite occurs with defective MMR, resulting in length changes in the microsatellite within the gene producing a frameshift mutation that renders the protein inactive. An analysis of repetitive sequences in tumor tissue led to the identification of inactivated genes in these tumors. For example, a repeat of 10 adenines in the TGFBR2 gene undergoes frameshift mutation in ~85% of colorectal tumors with MSI, inactivating this receptor36,81 and causing the tumor cells to escape the growth-suppressive effects of the ligand transforming growth factor (TGF)-β1. It is interesting to note that 15% of tumors with MSI carry monoallelic frameshift mutations of IGF2R at its polyguanine sequence, which may uncouple TGF-β1 growth suppression because the ligand cannot be activated by cleavage from its latent peptide.82 Another targeted gene is BAX, a member of the BCL2 gene family. BAX heterodimerizes with BCL2 within the cell, and the relative amounts of the heterodimer determine the commitment of the cell to programmed cell death. BAX contains a polyguanine (G)8 tract that shows monoallelic frameshift mutations in 50% of colorectal cancers with MSI.83,84 Mutation of BAX (by diminishing the ratio of BAX to BCL2) prevents programmed cell death, helping to immortalize cells.83,84 Mutation of TGFBR2 and BAX appear to be late in the adenoma to carcinoma progression because these mutations occur in high-grade dysplasia at the interface of defining malignancy.26,85 Additionally, mutations in TGFBR2 from MSI tumors of patients with stage III colon cancer are associated with improved survival.86 Other genetic targets identified include the MMR genes hMSH3 and hMSH6, which may broaden and accelerate the accumulation of mutations87 (Figure 3 and Table 2). A comprehensive search using a genetic database for coding microsatellites and subsequent colorectal tumor analysis revealed 9 genes that were mutated in more than 20% of the tumors.88 In addition to TGFBR2, BAX, and hMSH3, these include ACVR2 (the gene for the activin receptor type 2), SEC63 (human homologue of a yeast DnaJ-like endoplasmic reticulum translocon component protein gene), AIM2 (an interferon-inducible gene), reduced nicotin-amide adenine dinucleotide–ubiquinone oxidoreductase B14.5B subunit gene, KIAA0977 (probable human homologue of the mouse embryonal protein cordon-bleu), and PA2G4/EBP1 (homologue of the mouse cell cycle protein p38-2G4).88 Subsequently, ACVR2 has been shown to be mutated at an exon 10 polyadenine tract in the majority of microsatellite unstable colon cancers with loss of ACVR2 protein,37 and the loss of ACVR2 expression correlates with an increase in local tumor growth.89 Further, reconstitution of mutated ACVR2 leads to restitution of growth inhibitory signaling in colon cancer cells.90 The role of the other frequently mutated genes in the pathogenesis of tumors with MSI is not currently known.
Figure 3.
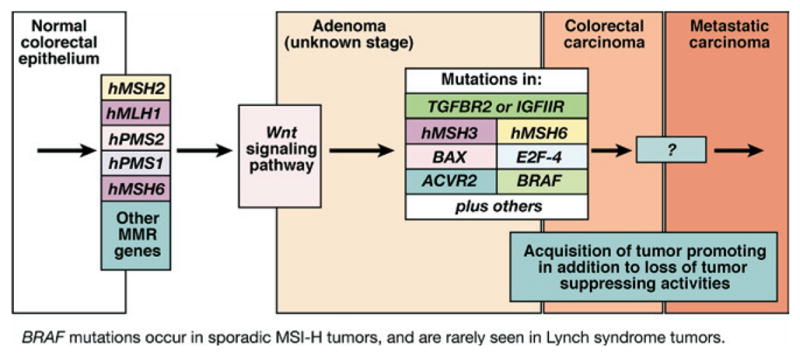
Progression of colorectal tumors with MSI. MSI tumors, whether sporadic or from patients with Lynch syndrome, lose MMR function early in the polyp → cancer progression sequence. Sporadic tumors almost uniformly lose MMR function due to hypermethylation of the promoter of hMLH1, whereas patients with Lynch syndrome have a germline mutation in one of the MMR genes. It is generally believed that Wnt signaling, the gatekeeper for colorectal neoplasia, is affected, but the full mechanisms are not clear and include mutation of CTNNB1, the gene encoding β-catenin in some cases of Lynch syndrome. The serrated adenoma may be one histologic form for MSI colorectal tumors. In the milieu of MMR absence, a hypermutable phenotype develops in which multiple mutations occur in DNA. Although most mutations occur in noncoding sequences such as intronic DNA microsatellites, certain genes such as TGFBR2, ACVR2, BAX, hMSH3, hMSH6, and others that have coding microsatellite sequences become frameshifted in the absence of DNA MMR. These mutations help drive the progression of the tumor. BRAF mutations are principally found in sporadic tumors with MSI and not tumors from patients with Lynch syndrome and can be used to differentiate these 2 groups of tumors. Notably, in MSI and non-MSI tumors, signaling through the TGF-β superfamily is altered to favor tumor promotion in a not yet fully understood fashion.
Table 2.
Some Targeted Genes in MMR-Deficient (or MSI) Colorectal Cancers and Their Reported Mutation Frequency
| Gene | Chromosome | Microsatellite tract | MSI colorectal cancer mutation frequency (%) | Normal function |
|---|---|---|---|---|
| TGFBR2 | 3p22 | A10 | 90 | TGF-β signaling |
| ACVR2 | 2q22-23 | A8 and A8 | 86 | Activin signaling |
| IGFIIR | 6q26-27 | G8 | 10 | Insulin-like growth factor and TGF-β signaling |
| BAX | 19q13.3-q13.4 | G8 | 50 | Apoptosis |
| hMSH3 | 5q11-q12 | A8 | 50 | DNA MMR |
| hMSH6 | 2p16 | C8 | 33 | DNA MMR |
| E2F-4 | 16q21-q22 | (CAG)13 | 65 | Cell cycle control |
| PTEN | 10q23.3 | A6 and A6 | 19–34 | Growth regulation |
| MBD4 (MED1) | 3q21-q22 | A10 | 40 | DNA repair and binding to methylated DNA |
| TCF4 | 10q25.3 | A9 | 39 | Growth regulation |
| CHK1 | 11q22-q23 | A9 | 10 | G2 cell cycle checkpoint |
| STK11 (LKB1) | 19p13.3 | C6 | <2 | Signal transduction |
| BLM | 15q26.1 | A9 | <18 | Chromosome stability/DNA repair; helicase |
| Caspase-5 (ICErel-III) | 11q22.2-22.3 | A10 | 62 | Apoptosis |
| CDX2 | 13q12.3 | C6 and G6 | <2 | Homeobox protein |
| TBP | 6q27 | (CAG)19 and (CAG)16 | 83 | TATA binding protein |
| RIZ | 1p36 | A8 and A9 | 26 | Interacts with RB |
| hRAD50 | 5q31 | A9 and A8 | 31 | DNA repair |
| SEC63 | 6q21 | A10 | 49 | ER chaperone protein |
| AIM2 | 1q22 | A10 | 48 | Interferon-inducible protein |
The BRAF gene encoding a downstream component from KRAS in the RAS/RAF/MAPK pathway is also often mutated in sporadic MSI tumors,91 although the mechanism is unknown because BRAF does not contain a coding microsatellite sequence. Mutations in BRAF are usually V600E missense mutations, which activate the protein to become an incessant signaling molecule.91
A number of clinicopathologic features of MSI colorectal tumors segregate it from tumors with CIN (although many of these features match CIMP tumors due to the overlap with hypermethylated hMLH1). These include correlation with the tumor’s location in the proximal (right) colon and an inverse relation to allelic loss.34,92,93 In addition, MSI colorectal tumors tend to be diploid, tend to possess a mucinous histology, have a better overall survival compared with microsatellite stable tumors,94 and have a lymphocytic reaction surrounding the tumor. The mechanisms responsible for the immune cell histologic differences are not fully understood but may involve truncated immunogenic proteins resulting from frameshift mutations.95 In comparison, CIN colorectal tumors are aneuploid and have an overall worse prognosis compared with MSI tumors.96,97
The Relationship Between Chemotherapy Responsiveness and DNA MMR Activity
In addition to its role in recognizing and directing repair of polymerase mistakes within DNA, the human MMR system is also capable of recognizing certain DNA adducts caused by exogenous alkylation damage.98–100 O6-methylguanine adducts are recognized by the MMR system and result in a G2/M cell cycle arrest and cell death.98 Similarly, when 6-thioguanine is incorporated into the DNA of MMR-proficient cells, a G2/M cell cycle arrest is induced.99 Both O6-methylguanine and 6-thioguanine are believed to be recognized by the MMR system because of mispairing with thymidine (or cytosine) with the altered nucleotide on the newly synthesized DNA strand. It is unclear how the selectivity of recognition of adducts occurs, although some of the selectivity may be mediated through effects on interstrand hydrogen bonding. For instance, 8-azoguanine does not interfere with classic Watson–Crick hydrogen bonding, which is in contrast to 6-thioguanine and O6-methylguanine, and thus it may escape recognition because of this. Furthermore, the MMR proteins can recognize specific intrastrand and interstrand cross-links induced by cisplatin or carboplatin and not those by its analogues. Because both cisplatin and its substituted amine analogues would be expected to cause distortion of double-stranded DNA, it has been suggested that lack of recognition of the cisplatin analogues by the MMR system might be due to steric hindrance of binding by the hMSH2 protein to the altered DNA.100,101
Notably, 5-fluorouracil (5-FU), the central agent used for the medical treatment of patients with advanced colorectal cancer, is recognized by the MMR system despite its inability to deform DNA.102,103 Because of the ability of 5-FU to inhibit thymidylate synthase, the enzyme required for thymidine triphosphate synthesis, DNA is subsequently synthesized with uridine triphosphate or 5-fluorodeoxyuridine triphosphate when thymidine triphosphate is depleted, allowing incorporation of the fluoropyrimidine into DNA.104 5-FU will selectively kill cells with intact MMR compared with cells with deficient MMR,102 and MMR-deficient cells resulting from hypermethylated hMLH1 that are 5-FU resistant become sensitive to 5-FU when hMLH1 is demethylated in vitro.105 Biochemical experiments have confirmed that human MMR proteins, particularly hMutSα, bind to 5-FU incorporated into DNA.103,106 Some studies of patient survival have shown a similar effect of 5-FU when separated by MSI status. Patients with stage II and III sporadic colorectal cancer with MSI tumors do not show a survival benefit with 5-FU therapy as compared with patients with MMR-proficient tumors in retrospective and prospective studies.107–109 Similarly, stage III patients with Lynch syndrome do not demonstrate a 5-year survival benefit with 5-FU treatment over untreated patients.110 These observations raise the possibility that MSI may be useful as a predictive marker for 5-FU treatment as well as a prognostic marker for the natural history of this molecular class of tumors.
It is not known how MMR signals the demise of the cancer cell after recognition of 5-FU incorporated into DNA. Data from O6-methylguanine–induced MMR recognition indicate p53 becomes phosphorylated at its serine residues 15 and 392,111 and this modulation may be one mechanism for p53 activation. The observed G2/M cell cycle arrest involves the ATM and Rad3-related (ATR) kinase, as demonstrated by the ablation of ATR (or inhibition of the ATR-activated checkpoint kinase CHK1) attenuating the cell cycle arrest.112 ATR can phosphorylate serine 15 of p53 after gamma irradiation or UV exposure.113 ATR-induced CHK1 kinase (or the ATM-induced CHK2 kinase) can phosphorylate cdc25A, a cell cycle phosphatase that is degraded upon its phosphorylation. Cdc25A controls the activation of CDK1 and CDK2 kinases that regulate the G1, intra-S, and G2 phases of the cell cycle.114 The G2 checkpoint activation was accompanied by the formation of nuclear foci containing the repair protein ATR as well as RPA.112 Taken together, ATR activation after O6-methylguanine formation is dependent on the presence of DNA MMR and posttranslationally modifies by phosphorylation the downstream targets CHK1 (and perhaps p53) to effect the proteasome-assisted destruction of cdc25A to cause cell cycle arrest. Apoptotic signaling by MMR recognition of O6-methylguanine lesions appears to be mediated by mitochondrial signaling and eventual caspase activation.115 The activation of cell death after the recognition of O6-methylguanine (1) is dependent on the presence of hMutSα, (2) can temporarily be blocked by overexpression of bcl-2 and bcl-XL, negative regulators of mitochondrial-regulated apoptosis, and (3) is not dependent on death receptor signaling.115 Neither the cell cycle arrest nor the cell death events demonstrate how DNA MMR responds to the O6-methylguanine lesion but highlight potential downstream effectors to prevent replication of damaged DNA.
Overall, the MMR system and other repair mechanisms such as BER have been implicated in mediating the response of cancer cells to chemotherapy. This has raised the possibility that the assessment of expression or activity of these proteins in colorectal cancer might be useful as a predictive marker for the response of patients with colorectal cancer to specific therapies and as a guide to the selection of optimal therapy.
CIN
Overview of CIN and Colon Cancer
CIN is the most common type of genomic instability observed in colon cancer and occurs in 80%–85% of colorectal tumors. Several forms of CIN can be observed in cancer in general. Recently proposed categories of genomic instability include the following: (1) subtle sequence changes, including base substitutions, deletions, or insertions as well as MSI; (2) alterations in chromosome number (aneuploidy, also termed CIN), (3) chromosome rearrangements, and (4) gene amplification.3,116 The genome of colon cancers, like many solid tumors, is often marked by a complex pattern of chromosome translocations that appears nearly random and is mediated at least in part by inactivation of proteins involved in homologous recombination–mediated events (eg, sister chromatid exchange and homologous recombination between nonallelic repetitive DNA sequences) and in nonhomologous end-joining events.116,117 This form of genomic instability, “translocation instability,” is likely mediated by proteins involved in double-strand DNA break repair, single-strand DNA break repair, and DNA replication, as well as by mechanisms that cause aneuploidy, which include mitotic spindle checkpoint regulators as well as proteins that regulate mitotic chromosome transmission.116 However, the role of chromosome rearrangements in solid tumors, like colon cancer, is still being defined.3,117,118 Finally, gene amplification occurs infrequently in colon cancer but does appear to play a role in driving stage transitions in at least some colon cancers.119,120
Despite the high frequency of CIN in colon cancer and the fact that aneuploidy is recognized as a hallmark of cancer, our understanding of the specific molecular events that result in this form of genomic instability is remarkably incomplete. Although there are clear examples of specific alterations of proteins that maintain DNA fidelity, causing CIN and predisposing to tumor formation, it is still debatable as to whether aneuploidy is merely a tolerated state rather than a fundamental pathogenic process of tumorigenesis. The identification of specific patterns of chromosome gains and losses that occur during the colon adenoma to carcinoma sequence and the demonstration that CIN is an early event in tumor formation that increases with tumor progression is consistent with the idea that CIN is a pathogenetic phenomenon in colon cancer (Figure 4).28,121,122 Furthermore, studies of mice heterozygous for the centromere-linked, kenisin-like motor protein CENP-E show that the cells from these mice missegregate chromosomes during mitosis due to a weakened mitotic checkpoint and develop lymphomas and lung adenomas at increased rates. Importantly, these mice have an intact DNA damage response, which implicates the occurrence of aneuploidy itself as the main factor leading to the predisposition to neoplasms.123
Figure 4.

Progression of colorectal tumors with CIN. The hallmark of the CIN pathway is aneuploidy. Initiation of neoplasia in this pathway occurs with interruption of components of the Wnt signaling pathway, including somatic mutation in one allele and loss of heterozygosity of the second normal allele of the APC gene, and is seen in dysplastic ACF histologically. Progression is then driven by successive waves of cellular clonal expansion that acquire enhanced growth characteristics and include mutational activation of the proto-oncogene KRAS and mutation of TP53 with subsequent loss of heterozygosity of the normal remaining TP53 allele to allow carcinoma formation. In some colorectal tumors, mutational activation of PIK3CA occurs late in the adenoma-carcinoma sequence. Several TGF-β signaling molecules are also affected in CIN progression, including mutations of the kinase domain of TGFBR2 and loss of heterozygosity at chromosome 18q, the site of SMAD4 and SMAD2. Several genes are believed to be involved in metastasis and include gene amplification of PRL3.
In addition and importantly for our understanding of CIN in colon cancer, Lengauer et al have shown that aneuploidy is indeed the result of an abnormally high rate of CIN that persists throughout the life of the tumor.3 Using cell fusion and chromosome transfer techniques, Lengauer et al also proved that the aneuploid state does not cause CIN and that CIN is an autosomal dominant trait, suggesting it can arise from gain-of-function mutations. Despite this insight into CIN, the mechanism(s) responsible for CIN has been identified in only a subset of colon cancers. TP53 mutations were initially suspected to be the leading cause of genomic instability in colon cancer, but it appears more likely that only specific TP53 mutations cause CIN and the remainder of the mutant forms of TP53 likely affect other processes such as apoptosis and create a permissive state that allows CIN to occur but does not actually cause the CIN.124
Mechanisms for CIN
In light of the appreciation of CIN as being a hallmark feature of the majority of colon cancers and of the likely contributory role of genomic instability in colon carcinogenesis, there has been intense interest in identifying the mechanisms responsible for the CIN. In an analogous approach to that used to determine the basis for MSI through the study of unicellular organisms that are MMR deficient, the mechanisms responsible for CIN are being first unraveled through the study of yeast that show CIN in order to identify proteins that regulate DNA fidelity during DNA replication and in response to genotoxic stress. These studies have proven to be extremely challenging because of the vast number of genes that regulate the fidelity of chromosomes. In fact, it is already appreciated that the list of candidate genes numbers more than 100 and includes genes used in kinetochore structure and function, centrosome and microtubule formation and behavior, chromosome condensation, sister chromatid cohesion, and cell cycle checkpoint control.24 Aneuploid cancers tend to show cytologic abnormalities during mitosis, including abnormal centrosome numbers, multipolar spindles, and lagging chromosomes, implicating deregulation of the processes that mediate the mitotic spindle checkpoint as a cause of CIN.24,125–127 In addition to inactivation of proteins that regulate the mitotic spindle checkpoint, inactivation of proteins that regulate DNA damage checkpoints, chromosome metabolism, and centrosome function have been directly shown in in vitro systems to influence chromosomal stability. Consequently, proteins involved in the regulation of the mitotic spindle checkpoints and DNA replication checkpoints have been the most closely scrutinized as potential causes of CIN in cancer (Figure 5).
Figure 5.

Schematic diagram of cell cycle checkpoints in relation to cell cycle phases. The DNA damage, DNA replication, and mitotic/spindle-kinetochore checkpoints are shown with a corresponding list of a subset of human and S cerevisiae genes that function at each of the checkpoints. The sensor proteins and downstream signal transduction and effector proteins are listed for the DNA replication checkpoints in S phase. The proteins listed at the other checkpoints operate as sensor, transducers, or effector proteins.
In light of the common occurrence of mutations in APC, KRAS, PIK3CA, SMAD4, TP53, and other CIN genes, assessment of the role of mutations in these genes as contributing factors to CIN in colorectal cancers has been performed.128,129 To date, there are no convincing data that mutations in any of these genes provide more than a permissive role for CIN, although there is a tight association between mutant APC and CIN mutant TP53 and CIN.24,130–132 Interestingly, although TP53 mutations have been implicated as the cause of some forms of genomic instability, no simple correlation between p53 mutation and the CIN phenotype was shown by Lengauer et al or by others.122,130 It remains to be determined if any of the other genes recently found to be mutated in colorectal cancer may play a more causal role in CIN.128
An important implication of gaining an improved understanding of the mechanisms responsible for mediating different forms of CIN is that there appears to be mechanism-specific susceptibilities to different classes of chemotherapy. Thus, identifying the mechanisms responsible for CIN in specific colorectal cancers is predicted to lead to a molecular typing strategy in which determination of the mechanism altered in the cancer will ultimately lead to the selection of chemotherapeutic agents that will be optimal for an individual’s specific tumor (ie, “personalized medicine”). The majority of the discussion of CIN in colorectal cancer that follows will focus on the possible mechanisms responsible for CIN in colorectal cancer.
BER defects and CIN
Inactivation of a second “DNA caretaker” mechanism, the BER system, is found in a subset of colorectal cancer cell lines and appears to lead to a predisposition to point mutations that contribute to colon cancer formation by causing genomic instability at the level of the base pair. Inactivation of one of the BER genes, MYH, is a cause of an autosomal recessive form of adenomatous polyposis, called the MAP syndrome.133 Germline mutations in MYH, which encodes for a protein involved in BER, is the cause of adenomatous polyposis in up to 5%–10% of individuals who have an adenomatous polyposis syndrome. MYH germline mutations were discovered as a cause of adenomatous polyposis when investigators identified an excessive number of somatic G:C → A:T mutations in neoplasms of people with adenomatous polyposis but who had no detectable germline mutations in APC.8,134,135 This type of mutation is commonly a consequence of oxidative damage to DNA that results in 8-oxo-7,8-dihydro2′ deoxyguanosine, which is one of the most stable deleterious products of oxidative DNA damage.133,136 The BER system is responsible for repairing this form of DNA damage, which led these investigators to assess candidate genes involved in this process: OGG1, MTHF1, and MYH. This assessment revealed biallelic germline MYH mutations in a subset of people with adenomatous polyposis but who did not have germline mutations in APC. The most common mutations are Tyr165Cys and Gly382Asp, which account for 82% of the mutant alleles detected to date.135 Despite the role of germline mutations in MYH as a cause of a colorectal cancer family syndrome, somatic MYH mutations are not common in sporadic colorectal cancer. A study of 1042 unselected patients with colorectal cancer in Finland revealed no somatic MYH mutations.133,137 Of interest, the tumors arising in the setting of biallelic MYH germline mutations do not show differences in the frequency of TP53, SMAD4, or TGFBR2 mutations but do appear to show CIN, suggesting they have a unique molecular pathogenesis compared with sporadic colorectal cancer.138,139 The role of MYH in causing CIN is not clear, but an analysis of adenomas arising in individuals with MAP compared with people with familial adenomatous polyposis (FAP) with array comparative genomic hybridization revealed chromosomal gains and losses in MAP and FAP adenomas. Cardoso et al found in 80% and 60% of MAP and FAP adenomas, respectively, that there were aneuploid changes, with frequent losses at chromosome 1p, 17, 19, and 22 and gains affecting chromosomes 7 and 13 in both groups of tumors.138 The discovery of MYH germline mutations in people with a hereditary colorectal cancer syndrome provides more evidence for the importance of genomic instability in cancer formation.
Mitotic spindle checkpoint genes and CIN
The proteins that regulate spindle-kinetochore interactions during mitosis are the best demonstrated to date to have a role in CIN in human cancers. Studies in model organisms have shown that the proteins encoded by the mitotic spindle checkpoint genes that control sister chromatid separation at the metaphase-anaphase transition can cause CIN. These genes are highly conserved, suggesting that they are a plausible functional class of genes that will be shown to be a common cause of CIN in human cancers.15,139 The mad and bub genes, in particular, are highly conserved key components that regulate the mitotic spindle checkpoint, and these genes have already been shown to be deregulated in a subset of colon cancers. The MAD and BUB gene products appear to operate as checkpoint sensors and signal transducers.15 Activation of MAD and BUB causes cell cycle arrest through the inhibition of the anaphase promoting complex/C (APC/C), a large ubiquitin-protein ligase. The APC/C normally becomes active at the metaphase-anaphase transition, resulting in the degradation of securin, a protein that inhibits anaphase. Securin interacts with separins, a class of caspase-related proteases that regulate cohesins, which create physical links between sister chromatids. MAD2 directly binds to the APC/C and blocks the APC/C-dependent ubiquitin-mediated degradation of securin. There is also a second MAD2-independent mechanism that involves BUBR1 and BUB3.140 In addition to MAD and BUB, there are additional, incompletely characterized factors involved in the regulation of the APC/C. For instance, studies to date indicate that the securin-separin complex is regulated by an autofeedback loop that prevents premature degradation of securin and that other proteins such as polo kinases (PLK) mediate the activity of this complex.141 All of these mechanisms serve to delay the progression to anaphase to allow enough time for the appropriate alignment of chromosomes on the metaphase plate so that the chromosomes appropriately sort during cell division. Thus, all of these mechanisms are candidates for mediating CIN in colorectal cancer.
Data showing mitotic checkpoint gene deregulation could cause CIN in colon cancer came from a landmark study by Cahill et al in 1998. These investigators found mutations in 2 members of this class of genes after recognizing that aneuploid colon cancer cell lines behave in a pattern consistent with having a spindle checkpoint defect.15 Mutations in 2 genes that control the human mitotic checkpoint, BUB1 and BUBR1, were found in a subset of CIN colon cancer cell lines. The mutant BUB1 functions in a dominant negative fashion, as would be predicted from previously published studies involving cell fusion experiments with CIN cancer cell lines.122 Mutations in other human mitotic checkpoint genes, MAD2 and MAD1, have been identified in breast cancer and leukemia, respectively, suggesting mutations in other genes that control chromosomal stability remain to be found in colorectal cancers.142,143 Furthermore, PTTG, the “pituitary tumor-transforming gene” and the human homologue of securin, is overexpressed in a variety of tumor types, including colon cancer.144–146 Importantly, MAD2, BUB1, and BUBR1 mutations generate the types of chromosome loss seen in naturally occurring human cancers, which has not been the case in studies of other candidate CIN-regulating genes such as components of the anaphase-promoting complex.15,147 Other genes involved in kinetochore function that have been found to be mutated in colon cancer include CDC4, hROD, hZW10, hZWILCH, and DING.125,148 Furthermore, some proteins that regulate kinetochore function have been found to be overexpressed in colon cancer, including CENP-A, CENP-H, HEC1, Aurora-B (AIM1), and INCENP.125,149–152 Of note, despite the initial success of this candidate approach, mutation analysis of other mitotic spindle checkpoint genes, MAD1L1, MAD2, MAD2B, BUB3, MPS1L1, and CDC20, has not revealed any mutations in a panel of 19 aneuploid colorectal cancer cell lines.15
Centrosome regulation and CIN
In addition to the deregulation of proteins that directly govern the mitotic spindle checkpoint being a candidate mechanism for CIN, abnormal centrosome number and function have been proposed as a candidate mechanism for aneuploidy since Boveri first observed centrosomes more than 100 years ago.153 Most solid cancers are characterized by centrosome amplification as well as by aneuploidy, and there is a strong association between abnormal centrosome number and aneuploidy in cancers.154–156 Because the centrosome plays a central role in chromosome segregation during mitosis, the centrosome amplification observed in cancers is postulated to increase the frequency of abnormal mitoses and chromosome missegregation.157–160 More concrete evidence of abnormal centrosome function causing CIN in colon cancer became available as the result of assessment of the genes in a locus on chromosome 20q13, which is frequently amplified in a variety of cancers including breast, colon, and pancreatic cancer.161 Sen et al cloned STK15/BTAK/ARK1/aurora 2, a centrosome-associated serine-threonine kinase, from this region and detected overexpression in breast cancer cell lines.162 Subsequent studies revealed STK15 amplification and overexpression in colon cancer cell lines as well and demonstrated that overexpression of this gene can induce aneuploidy in NIH3T3 cells.161,163 Studies in Drosophila and Saccharomyces cerevisiae indicate that STK15 has a role in centrosome maturation, spindle assembly, and chromosome segregation and that its over-expression may lead to abnormal mitotic spindle assembly and CIN.153,164,165 Furthermore, studies in prostate and breast cancer have shown an association between aneuploidy, abnormal centrosome number, and clinically aggressive disease.158,159 Thus, STK15 amplification appears to be a promising potential cause of CIN. In addition to STK15 playing a role in abnormal centrosome function in colon cancer, 2 other cell cycle–regulated proteins, polo-like kinase 2 and 4 (PLK2 and PLK4), are overexpressed in some cases of colon cancer and have been implicated in aneuploidy and abnormal centrosome function.166 Beyond these proteins that directly interact with centrosomes, many proteins that directly or indirectly regulate the number and/or function of centromeres are abnormal in colon cancer. Some proteins that are involved with regulating centrosome number, including nucleophosmin, MPS1, and mortalin, are targets of transcriptional or posttranslational modification by cdk2/cyclin E and are overexpressed in cancer.157,167,168 It is also predicted that overactivation of CDK4/CDK6 and cyclin D contributes to tumorigenesis by favoring the generation of amplified centrosomes.169 The results of these studies create a strong argument that the abnormal regulation of centrosomes is a cause of CIN in colon cancer, but further studies are needed to develop a better understanding of the role of centrosome amplification and centrosome-associated kinase activity in CIN in primary human colon cancer.
DNA checkpoint genes and CIN
Genomic stability is maintained not only by checkpoints that regulate centrosome function and number, mitosis, and sister chromatid separation but also by checkpoints that regulate the maintenance of DNA fidelity in response to genotoxic stress. In fact, cell cycle checkpoints were originally recognized as pathways that mediate cell cycle progression in response to DNA damage. Mutations that disrupt replication checkpoint genes, such as rfc5-1, dpb11-1, mec1Δ, ddc2Δ, and dun1Δ, substantially increase the rate of genome rearrangements.170 These DNA damage checkpoints are traditionally considered in 2 classes: (1) the DNA damage checkpoints that recognize and respond to DNA damage from genotoxic agents and (2) the DNA replication checkpoints that monitor the fidelity of replicated DNA.171 The proteins involved in the DNA checkpoints act in one or more of the following roles: (1) sensors of DNA damage, (2) transducers of the DNA damage signal to the effector proteins, or (3) effectors that regulate cell cycle arrest/progression and DNA repair. The proteins are also commonly characterized based on the type of DNA damage they most commonly repair.172 A number of these DNA repair proteins have been identified in yeast, and some have already been shown to have a role in human cancer, including TP53, ATM, the ATM-related gene ATR, BRCA1 and BRCA2, which interact with the human Rad51 homologue, and hRAD17173–177 (Table 3). In yeast, Mec1 and Tel1, which are homologous to ATM and ATR, appear to operate as the transducer/effector components of an important DNA damage-signaling network that regulates the cellular response to a variety of DNA damage events. These proteins belong to a family of protein kinases termed PIKKs (phosphatidyl-inositol 3-kinase-like protein kinase), and they phosphorylate several key effector proteins (eg, Rad53p/hCHK2, Chk1p/hCHK1) in response to DNA damage. These effector proteins require additional factors to form a pentameric replication factor C–like complex that can associate with sites of DNA damage and elicit the appropriate cellular response.178 These DNA replication checkpoints operate during S phase to either arrest or slow replication in response to DNA damage and consist of both an S-phase checkpoint as well as an intra–S-phase checkpoint. These checkpoints are redundant, but if both are inactivated there is a synergistic increase in the rate of genomic rearrangements. The kinds of rearrangements observed in primary human cancer are similar to those seen when these checkpoint proteins are inactivated in yeast, providing indirect evidence for a role of the replication checkpoints in the genomic instability found in colon cancer.179 Of all these checkpoint proteins, TP53 has been most clearly shown to have a role in human colon cancer and to at least play a permissive role for the development of CIN.
Table 3.
DNA Damage Repair Genes and CIN
| Human gene | Protein function | Gene alteration | Syndrome |
|---|---|---|---|
| BRCA1 | Sensor: BRCT containing | BRCA1: germline mutation BRCA2: germline mutation |
Hereditary breast-ovarian cancer |
| MRE11 | Sensor: DSB recognition/repair | Germline mutation Somatic mutation in MSI colon cancer |
Ataxia-telangiectasia–like disorder |
| BLM | Sensor: replication proteins | BLMAsh: increased risk of colon cancer | Bloom syndrome |
| WRN | Aberrant DNA methylation | Werner syndrome | |
| RTS | Rothmund–Thomson syndrome | ||
| ATM | Transducer: PI3-kinase (PIKK) | Germline mutation | Ataxia-telangiectasia |
| TP53 | Transducer: effector kinase | TP53: germline mutation | Li–Fraumeni syndrome |
| Somatic mutations | |||
| CHK2 | Transducer: effector kinase | Germline mutation | Li–Fraumeni–like syndrome |
| MYH | BER | MYH: germline mutation | MAP |
| MGMT | DNA damage repair | Aberrant DNA methylation | |
| BUB1 | Kinetochore regulation | Somatic mutation | |
| BUBR1 | Somatic mutation | ||
| MAD2 | Abnormal expression | ||
| hRod | Rare somatic mutation | ||
| hZw10 | Rare somatic mutation | ||
| hZwilch | Rare somatic mutation | ||
| CENP-A | Gene overexpression | ||
| CENP-H | Gene overexpression | ||
| HEC1 | Gene overexpression | ||
| AIM1 | Gene overexpression | ||
| INCENP | Gene overexpression | ||
| Ku70 | Nonhomologous end-joining | Loss of expression | |
| Ku86 | Loss of expression in rectal cancer | ||
| AURKA | Chromosome segregation defects | Gene amplification and overexpression | |
| TERC | Telomere regulation | Overexpression | |
| CCNE1 | Cell cycle regulation | Overexpression | |
| FBXW7/CDC4 | |||
| CDC25A ? | |||
| CDKN2A | Aberrant DNA methylation |
NOTE. Some genes that have been identified to cause cancer family syndromes or to be deregulated in human colon cancer. BRCT, Brca1 carboxy terminus; DSB, double-strand break.
In addition, mutations or amplification of other DNA checkpoint genes known to cause experimental forms of CIN have been identified in cancer, including hBUB1, ATM, ATR, BRCA1, BRCA2, STK15, PLK1, and CDC4.15,161,163,174–176,180 More recently, Wang et al used computational analysis to identify >1000 possible genes that could cause genomic instability based on their homology to genes in yeast and Drosophila melanogaster. They performed mutation analysis of 100 of these genes, which were selected based on the strength of the phenotypes observed in the model systems and on sequence similarity to the human homologue, in 24 early-passage colon cancer cell lines and found 19 somatic mutations in 5 genes.16 MRE11, which is involved in double-strand DNA break repair, is one of the genes that was identified in this screen for regulators of CIN in human cancers. MRE11 is commonly mutated in MSI colon cancers, although its functional role in colon cancer is a subject of debate.181
Cell cycle proteins and CIN
Recently, evidence has accrued that has implicated cycle proteins in the maintenance of chromosome fidelity in human cancers. Somatic mutations in hCDC4, which is also known as Fbw7 or Archipelago, were found in 11.5% of human colon cancers (n = 22/190) and in 4 of 58 adenomas.180 CDC4 is an evolutionarily conserved E3 ubiquitin ligase that regulates the G1-S checkpoint by targeting proteins for destruction by the SCF complex of proteins. After identifying CDC4 mutations in a subset of colon cancers, Rajagopalan et al assessed the functional consequence of CDC4 inactivation by disrupting both alleles through homologous recombination and in the karyotypically stable colon cancer cell lines HCT116 and DLD1. They observed nuclear atypia, an increased frequency of multipolar spindles, and chromosome instability. Notably, this effect was dependent on an increase in cyclin E, which is a substrate of CDC4.180 The significance of deregulation of the CDC4/cyclin E pathway has been substantiated by other investigators, who have shown that inhibition of degradation of cyclin E in mice leads to tumors that display genomic instability and that Fbxw7/hCDC4 cooperates with p53 to suppress tumor formation.182,183 It appears that Cdc4 is regulated by p53 and is one of the downstream effectors of p53-mediated regulation of genome stability, especially in the setting of genomic stress. Notably, Cdc4 may affect genome stability not only through the regulation of cyclin E but also through effects on Notch and/or c-Jun.183 Yoon et al have also shown that KLF4 is a p53-regulated protein that can prevent centrosome amplification after γ-irradiation–induced DNA damage.184 Thus, KLF4 may be one of the ultimate effectors of CIN that results from Cdc4 deregulation. In aggregate, these data provide further support for an underlying deregulation of cell cycle proteins as being a basis for CIN and for the hypothesis that deregulation of mechanisms that mediate DNA fidelity lead to complex consequences that ultimately induce CIN.
Telomeres and telomerase
Telomeres are special chromatin structures at the ends of linear chromosomes that are believed to play a fundamentally important role in protecting these regions from degradation and recombination. The regulation of telomeres is tightly associated with the regulation of senescence. In vertebrates, telomeres are composed of tandem repeats of the TTAGGG sequence and are bound by specific proteins.185 Conventional DNA polymerases are not able to completely synthesize chromosomal ends, resulting in the gradual shortening of telomeres with successive rounds of cell divisions until a critical short length is reached that elicits the activation of cellular checkpoints similar to those activated by DNA damage.186 In human cells, this telomere shortening culminates in activation of the Hayflick limit and the cessation of cell divisions. However, if p53 or Rb is inactivated, then the cells can continue to divide and will go through a period of massive cell death termed “cellular crisis.” Cells that survive crisis appear to activate mechanisms to maintain the telomeres and do this most frequently by increasing the expression of telomerase, a specialized ribonucleoprotein complex that consists of a catalytic telomerase reverse transcriptase (TERT) and an RNA subunit encoded by TERC. Most human cancers express telomerase, and it appears that during the evolution of normal cells to cancer cells the cells progress through a period of severe telomere dysfunction before regaining mechanisms to maintain telomere length.187 Now data from experiments from Terc−/− mice suggest that this period of telomere dysfunction may cause marked CIN that promotes the formation of cancer cells.186 Consistent with this model, human colon cancers show a peak in the anaphase bridge index, which is a measure of the number of metaphases that contain anaphase bridges, in early high-grade dysplastic lesions and less in more advanced carcinoma stages.188 However, the identification of chromosomal abnormalities in early colon adenomas, at a stage that precedes telomere dysfunction, suggests that telomere dysfunction cannot be the sole mechanism that induces genomic instability in colon cancer and that its role in tumor formation may depend on the nature of concurrent genetic alterations in the tumor cells. In support of this model, mTerc−/−/Min (Apc−/+), mice form fewer macroadenomas than mice with wild-type mTerc, but mTerc−/−/Tp53−/− mice show a high level of genomic instability similar to that seen in human cancers and reduced tumor latency in late-generation mice.186,189,190 Thus, it appears that telomere dysfunction may also contribute to genomic instability seen in cancer, although more definitive evidence of its role in human cancer remains to be shown.
Other factors that may participate in the development of CIN in colon cancer
Other factors than genetic inactivation of the proteins controlling the replication checkpoints have also been proposed to play a role in inducing genomic instability in colon cancer through effects on cell cycle regulation and DNA replication. These mechanisms include TGF-β resistance, JC virus infection, and C-MYC overexpression.191,192 For example, MYC overexpression has been implicated in attenuating the DNA damage checkpoint at G1-S phase and contributing to tumorigenesis through the induction of aneuploidy by this mechanism.193 Nonetheless, the data supporting these mechanisms having a role in genomic instability are confined to results from in vitro systems and have not been generated from in vivo models or in primary human cancers to date.
Epigenetic Alterations and Epigenetic Instability
In addition to genomic DNA instability being a common phenomenon in colorectal cancer, epigenetic instability also appears to be common in colorectal neoplasms. Although recognized more than 2 decades ago, epigenetic alterations, specifically aberrant DNA methylation, have not been intensively studied until recently because of controversy surrounding the significance of these alterations to the pathogenesis of colorectal cancer. DNA methylation is present throughout the majority of the human genome and is maintained in relatively stable patterns that are established during development.194 In humans, approximately 70% of CpG dinucleotides carry this epigenetic modification. However, there are regions that are enriched for CpG dinucleotides, called CpG islands, that are present in the 5′ region of approximately 50%–60% of genes and are normally maintained in an unmethylated state. In cancers, many of these CpG islands become aberrantly methylated, and this aberrant methylation can be accompanied by transcriptional repression195,196 (examples in Table 4).
Table 4.
Selected Genes Affected by Aberrant Methylation in Colorectal Cancer
| Gene | CIMP association | Age-related methylation vs cancer-related methylation | Concurrent mutations | Methylation frequency in colorectal cancer (%) |
|---|---|---|---|---|
| MLH1 | CIMP1 | C-type |
BRAF V600E MSI |
10–20 |
| TIMP3 | CIMP1 | C-type |
BRAF V600E MSI |
20–30 |
| MINT17 | CIMP1 |
BRAF V600E MSI |
— | |
| P14 | CIMP1 | C-type |
BRAF V600E MSI |
10–20 |
| RIZ1 | CIMP1 | C-type |
BRAF V600E MSI |
10–20 |
| CDKN2A | CIMP1 | C-type |
BRAF V600E MSI |
20–30 |
| MINT12 | CIMP1 |
BRAF V600E MSI |
20–40 | |
| RUNX3 | CIMP1 and “New”/Laird | C-type |
BRAF V600E MSI |
>20 |
| SOCS1 | CIMP1 and “New”/Laird | C-type |
BRAF V600E MSI |
20 |
| MINT1 | CIMP1 and CIMP2 | 0–20 | ||
| MINT31 | CIMP1 and CIMP2 | 0–20 | ||
| MINT27 | CIMP2 | KRAS mutant | — | |
| MSS/MSI-L | ||||
| MINT2 | CIMP2 | KRAS mutant | 0–20 | |
| MSS/MSI-L | ||||
| MEGALIN | CIMP2 | C-type | KRAS mutant | 10–20 |
| MSS/MSI-L | ||||
| NEUROG1 | CIMP2 and “New”/Laird | C-type | BRAF V600E | 20 |
| CACNA1G | “New”/Laird | C-type | BRAF V600E | 20 |
| IGF2 | “New”/Laird | A-type | BRAF V600E | 100 |
| APC | C-type | 10–20 | ||
| CALCA | C-type | 50 | ||
| MiR342/EVL | C-type | 70–80 | ||
| TSP1 | C-type | 20–30 | ||
| HIC1 | C-type | 50 | ||
| HLTF | C-type | 50 | ||
| VIM | C-type | 60–80 | ||
| MGMT | C-type | 30–40 | ||
| SFRP2 | C-type | 60–80 | ||
| SLC5A8 | C-type | 40–60 | ||
| ESR1 | A-type | >90 | ||
| MYOD1 | A-type | >90 | ||
| N33 | A-type | 80–90 | ||
| PAX6 | A-type | 60–70 | ||
| RASSF1 | A-type | 50 | ||
| RARβ2 | A-type | 10–40 |
A-type, age-related methylation; C-type, cancer-related methylation.
As mentioned previously, the significance of these epigenetic alterations in the pathogenesis of cancer has been a point of substantial controversy.197,198 Nonetheless, there are now sufficient data to show that the aberrant methylation of at least some of these genes, such as MLH1, MGMT, and HIC1, can be pathogenetic in cancer52–54,199,200 (Figure 6). The aberrant methylation of MLH1 occurs in >80% of sporadic MSI colorectal cancers, and the restoration of MLH1 expression and function by demethylating the MLH1 promoter in MSI colorectal cancer cell lines strongly suggests that such aberrant methylation is a cause rather than a consequence of colorectal carcinogenesis.52–54 Moreover, it is likely that the aberrant hypermethylation of 5′ CpG dinucleotides that has been shown to silence a variety of tumor suppressor genes in colorectal cancer, including CDKN2A/p16, MGMT, p14ARF, and HLTF, may be similarly pathogenetic in colorectal cancer.52–54,195,201–203 Of specific note, methylation of CDKN2A/p16, a canonical tumor suppressor gene, is detected in 40% of colorectal cancers202 and has been found in not only colorectal cancer but also in colorectal adenomas, as have other aberrantly methylated genes.204,205 This observation, as well as the detection of aberrantly methylated genes HLTF, SLC5A8, MGMT, MINT1, and MINT31 in ACF, shows that aberrant promoter methylation is occurring early in the adenoma sequence, although it does not confirm that the aberrant methylation is a primary rather than a secondary event in the tumorigenesis process206–208 (Figure 6).
Figure 6.
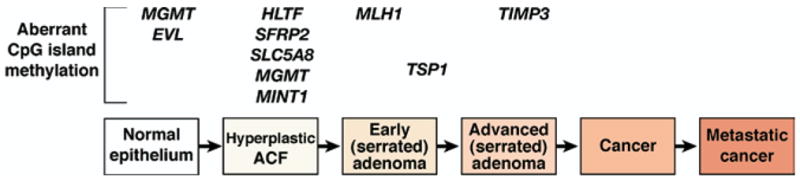
Conceptual progression of colorectal tumors with CpG island Methylator Phenotype. The hallmark of the CIMP pathway is abnormal methylation of several promoter sequences of tumor suppressor genes. This pathway in part overlaps with sporadic MSI tumors because the MMR gene hMLH1 is targeted for hypermethylation, which causes loss of MMR function. Although a full understanding of the CIMP is not clear, including the mechanism for hypermethylation of genes that is observed, we propose the following adenoma-carcinoma sequence based on studies published to date. Instead of dysplastic ACF with interrupted Wnt signaling, hyperplastic ACF may be the initial lesion in this pathway. Methylated promoters of MGMT, EVL, HLTF, SFRP2, SLC5A8, and MINT1 develop during the initiation phase of colorectal tumor development. hMLH1 promoter hypermethylation might correspond to the development of a serrated adenoma, with methylated TSP1 and TIMP3 helping to drive the progression of the CIMP tumor.
In addition, some studies have suggested that there is a subgroup of colorectal cancers that hypermethylate a high proportion of genes that may belong to a distinct subclass of colorectal cancers, termed CIMP. The criteria for defining CIMP colorectal cancers is in evolution, which has contributed to some of the controversies in this field of study.209–211 If definitively proven to be a unique molecular subgroup of colorectal cancers, these tumors will be defined by a high proportion of aberrantly methylated gene promoters that arise by distinct and unique mechanisms compared with non-CIMP tumors.202,203,210,212 The concept of CIMP tumors has been surrounded by substantial controversy because it is not clear whether tumors that display the CIMP molecular phenotype are truly a unique molecular subgroup of tumors or a group of tumors that are on the extreme part of a normal distribution with regard to aberrant DNA methylation.209,211 The mechanism responsible for causing CIMP tumors is not known at this time, but it is known that this class of tumors commonly possesses mutant BRAF V600E.210 Furthermore, the existence of other classes of CIMP tumors (eg, CIMP2, CIMP-low, CIMP-high) has been suggested recently, raising the possibility that in the future tumors may be able to be classified by both the type of genomic instability and the type of epigenomic instability they display.211,213 There has also been considerable research activity regarding markers that will accurately identify CIMP tumors (Table 4). These studies have led to different classification schemes and to the identification of different panels of markers that associate with excessive DNA methylation in colorectal cancer.210,211 It is not clear at this time which scheme and marker panel will ultimately be the most accurate for identifying CIMP tumors, assuming that this class of colorectal cancers is ultimately shown to arise via a process that is distinct from non-CIMP tumors.
Also worthy of note is recent progress in our understanding of mechanisms through which DNA methylation may affect transcription, which is tightly allied with mechanisms that regulate chromatin structure. DNA methylation may impair transcription by direct inhibition between methylated promoters and transcription factors, such as AP-2, CREB, E2F, CBF, and nuclear factor κB.194,214 CpG island methylation also can mediate transcriptional silencing by recruiting methyl binding proteins MeCP2, MBD2, and MBD3, which recognize methylated sequence and recruit histone deacetylases. The histone deacetylases then induce changes in chromatin structure that impede the access of transcription factors to the promoter.195,214 Indeed, the posttranslational modification state of the histones, which includes modifications such as acetylation of histone 3 (H3) at lysines 9 (K9) and 18 (K18), acetylation of histone 4 at lysine 12 (K12), dimethylation of histone 4 at arginine 3 (di-me R3), and dimethylation of histone 3 at lysine 4, among others, appears to regulate the chromatin state in a transcriptionally active (euchromatin) or transcriptionally repressed state (heterochromatin) through a “histone code.”215 This histone code is altered from the normal state in cancer and appears to cooperate with aberrant methylation to alter the expression of tumor suppressor genes in cancer.216,217 It is noteworthy that the relationship between DNA methylation and posttranslational modification of histones is complex and only partially understood at this time. Studies have shown changes in the methylation state of H3/lysine 9 and H3/lysine 4 precede changes in DNA methylation, suggesting that the histone modification state and chromatin structure may cause the DNA methylation changes as opposed to the DNA methylation directing alterations in the histones as noted previously.194
Thus, alterations in both the epigenome and genome occur commonly in colorectal cancer and likely drive the tumorigenesis process through activating oncogenes and inactivating tumor suppressor genes. The specifics of the process(es) through which genomic and epigenomic instability contribute to gene-specific alterations that drive colorectal cancer formation are under intense investigation. It is widely believed that the instability will ultimately be shown to play an important role in cancer formation through the creation of a permissive state that leads to gene mutations and epigenetic alterations that create opportunities for neoplastic clone progression.
Conclusions
The study of colon cancer genetics and epigenetics has yielded a host of new insights and paradigms that have broadly informed the studies of most solid tumors and the role that genomic instability plays in carcinogenesis in general. Recent progress in the investigation of genomic instability in colorectal cancer has provided support for the following: (1) the role of genomic instability early in the cancer formation process, even in the premalignant phase of the adenoma-carcinoma sequence, (2) the genetic and epigenetic basis for genomic instability in at least a subset of colon cancer (eg, inactivation of DNA repair genes, such as MLH1 and MYH), and (3) the complex mechanisms through which genomic instability arises as the result of impaired function of genes involved with the maintenance of DNA fidelity. Nonetheless, many challenges remain. A more complete understanding of the basis of CIN, aneuploidy, and aberrant methylation of the cancer genome has yet to be achieved. Furthermore, the translation of molecular genetics to new diagnostic, prognostic, and therapeutic modalities remains a challenge but carries the promise of more effective medical treatment strategies for colon cancer.
Acknowledgments
Supported by the US Public Health Service (CA11518 to W.M.G. and DK067287 and DK080506 to J.M.C.) and the VA Research Service (W.M.G. and J.M.C.).
Abbreviations used in this paper
- ACF
aberrant crypt focus
- APC/C
anaphase promoting complex/C
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia mutated and Rad3 related
- BER
base excision repair
- CIMP
CpG island methylator phenotype
- CIN
chromosomal instability
- FAP
familial adenomatous polyposis
- 5-FU
5-fluorouracil
- MAP
mut Y homologue–associated polyposis
- MMR
mismatch repair
- MSI
microsatellite instability
- MYH
mut Y homologue
- RPA
replication protein A
- TGF
transforming growth factor
Footnotes
The authors express their regrets at not being able to note the outstanding work of many investigators studying genomic and epigenomic instability in cancer. Space limitations prevented inclusion of these studies in this review.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 3.Lengauer C, Kinzler K, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 4.Grady WM. Genomic instability and colon cancer. Cancer Metastasis Rev. 2004;23:11–27. doi: 10.1023/a:1025861527711. [DOI] [PubMed] [Google Scholar]
- 5.Gollin SM. Mechanisms leading to chromosomal instability. Semin Cancer Biol. 2005;15:33–42. doi: 10.1016/j.semcancer.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Boland CR, Koi M, Chang DK, et al. The biochemical basis of microsatellite instability and abnormal immunohistochemistry and clinical behavior in Lynch Syndrome: from bench to bedside. Familial Cancer. 2008;7:41–52. doi: 10.1007/s10689-007-9145-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sieber OM, Lipton L, Crabtree M, et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 2003;348:791–799. doi: 10.1056/NEJMoa025283. [DOI] [PubMed] [Google Scholar]
- 8.Sampson JR, Dolwani S, Jones S, et al. Autosomal recessive colorectal adenomatous polyposis due to inherited mutations of MYH. Lancet. 2003;362:39–41. doi: 10.1016/S0140-6736(03)13805-6. [DOI] [PubMed] [Google Scholar]
- 9.Sieber OM, Heinimann K, Tomlinson IP. Genomic instability—the engine of tumorigenesis? Nat Rev Cancer. 2003;3:701–708. doi: 10.1038/nrc1170. [DOI] [PubMed] [Google Scholar]
- 10.Cheadle JP, Sampson JR. Exposing the MYtH about base excision repair and human inherited disease. Hum Mol Genet. 2003;12:R159–R165. doi: 10.1093/hmg/ddg259. [DOI] [PubMed] [Google Scholar]
- 11.De Jong AE, Morreau H, Van Puijenbroek M, et al. The role of mismatch repair gene defects in the development of adenomas in patients with HNPCC. Gastroenterology. 2004;126:42–48. doi: 10.1053/j.gastro.2003.10.043. [DOI] [PubMed] [Google Scholar]
- 12.Kirchhoff T, Satagopan JM, Kauff ND, et al. Frequency of BRCA1 and BRCA2 mutations in unselected Ashkenazi Jewish patients with colorectal cancer. J Natl Cancer Inst. 2004;96:68–70. doi: 10.1093/jnci/djh006. [DOI] [PubMed] [Google Scholar]
- 13.Gruber SB, Ellis NA, Scott KK, et al. BLM heterozygosity and the risk of colorectal cancer. Science. 2002;297:2013. doi: 10.1126/science.1074399. [DOI] [PubMed] [Google Scholar]
- 14.Herrmann JL, Rastelli L, Burgess CE, et al. Implications of oncogenomics for cancer research and clinical oncology. Cancer J. 2001;7:40–51. [PubMed] [Google Scholar]
- 15.Cahill DP, Lengauer C, Yu J, et al. Mutations of mitotic checkpoint genes in human cancers. Nature. 1998;392:300–303. doi: 10.1038/32688. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, Cummins JM, Shen D, et al. Three classes of genes mutated in colorectal cancers with chromosomal instability. Cancer Res. 2004;64:2998–3001. doi: 10.1158/0008-5472.can-04-0587. [DOI] [PubMed] [Google Scholar]
- 17.Storchová Z, Breneman A, Cande J, et al. Genome-wide genetic analysis of polyploidy in yeast. Nature. 2006;443:541–547. doi: 10.1038/nature05178. [DOI] [PubMed] [Google Scholar]
- 18.Grady WM, Markowitz SD. Genetic and epigenetic alterations in colon cancer. Annu Rev Genomics Hum Genet. 2002;3:101–128. doi: 10.1146/annurev.genom.3.022502.103043. [DOI] [PubMed] [Google Scholar]
- 19.Carethers JM. The cellular and molecular pathogenesis of colorectal cancer. Gastroenterol Clin North Am. 1996;25:737–754. doi: 10.1016/s0889-8553(05)70272-7. [DOI] [PubMed] [Google Scholar]
- 20.Michor F, Iwasa Y, Lengauer C, et al. Dynamics of colorectal cancer. Semin Cancer Biol. 2005;15:484–493. doi: 10.1016/j.semcancer.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 21.Rajagopalan H, Nowak MA, Vogelstein B, et al. The significance of unstable chromosomes in colorectal cancer. Nat Rev Cancer. 2003;3:695–701. doi: 10.1038/nrc1165. [DOI] [PubMed] [Google Scholar]
- 22.Muhua L, Adames NR, Murphy MD, et al. A cytokinesis checkpoint requiring the yeast homologue of an APC-binding protein. Nature. 1998;393:487–491. doi: 10.1038/31014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fodde R, Kuipers J, Rosenberg C, et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol. 2001;3:433–438. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- 24.Jallepalli PV, Lengauer C. Chromosome segregation and cancer: cutting through the mystery. Nat Rev Cancer. 2001;1:109–117. doi: 10.1038/35101065. [DOI] [PubMed] [Google Scholar]
- 25.Aaltonen LA, Peltomäki P, Mecklin JP, et al. Replication errors in benign and malignant tumors from hereditary nonpolyposis colorectal cancer patients. Cancer Res. 1994;54:1645–1648. [PubMed] [Google Scholar]
- 26.Grady WM, Rajput A, Myeroff L, et al. Mutation of the type II transforming growth factor-b receptor is coincident with the transformation of human colon adenomas to malignant carcinomas. Cancer Res. 1998;58:3101–3104. [PubMed] [Google Scholar]
- 27.Shih IM, Zhou W, Goodman SN, et al. Evidence that genetic instability occurs at an early stage of colorectal tumorigenesis. Cancer Res. 2001;61:818–822. [PubMed] [Google Scholar]
- 28.Stoler DL, Chen N, Basik M, et al. The onset and extent of genomic instability in sporadic colorectal tumor progression. Proc Natl Acad Sci U S A. 1999;96:15121–15126. doi: 10.1073/pnas.96.26.15121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nowak MA, Komarova NL, Sengupta A, et al. The role of chromosomal instability in tumor initiation. Proc Natl Acad Sci U S A. 2002;99:16226–16231. doi: 10.1073/pnas.202617399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Michor F, Iwasa Y, Nowak MA. Dynamics of cancer progression. Nat Rev Cancer. 2004;4:197–205. doi: 10.1038/nrc1295. [DOI] [PubMed] [Google Scholar]
- 31.Michor F, Iwasa Y, Vogelstein B, et al. Can chromosomal instability initiate tumorigenesis? Semin Cancer Biol. 2005;15:43–49. doi: 10.1016/j.semcancer.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 32.Bartkova J, Horejsí Z, Koed K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–870. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 33.Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 34.Aaltonen LA, Peltomaki P, Leach FS, et al. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 35.Tsao JL, Yatabe Y, Salovaara R, et al. Genetic reconstruction of individual colorectal tumor histories. Proc Natl Acad Sci U S A. 2000;97:1236–1241. doi: 10.1073/pnas.97.3.1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Markowitz S, Wang J, Myeroff L, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 37.Jung B, Doctolero RT, Tajima A, et al. Loss of activin receptor type 2 protein expression in microsatellite unstable colon cancers. Gastroenterology. 2004;126:654–659. doi: 10.1053/j.gastro.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 38.Marra G, Boland CR. DNA repair and colorectal cancer. Gastroenterol Clin North Am. 1996;25:755–772. doi: 10.1016/s0889-8553(05)70273-9. [DOI] [PubMed] [Google Scholar]
- 39.Fishel R. Mismatch repair, molecular switches, and signal transduction. Genes Dev. 1998;12:2096–2101. doi: 10.1101/gad.12.14.2096. [DOI] [PubMed] [Google Scholar]
- 40.Hare JT, Taylor JH. One role for DNA methylation in vertebrate cells is strand discrimination in mismatch repair. Proc Natl Acad Sci U S A. 1985;82:7350–7354. doi: 10.1073/pnas.82.21.7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thomas DC, Roberts JD, Kunkel TA. Heteroduplex repair in extracts of human HeLa cells. J Biol Chem. 1990;266:3744–3751. [PubMed] [Google Scholar]
- 42.Fishel R, Ewel A, Lescoe MK. Purified human MSH2 protein binds to DNA containing mismatched nucleotides. Cancer Res. 1994;54:5539–5542. [PubMed] [Google Scholar]
- 43.Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- 44.Fishel R, Lescoe MK, Rao MRS, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 45.Papadopoulos N, Nicolaides NC, Wei Y-F, et al. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994;263:1625–1629. doi: 10.1126/science.8128251. [DOI] [PubMed] [Google Scholar]
- 46.Bronner CE, Baker SM, Morrison PT, et al. Mutation in the DNA mismatch repair homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–261. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- 47.Akiyama Y, Sato H, Yamada T, et al. Germ-line mutation of the hMSH6/GTBP gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Cancer Res. 1997;57:3920–3923. [PubMed] [Google Scholar]
- 48.Wijnen J, de Leeuw W, Vasen H, et al. Familial endometrial cancer in female carriers of MSH6 germline mutations. Nat Genet. 1999;23:142–144. doi: 10.1038/13773. [DOI] [PubMed] [Google Scholar]
- 49.Kolodner RD, Tytell JD, Schmeits J, et al. Germline msh6 mutations in colorectal cancer families. Cancer Res. 1999;59:5068–5074. [PubMed] [Google Scholar]
- 50.Nicolaides NC, Papadopoulos N, Liu B, et al. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature. 1994;371:75–80. doi: 10.1038/371075a0. [DOI] [PubMed] [Google Scholar]
- 51.Chang DK, Ricciardiello L, Goel A, et al. Steady-state regulation of the human DNA mismatch repair system. J Biol Chem. 2000;275:18424–18431. doi: 10.1074/jbc.M001140200. [DOI] [PubMed] [Google Scholar]
- 52.Kane MF, Loda M, Gaida GM, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–811. [PubMed] [Google Scholar]
- 53.Herman JG, Umar A, Polyak K. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci U S A. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Veigl ML, Kasturi L, Olechnowicz J, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci U S A. 1998;95:8698–8702. doi: 10.1073/pnas.95.15.8698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hemminki A, Peltomaki P, Mecklin J-K. Loss of the wild type MLH1 gene is a feature of hereditary nonpolyposis colorectal cancer. Nat Genet. 1994;8:405–410. doi: 10.1038/ng1294-405. [DOI] [PubMed] [Google Scholar]
- 56.Drummond JT, Li G-M, Longley MJ, et al. Isolation of an hMSH2-p160 heterodimer that restores DNA mismatch repair to tumor cells. Science. 1995;268:1909. doi: 10.1126/science.7604264. [DOI] [PubMed] [Google Scholar]
- 57.Palombo F, Gallinari P, Iaccarino I, et al. GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science. 1995;268:1912–1914. doi: 10.1126/science.7604265. [DOI] [PubMed] [Google Scholar]
- 58.Papadopoulos N, Nicolaides NC, Liu B, et al. Mutations of GTBP in genetically unstable cells. Science. 1995;268:1915–1917. doi: 10.1126/science.7604266. [DOI] [PubMed] [Google Scholar]
- 59.Fishel R, Ewel A, Lescoe MK. Purified human MSH2 protein binds to DNA containing mismatched nucleotides. Cancer Res. 1994;54:5539–5542. [PubMed] [Google Scholar]
- 60.Palombo F, Iaccarino I, Nakajima E, et al. hMutSbeta, a heterodimer of hMSH2 and hMSH3, binds to insertion/deletion loops in DNA. Curr Biol. 1996;6:1181–1184. doi: 10.1016/s0960-9822(02)70685-4. [DOI] [PubMed] [Google Scholar]
- 61.Genschel J, Littman SJ, Drummond JT, et al. Isolation of MutSb from human cells and comparison of the mismatch repair specificities of MutSb and MutSa. J Biol Chem. 1998;273:19895–19901. doi: 10.1074/jbc.273.31.19895. [DOI] [PubMed] [Google Scholar]
- 62.McCulloch SD, Gu L, Li GM. Bi-directional processing of DNA loops by mismatch repair-dependent and -independent pathways in human cells. J Biol Chem. 2003;278:3891–3896. doi: 10.1074/jbc.M210687200. [DOI] [PubMed] [Google Scholar]
- 63.Littman SJ, Fang WH, Modrich P. Repair of large insertion/deletion heterologies in human nuclear extracts is directed by a 5′ single-strand break and is independent of the mismatch repair system. J Biol Chem. 1999;274:7474–7481. doi: 10.1074/jbc.274.11.7474. [DOI] [PubMed] [Google Scholar]
- 64.Gradia S, Subramanian D, Wilson T, et al. hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol Cell. 1999;3:255–261. doi: 10.1016/s1097-2765(00)80316-0. [DOI] [PubMed] [Google Scholar]
- 65.Gradia S, Acharya S, Fishel R. The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell. 1997;91:995–1005. doi: 10.1016/s0092-8674(00)80490-0. [DOI] [PubMed] [Google Scholar]
- 66.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 67.Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu Rev Biochem. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 68.Schmutte C, Marinescu RC, Sadoff MM, et al. Human exonuclease I interacts with the mismatch repair protein hMSH2. Cancer Res. 1998;58:4537–4542. [PubMed] [Google Scholar]
- 69.Genschel J, Modrich P. Mechanism of 5′-directed excision in human mismatch repair. Mol Cell. 2003;12:1077–1086. doi: 10.1016/s1097-2765(03)00428-3. [DOI] [PubMed] [Google Scholar]
- 70.Dzantiev L, Constantin N, Genschel J, et al. A defined huamn system that supports bidirectional mismatch-provoked excision. Mol Cell. 2004;15:31–41. doi: 10.1016/j.molcel.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 71.Longley MJ, Pierce AJ, Modrich P. DNA polymerase delta is required for human mismatch repair in vitro. J Biol Chem. 1997;272:10917–10921. doi: 10.1074/jbc.272.16.10917. [DOI] [PubMed] [Google Scholar]
- 72.Umar A, Buermeyer AB, Simon JA, et al. Requirement for PCNA in DNA mismatch repair at a step preceding DNA resynthesis. Cell. 1996;87:65–73. doi: 10.1016/s0092-8674(00)81323-9. [DOI] [PubMed] [Google Scholar]
- 73.Zhang Y, Yuan F, Presnell SR, et al. Reconstitution of 5′-directed human mismatch repair in a purified system. Cell. 2005;122:693–705. doi: 10.1016/j.cell.2005.06.027. [DOI] [PubMed] [Google Scholar]
- 74.Clark AB, Valle F, Drotschmann K, et al. Functional interaction of proliferating cell nuclar antigen with MSH2-MSH6 and MSH2-MSH3 complexes. J Biol Chem. 2000;275:36498–36501. doi: 10.1074/jbc.C000513200. [DOI] [PubMed] [Google Scholar]
- 75.Flores-Rozas H, Clark D, Kolodner RD. Proliferating cell nuclear antigen and Msh2p-Msh6p interact to form an active mispair recognition complex. Nat Genet. 2000;26:375–378. doi: 10.1038/81708. [DOI] [PubMed] [Google Scholar]
- 76.Kleezkowska HE, Marra G, Lettieri T, et al. hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev. 2001;15:724–736. doi: 10.1101/gad.191201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kadyrov FA, Dzantiev L, Constantin N, et al. Endonucleolic function of MutLalpha in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 78.Ramilo C, Gu L, Guo S, et al. Partial reconstitution of human DNA mismatch repair in vitro: characterization of the role of human replication protein A. Mol Cell Biol. 2002;22:2037–2046. doi: 10.1128/MCB.22.7.2037-2046.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Guo S, Zhang Y, Yuan F, et al. Regulation of replication protein A functions in mismatch repair by phosphorylation. J Biol Chem. 2006;281:21607–21616. doi: 10.1074/jbc.M603504200. [DOI] [PubMed] [Google Scholar]
- 80.Yan F, Gu L, Guo S, Wang C, et al. Evidence for involvement of HMGB1 protein in human DNA mismatch repair. J Biol Chem. 2004;279:20935–20940. doi: 10.1074/jbc.M401931200. [DOI] [PubMed] [Google Scholar]
- 81.Parsons R, Myeroff LL, Liu B, et al. Microsatellite instability and mutations of the transforming growth factor beta type II receptor gene in colorectal cancer. Cancer Res. 1995;55:5548–5550. [PubMed] [Google Scholar]
- 82.Souza RF, Appel R, Yin J, et al. Microsatellite instability in the insulin-like growth factor II receptor gene in gastrointestinal tumours. Nat Genet. 1996;14:255–257. doi: 10.1038/ng1196-255. [DOI] [PubMed] [Google Scholar]
- 83.Rampino N, Yamamoto H, Ionov Y, et al. Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science. 1997;275:967–969. doi: 10.1126/science.275.5302.967. [DOI] [PubMed] [Google Scholar]
- 84.Yamamoto H, Sawai H, Perucho M. Frameshift somatic mutations in gastrointestinal cancer of the microsatellite mutator phenotype. Cancer Res. 1997;57:4420–4426. [PubMed] [Google Scholar]
- 85.Yagi OK, Yoshimitsu A, Nomizu T, et al. Proapoptotic gene BAX is frequently mutated in hereditary nonpolyposis colorectal cancers but not in adenomas. Gastroenterology. 1998;114:268–274. doi: 10.1016/s0016-5085(98)70477-9. [DOI] [PubMed] [Google Scholar]
- 86.Watanabe T, Wu TT, Catalano PJ, et al. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N Engl J Med. 2001;344:1196–1206. doi: 10.1056/NEJM200104193441603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Malkhosyan S, Rampino N, Yamamoto H, et al. Frameshift mutator mutations. Nature. 1996;382:499–500. doi: 10.1038/382499a0. [DOI] [PubMed] [Google Scholar]
- 88.Mori Y, Yin J, Rashid A, et al. Instabilotyping: comprehensive identification of frameshift mutations caused by coding region microsatellite instability. Cancer Res. 2001;61:6046–6049. [PubMed] [Google Scholar]
- 89.Jung B, Smith EJ, Doctolero RT, et al. Influence of target gene mutations on survival, stage and histology in sporadic microsatellite unstable colon cancers. Int J Cancer. 2006;118:2509–2513. doi: 10.1002/ijc.21710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jung BH, Beck SE, Cabral J, et al. Activin type 2 receptor restoration in MSI-H colon cancer suppresses growth and enhances migration with activin. Gastroenterology. 2007;132:633–644. doi: 10.1053/j.gastro.2006.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Deng G, Bell I, Crawley S, et al. BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin Cancer Res. 2004;10:191–195. doi: 10.1158/1078-0432.ccr-1118-3. [DOI] [PubMed] [Google Scholar]
- 92.Ionov Y, Peinado MA, Malkhosyan S, et al. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993;363:558–561. doi: 10.1038/363558a0. [DOI] [PubMed] [Google Scholar]
- 93.Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993;260:816–819. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- 94.Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–618. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 95.Schwitalle Y, Kloor M, Eiermann S, et al. Immune response against frameshift-induced neopeptides in HNPCC patients and healthy HNPCC mutation carriers. Gastroenterology. 2008;134:988–997. doi: 10.1053/j.gastro.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 96.Gryfe R, Kim H, Hsieh ET, et al. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med. 2000;342:69–77. doi: 10.1056/NEJM200001133420201. [DOI] [PubMed] [Google Scholar]
- 97.Walther A, Houlston R, Tomlinson I. Association between chromosomal instability and prognosis in colorectal cancer: a meta-analysis. Gut. 2008;57:941–950. doi: 10.1136/gut.2007.135004. [DOI] [PubMed] [Google Scholar]
- 98.Carethers JM, Hawn MT, Chauhan DP, et al. Competency in mismatch repair prohibits clonal expansion of cancer cells treated with N-methyl-N′-nitro-N-nitrosoguanidine. J Clin Invest. 1996;98:199–206. doi: 10.1172/JCI118767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hawn MT, Umar A, Carethers JM, et al. Evidence for a connection between the mismatch repair system and the G2 cell cycle checkpoint. Cancer Res. 1995;55:3721–3725. [PubMed] [Google Scholar]
- 100.Fink D, Nebel S, Aebi S, et al. The role of DNA mismatch repair in platinum drug resistance. Cancer Res. 1996;56:4881–4886. [PubMed] [Google Scholar]
- 101.Papouli E, Cejka P, Jiricny J. Dependence of the cytotoxicity of DNA-damaging agents on the mismatch repair status of human cells. Cancer Res. 2004;64:3391–3394. doi: 10.1158/0008-5472.CAN-04-0513. [DOI] [PubMed] [Google Scholar]
- 102.Carethers JM, Chauhan DP, Fink D, et al. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology. 1999;117:123–131. doi: 10.1016/s0016-5085(99)70558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tajima A, Hess MT, Cabrera BL, et al. The mismatch repair complex hMutSa recognizes 5-fluoruracil-modified DNA: implications for chemosensitivity and resistance. Gastroenterology. 2004;127:1678–1684. doi: 10.1053/j.gastro.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 104.Major PP, Egan E, Herrick D, et al. 5-Fluorouracil incorporation in DNA of human breast carcinoma cells. Cancer Res. 1982;42:3005–3009. [PubMed] [Google Scholar]
- 105.Arnold CN, Goel A, Boland CR. Role of hMLH1 promoter hypermethylation in drug resistance to 5-fluoruracil in colorectal cancer cell lines. Int J Cancer. 2003;106:66–73. doi: 10.1002/ijc.11176. [DOI] [PubMed] [Google Scholar]
- 106.Meyers M, Wagner MW, Mazurek A, et al. DNA mismatch repair-dependent response to fluoropyrimidine-generated damage. J Biol Chem. 2005;280:5516–5526. doi: 10.1074/jbc.M412105200. [DOI] [PubMed] [Google Scholar]
- 107.Carethers JM, Smith EJ, Behling CA, et al. Use of 5-fluorouracil and survival in patients with microsatellite unstable colorectal cancer. Gastroenterology. 2004;126:394–401. doi: 10.1053/j.gastro.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 108.Ribic CM, Sargent DJ, Moore MJ, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349:247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jover R, Zapater P, Castells A, et al. Mismatch repair status in the prediction of benefit from adjuvant fluorouracil chemotherapy in colorectal cancer. Gut. 2006;55:848–855. doi: 10.1136/gut.2005.073015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.de Vos tot Nedervee Cappel WH, Neulenbeld HJ, Keibeuker JH, et al. Survival after adjuvant 5-FU treatment for stage III coon cancer in hereditary nonpolyposis colorectal cancer. In J Cancer. 2004;109:468–471. doi: 10.1002/ijc.11712. [DOI] [PubMed] [Google Scholar]
- 111.Duckett DR, Bronstein SM, Taya Y, et al. hMutSa- and hMutLa-dependent phosphorylation of p53 in response to DNA methylator damage. Proc Natl Acad Sci U S A. 1999;96:12384–12388. doi: 10.1073/pnas.96.22.12384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stojic L, Mojas N, Cejka P, et al. Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 2004;19:1331–1344. doi: 10.1101/gad.294404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tibbetts RS, Brumbaugh KM, Williams JM, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mailand N, Podtelejnikov AV, Groth A, et al. Regulation of G2/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J. 2002;21:5911–5920. doi: 10.1093/emboj/cdf567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hickman MJ, Samson LD. Apoptotic signaling in response to a single type of DNA lesion, O6-methylguanine. Mol Cell. 2004;14:105–116. doi: 10.1016/s1097-2765(04)00162-5. [DOI] [PubMed] [Google Scholar]
- 116.Aguilera A, Gómez-González B. Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet. 2008;9:204–217. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- 117.Bardi G, Sukhikh T, Pandis N, et al. Karyotypic characterization of colorectal adenocarcinomas. Genes Chromosomes Cancer. 1995;12:97–109. doi: 10.1002/gcc.2870120204. [DOI] [PubMed] [Google Scholar]
- 118.Tomlins SA, Laxman B, Dhanasekaran SM, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature. 2007;448:595–599. doi: 10.1038/nature06024. [DOI] [PubMed] [Google Scholar]
- 119.Saha S, Bardelli A, Buckhaults P, et al. A phosphatase associated with metastasis of colorectal cancer. Science. 2001;294:1343–1346. doi: 10.1126/science.1065817. [DOI] [PubMed] [Google Scholar]
- 120.Bardelli A, Saha S, Sager JA, et al. PRL-3 expression in meta-static cancers. Clin Cancer Res. 2003;9:5607–5615. [PubMed] [Google Scholar]
- 121.Shin HJ, Baek KH, Jeon AH, et al. Dual roles of human BubR1, a mitotic checkpoint kinase, in the monitoring of chromosomal instability. Cancer Cell. 2003;4:483–497. doi: 10.1016/s1535-6108(03)00302-7. [DOI] [PubMed] [Google Scholar]
- 122.Lengauer C, Kinzler K, Vogelstein B. Genetic instability in colorectal cancers. Nature. 1997;386:623–627. doi: 10.1038/386623a0. [DOI] [PubMed] [Google Scholar]
- 123.Weaver BA, Silk AD, Montagna C, et al. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 124.Liu G, Parant JM, Lang G, et al. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet. 2004;36:63–68. doi: 10.1038/ng1282. [DOI] [PubMed] [Google Scholar]
- 125.Yuen KW, Warren CD, Chen O, et al. Systematic genome instability screens in yeast and their potential relevance to cancer. Proc Natl Acad Sci U S A. 2007;104:3925–3930. doi: 10.1073/pnas.0610642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sato N, Mizumoto K, Nakamura M, et al. Correlation between centrosome abnormalities and chromosomal instability in human pancreatic cancer cells. Cancer Genet Cytogenet. 2001;126:13–19. doi: 10.1016/s0165-4608(00)00384-8. [DOI] [PubMed] [Google Scholar]
- 127.Saunders WS, Shuster M, Huang X, et al. Chromosomal instability and cytoskeletal defects in oral cancer cells. Proc Natl Acad Sci U S A. 2000;97:303–308. doi: 10.1073/pnas.97.1.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 129.Chittenden TW, Howe EA, Culhane AC, et al. Functional classification analysis of somatically mutated genes in human breast and colorectal cancers. Genomics. 2008;91:508–511. doi: 10.1016/j.ygeno.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Eshleman JR, Casey G, Kochera ME, et al. Chromosome number and structure both are markedly stable in RER colorectal cancers and are not destabilized by mutation of p53. Oncogene. 1998;17:719–725. doi: 10.1038/sj.onc.1201986. [DOI] [PubMed] [Google Scholar]
- 131.Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004;432:338–341. doi: 10.1038/nature03099. [DOI] [PubMed] [Google Scholar]
- 132.Sieber OM, Heinimann K, Gorman P, et al. Analysis of chromosomal instability in human colorectal adenomas with two mutational hits at APC. Proc Natl Acad Sci U S A. 2002;99:16910–16915. doi: 10.1073/pnas.012679099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Chow E, Thirlwell C, Macrae F, et al. Colorectal cancer and inherited mutations in base-excision repair. Lancet Oncol. 2004;5:600–606. doi: 10.1016/S1470-2045(04)01595-5. [DOI] [PubMed] [Google Scholar]
- 134.Al-Tassan N, Chmiel NH, Maynard J, et al. Inherited variants of MYH associated with somatic G:C → T:A mutations in colorectal tumors. Nat Genet. 2002;30:227–232. doi: 10.1038/ng828. [DOI] [PubMed] [Google Scholar]
- 135.Sieber OM, Lipton L, Crabtree M, et al. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. N Engl J Med. 2003;348:791–799. doi: 10.1056/NEJMoa025283. [DOI] [PubMed] [Google Scholar]
- 136.Olinski R, Zastawny T, Budzbon J, et al. DNA base modifications in chromatin of human cancerous tissues. FEBS Lett. 1992;309:193–198. doi: 10.1016/0014-5793(92)81093-2. [DOI] [PubMed] [Google Scholar]
- 137.Halford SE, Rowan AJ, Lipton L, et al. Germline mutations but not somatic changes at the MYH locus contribute to the pathogenesis of unselected colorectal cancers. Am J Pathol. 2003;162:1545–1548. doi: 10.1016/S0002-9440(10)64288-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cardoso J, Molenaar L, de Menezes RX, et al. Chromosomal instability in MYH- and APC-mutant adenomatous polyps. Cancer Res. 2006;66:2514–2519. doi: 10.1158/0008-5472.CAN-05-2407. [DOI] [PubMed] [Google Scholar]
- 139.Amon A. The spindle checkpoint. Curr Opin Genet Dev. 1999;9:69–75. doi: 10.1016/s0959-437x(99)80010-0. [DOI] [PubMed] [Google Scholar]
- 140.Tang Z, Bharadwaj R, Li B, et al. Mad2-Independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev Cell. 2001;1:227–237. doi: 10.1016/s1534-5807(01)00019-3. [DOI] [PubMed] [Google Scholar]
- 141.Jallepalli PV, Waizenegger IC, Bunz F, et al. Securin is required for chromosomal stability in human cells. Cell. 2001;105:445–457. doi: 10.1016/s0092-8674(01)00340-3. [DOI] [PubMed] [Google Scholar]
- 142.Li Y, Benezra R. Identification of a human mitotic checkpoint gene: hsMAD2. Science. 1996;274:246–248. doi: 10.1126/science.274.5285.246. [DOI] [PubMed] [Google Scholar]
- 143.Jin D, Spencer F, Jeang K. Human T cell leukemia virus type 1 oncoprotein Tax targets the human mitotic checkpoint protein MAD1. Cell. 1998;93:81–91. doi: 10.1016/s0092-8674(00)81148-4. [DOI] [PubMed] [Google Scholar]
- 144.Domínguez A, Ramos-Morales F, Romero F, et al. hpttg, a human homologue of rat pttg, is overexpressed in hematopoietic neoplasms. Evidence for a transcriptional activation function of hPTTG. Oncogene. 1998;17:2187–2193. doi: 10.1038/sj.onc.1202140. [DOI] [PubMed] [Google Scholar]
- 145.Sáez C, Japón MA, Ramos-Morales F, et al. hpttg is over-expressed in pituitary adenomas and other primary epithelial neoplasias. Oncogene. 1999;18:5473–5476. doi: 10.1038/sj.onc.1202914. [DOI] [PubMed] [Google Scholar]
- 146.Heaney AP, Singson R, McCabe CJ, et al. Expression of pituitary-tumour transforming gene in colorectal tumours. Lancet. 2000;355:716–719. doi: 10.1016/S0140-6736(99)10238-1. [DOI] [PubMed] [Google Scholar]
- 147.Michel LS, Liberal V, Chatterjee A, et al. MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature. 2001;409:355–359. doi: 10.1038/35053094. [DOI] [PubMed] [Google Scholar]
- 148.Wang X, Cheung HW, Chun AC, et al. Mitotic checkpoint defects in human cancers and their implications to chemotherapy. Front Biosci. 2008;13:2103–2114. doi: 10.2741/2827. [DOI] [PubMed] [Google Scholar]
- 149.Tomonaga T, Matsushita K, Yamaguchi S, et al. Overexpression and mistargeting of centromere protein-A in human primary colorectal cancer. Cancer Res. 2003;63:3511–3516. [PubMed] [Google Scholar]
- 150.Tomonaga T, Matsushita K, Ishibashi M, et al. Centromere protein H is up-regulated in primary human colorectal cancer and its overexpression induces aneuploidy. Cancer Res. 2005;65:4683–4689. doi: 10.1158/0008-5472.CAN-04-3613. [DOI] [PubMed] [Google Scholar]
- 151.Tatsuka M, Katayama H, Ota T, et al. Multinuclearity and increased ploidy caused by overexpression of the aurora- and Ipl1-like midbody-associated protein mitotic kinase in human cancer cells. Cancer Res. 1998;58:4811–4816. [PubMed] [Google Scholar]
- 152.Adams RR, Eckley DM, Vagnarelli P, et al. Human INCENP colocalizes with the Aurora-B/AIRK2 kinase on chromosomes and is overexpressed in tumour cells. Chromosoma. 2001;110:65–74. doi: 10.1007/s004120100130. [DOI] [PubMed] [Google Scholar]
- 153.Doxsey S. The centrosome-a tiny organelle with big potential. Nat Genet. 1998;20:104–106. doi: 10.1038/2392. [DOI] [PubMed] [Google Scholar]
- 154.Oikawa T, Okuda M, Ma Z, et al. Transcriptional control of BubR1 by p53 and suppression of centrosome amplification by BubR1. Mol Cell Biol. 2005;25:4046–4061. doi: 10.1128/MCB.25.10.4046-4061.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.D’Assoro AB, Lingle WL, Salisbury JL. Centrosome amplification and the development of cancer. Oncogene. 2002;21:6146–6153. doi: 10.1038/sj.onc.1205772. [DOI] [PubMed] [Google Scholar]
- 156.Brinkley BR. Managing the centrosome numbers game: from chaos to stability in cancer cell division. Trends Cell Biol. 2001;11:18–21. doi: 10.1016/s0962-8924(00)01872-9. [DOI] [PubMed] [Google Scholar]
- 157.Fukasawa K. Oncogenes and tumour suppressors take on centrosomes. Nat Rev Cancer. 2007;7:911–924. doi: 10.1038/nrc2249. [DOI] [PubMed] [Google Scholar]
- 158.Pihan GA, Purohit A, Wallace J, et al. Centrosome defects can account for cellular and genetic changes that characterize prostate cancer progression. Cancer Res. 2001;61:2212–2219. [PubMed] [Google Scholar]
- 159.Lingle WL, Barrett SL, Negron VC, et al. Centrosome amplification drives chromosomal instability in breast tumor development. Proc Natl Acad Sci U S A. 2002;99:1978–1983. doi: 10.1073/pnas.032479999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhou M, Sheldon S, Akel N, et al. Chromosomal aneuploidy in leukemic blast crisis: a potential source of error in interpretation of bone marrow engraftment analysis by VNTR amplification. Mol Diagn. 1999;4:153–157. doi: 10.1016/s1084-8592(99)80039-3. [DOI] [PubMed] [Google Scholar]
- 161.Zhou H, Kuang J, Zhong L, et al. Tumour amplified kinase STK15/BTAK induces centrosome amplification, aneuploidy and transformation. Nat Genet. 1998;20:189–193. doi: 10.1038/2496. [DOI] [PubMed] [Google Scholar]
- 162.Sen S, Zhou H, White RA. A putative serine/threonine kinase encoding gene BTAK on chromosome 20q13 is amplified and overexpressed in human breast cancer cell lines. Oncogene. 1997;14:2195–2200. doi: 10.1038/sj.onc.1201065. [DOI] [PubMed] [Google Scholar]
- 163.Bischoff JR, Anderson L, Zhu Y, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998;17:3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Glover DM, Leibowitz MH, McLean DA, et al. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell. 1995;81:95–105. doi: 10.1016/0092-8674(95)90374-7. [DOI] [PubMed] [Google Scholar]
- 165.Francisco L, Wang W, Chan CS. Type 1 protein phosphatase acts in opposition to IpL1 protein kinase in regulating yeast chromosome segregation. Mol Cell Biol. 1994;14:4731–4740. doi: 10.1128/mcb.14.7.4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Macmillan JC, Hudson JW, Bull S, et al. Comparative expression of the mitotic regulators SAK and PLK in colorectal cancer. Ann Surg Oncol. 2001;8:729–740. doi: 10.1007/s10434-001-0729-6. [DOI] [PubMed] [Google Scholar]
- 167.Grisendi S, Mecucci C, Falini B, et al. Nucleophosmin and cancer. Nat Rev Cancer. 2006;6:493–505. doi: 10.1038/nrc1885. [DOI] [PubMed] [Google Scholar]
- 168.Czarnecka AM, Campanella C, Zummo G, et al. Mitochondrial chaperones in cancer: from molecular biology to clinical diagnostics. Cancer Biol Ther. 2006;5:714–720. doi: 10.4161/cbt.5.7.2975. [DOI] [PubMed] [Google Scholar]
- 169.Ortega S, Malumbres M, Barbacid M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta. 2002;1602:73–87. doi: 10.1016/s0304-419x(02)00037-9. [DOI] [PubMed] [Google Scholar]
- 170.Myung K, Chen C, Kolodner RD. Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature. 2001;411:1073–1076. doi: 10.1038/35082608. [DOI] [PubMed] [Google Scholar]
- 171.Nyberg KA, Michelson RJ, Putnam CW, et al. Toward maintaining the genome: DNA Damage and Replication Checkpoints. Annu Rev Genet. 2002;36:617–656. doi: 10.1146/annurev.genet.36.060402.113540. [DOI] [PubMed] [Google Scholar]
- 172.Kennedy RD, D’Andrea AD. DNA repair pathways in clinical practice: lessons from pediatric cancer susceptibility syndromes. J Clin Oncol. 2006;24:3799–3808. doi: 10.1200/JCO.2005.05.4171. [DOI] [PubMed] [Google Scholar]
- 173.Bao S, Chang M-S, Auclair D, et al. HRad17, a human homologue of the Schizosaccharomyces pombe checkpoint gene rad17, is overexpressed in colon carcinoma. Cancer Res. 1999;59:2023–2028. [PubMed] [Google Scholar]
- 174.Rotman G, Shiloh Y. ATM: from gene to function. Hum Mol Genet. 1998;7:1555–1563. doi: 10.1093/hmg/7.10.1555. [DOI] [PubMed] [Google Scholar]
- 175.Smith L. Duplication of ATR inhibits MyoD, induces aneuploidy and eliminates radiation-induced G1 arrest. Nat Genet. 1998;19:39–46. doi: 10.1038/ng0598-39. [DOI] [PubMed] [Google Scholar]
- 176.Zhang H, Tombline G, Weber B. BRCA1, BRCA2, and DNA damage response: collision or collusion. Cell. 1998;92:433–436. doi: 10.1016/s0092-8674(00)80936-8. [DOI] [PubMed] [Google Scholar]
- 177.Lane D. Awakening angels. Nature. 1998;394:616–617. doi: 10.1038/29166. [DOI] [PubMed] [Google Scholar]
- 178.Rouse J, Jackson SP. Interfaces between the detection, signaling, and repair of DNA damage. Science. 2002;297:547–551. doi: 10.1126/science.1074740. [DOI] [PubMed] [Google Scholar]
- 179.Kolodner RD, Putnam CD, Myung K. Maintenance of genome stability in Saccharomyces cerevisiae. Science. 2002;297:552–557. doi: 10.1126/science.1075277. [DOI] [PubMed] [Google Scholar]
- 180.Rajagopalan H, Jallepalli PV, Rago C, et al. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004;428:77–81. doi: 10.1038/nature02313. [DOI] [PubMed] [Google Scholar]
- 181.van der Heijden MS, Brody JR, Elghalbzouri-Maghrani E, et al. Does tumorigenesis select for or against mutations of the DNA repair-associated genes BRCA2 and MRE11?: considerations from somatic mutations in microsatellite unstable (MSI) gastrointestinal cancers. BMC Genet. 2006;7:3. doi: 10.1186/1471-2156-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Loeb KR, Kostner H, Firpo E, et al. A mouse model for cyclin E-dependent genetic instability and tumorigenesis. Cancer Cell. 2005;8:35–47. doi: 10.1016/j.ccr.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 183.Perez-Losada J, Mao JH, Balmain A. Control of genomic instability and epithelial tumor development by the p53-Fbxw7/Cdc4 pathway. Cancer Res. 2005;65:6488–6492. doi: 10.1158/0008-5472.CAN-05-1294. [DOI] [PubMed] [Google Scholar]
- 184.Yoon HS, Ghaleb AM, Nandan MO, et al. Krüppel-like factor 4 prevents centrosome amplification following gamma-irradiation-induced DNA damage. Oncogene. 2005;24:4017–4025. doi: 10.1038/sj.onc.1208576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet. 2005;6:611–622. doi: 10.1038/nrg1656. [DOI] [PubMed] [Google Scholar]
- 186.Maser RS, DePinho RA. Connecting chromosomes, crisis, and cancer. Science. 2002;297:565–569. doi: 10.1126/science.297.5581.565. [DOI] [PubMed] [Google Scholar]
- 187.Kim NW, Piatyszek MA, Prowse KR, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 188.Rudolph KL, Millard M, Bosenberg MW, et al. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet. 2001;28:155–159. doi: 10.1038/88871. [DOI] [PubMed] [Google Scholar]
- 189.O’Hagan RC, Chang S, Maser RS, et al. Telomere dysfunction provokes regional amplification and deletion in cancer genomes. Cancer Cell. 2002;2:149–155. doi: 10.1016/s1535-6108(02)00094-6. [DOI] [PubMed] [Google Scholar]
- 190.Artandi SE, Chang S, Lee SL, et al. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–645. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- 191.Glick AB, Weinberg WC, Wu IH, et al. Transforming growth factor beta 1 suppresses genomic instability independent of a G1 arrest, p53, and Rb. Cancer Res. 1996;56:3645–3650. [PubMed] [Google Scholar]
- 192.Shadan FF, Cunningham C, Boland CR. JC virus: a biomarker for colorectal cancer? Med Hypotheses. 2002;59:667–669. doi: 10.1016/s0306-9877(02)00166-4. [DOI] [PubMed] [Google Scholar]
- 193.Felsher DW, Bishop JM. Transient excess of MYC activity can elicit genomic instability and tumorigenesis. Proc Natl Acad Sci U S A. 1999;96:3940–3944. doi: 10.1073/pnas.96.7.3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Kondo Y, Issa JP. Epigenetic changes in colorectal cancer. Cancer Metastasis Rev. 2004;23:29–39. doi: 10.1023/a:1025806911782. [DOI] [PubMed] [Google Scholar]
- 195.Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trends Genet. 2000;16:168–174. doi: 10.1016/s0168-9525(99)01971-x. [DOI] [PubMed] [Google Scholar]
- 196.Jones P, Laird P. Cancer epigenetics comes of age. Nat Genet. 1999;21:163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- 197.Jubb AM, Bell SM, Quirke P. Methylation and colorectal cancer. J Pathol. 2001;195:111–134. doi: 10.1002/path.923. [DOI] [PubMed] [Google Scholar]
- 198.Baylin SB, Bestor TH. Altered methylation patterns in cancer cell genomes: cause or consequence. Cancer Cell. 2002;1:299–305. doi: 10.1016/s1535-6108(02)00061-2. [DOI] [PubMed] [Google Scholar]
- 199.Esteller M, Fraga MF, Guo M, et al. DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum Mol Genet. 2001;10:3001–3007. doi: 10.1093/hmg/10.26.3001. [DOI] [PubMed] [Google Scholar]
- 200.Chen WY, Zeng X, Carter MG, et al. Heterozygous disruption of Hic1 predisposes mice to a gender-dependent spectrum of malignant tumors. Nat Genet. 2003;33:197–202. doi: 10.1038/ng1077. [DOI] [PubMed] [Google Scholar]
- 201.Herman JG, Merlo A, Mao L, et al. Inactivation of the CDKN2/p16/MTS1 gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995;55:4525–4530. [PubMed] [Google Scholar]
- 202.Toyota M, Ho C, Ahuja N, et al. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res. 1999;59:2307–2312. [PubMed] [Google Scholar]
- 203.Toyota M, Ahuja N, Ohe-Toyota M, et al. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Rashid A, Shen L, Morris JS, et al. CpG island methylation in colorectal adenomas. Am J Pathol. 2001;159:1129–1135. doi: 10.1016/S0002-9440(10)61789-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Petko Z, Ghiassi M, Shuber A, et al. Aberrantly methylated CDKN2A, MGMT, and MLH1 in colon polyps and in fecal DNA from patients with colorectal polyps. Clin Cancer Res. 2005;11:1203–1209. [PubMed] [Google Scholar]
- 206.Li H, Myeroff L, Smiraglia D, et al. SLC5A8, a sodium transporter, is a tumor suppressor gene silenced by methylation in human colon aberrant crypt foci and cancers. Proc Natl Acad Sci U S A. 2003;100:8412–8417. doi: 10.1073/pnas.1430846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chan AO, Broaddus RR, Houlihan PS, et al. CpG island methylation in aberrant crypt foci of the colorectum. Am J Pathol. 2002;160:1823–1830. doi: 10.1016/S0002-9440(10)61128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Moinova HR, Chen WD, Shen L, et al. HLTF gene silencing in human colon cancer. Proc Natl Acad Sci U S A. 2002;99:4562–4567. doi: 10.1073/pnas.062459899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Yamashita K, Dai T, Dai Y, et al. Genetics supersedes epigenetics in colon cancer phenotype. Cancer Cell. 2003;4:121–131. doi: 10.1016/s1535-6108(03)00190-9. [DOI] [PubMed] [Google Scholar]
- 210.Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet. 2006;38:787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 211.Shen L, Toyota M, Kondo Y, et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci U S A. 2007;104:18654–18659. doi: 10.1073/pnas.0704652104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Samowitz WS, Albertsen H, Herrick J, et al. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology. 2005;129:837–845. doi: 10.1053/j.gastro.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 213.Ogino S, Kawasaki T, Kirkner GJ, et al. Molecular correlates with MGMT promoter methylation and silencing support CpG island methylator phenotype-low (CIMP-low) in colorectal cancer. Gut. 2007;56:1564–1571. doi: 10.1136/gut.2007.119750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Deng G, Chen A, Pong E, et al. Methylation in hMLH1 promoter interferes with its binding to transcription factor CBF and inhibits gene expression. Oncogene. 2001;20:7120–7127. doi: 10.1038/sj.onc.1204891. [DOI] [PubMed] [Google Scholar]
- 215.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 216.Seligson DB, Horvath S, Shi T, et al. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435:1262–1266. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- 217.Fraga MF, Esteller M. Towards the human cancer epigenome: a first draft of histone modifications. Cell Cycle. 2005;4:1377–1381. doi: 10.4161/cc.4.10.2113. [DOI] [PubMed] [Google Scholar]