

Abstract
Thrombus formation at sites of disrupted atherosclerotic plaques is a leading cause of death and disability worldwide. Although the platelet is now recognized to be a central regulator of thrombus formation, development of antiplatelet reagents that selectively target thrombosis over hemostasis represents a challenge. Existing prophylactic antiplatelet therapies are centered on the use of aspirin, an irreversible cyclo-oxygenase inhibitor, and a thienopyridine such as clopidogrel (Plavix), which inactivates the ADP-stimulated P2Y12 receptor. Whilst these compounds are widely used and have beneficial effects for patients, their antithrombotic benefit is complicated by an elevated bleeding risk and substantial or partial “resistance”. Moreover, combination therapy with these two drugs increases the hemorrhagic risk even further. This review explores the possibility of inhibiting the platelet-surface ionotropic P2X1 receptor and/or elevating CD39/NTPDase1 activity as new therapeutic approaches to reduce overall platelet reactivity and recruitment of surrounding platelets at pro-thrombotic locations. Since both proteins affect platelet activation at an early stage in the events leading to thrombosis, but are less crucial in hemostasis, they provide new strategies to widen the cardiovascular therapeutic window without compromising safety.
Keywords: CD39, NTPDase1, apyrase, P2X1, platelet, thrombosis, drug therapy
Platelet Inhibition and Activation
Platelets are small, anuclear cellular components of the blood whose physiological role is to seal and initiate repair of damaged areas of the vasculature. In the intact circulation, platelets are normally kept quiescent by inhibitory factors promulgated by the intact endothelium (Marcus et al. 2005). Two of the major endothelial-derived inhibitors – prostacyclin (PGI2) and nitric oxide (NO) – are short-lived, membrane-permeable autacoids that function by increasing intra-platelet cyclic AMP (cAMP) and cyclic GMP (cGMP), respectively (Schwarz et al. 2001). An additional inhibitory system is presented by CD39/NTPDase1 (an apyrase) and CD73 (a 5′-nucleotidase). CD39 and CD73 are transmembrane proteins present on the surface of a variety of cells, including both endothelium and leukocytes. CD39 hydrolyzes luminal ATP and ADP to form AMP, which is then converted to adenosine via CD73 (reviewed in (Marcus et al. 2003)) [Figure 1]. Generated adenosine acts as an endogenous platelet inhibitor via the platelet A2A receptor to further dampen platelet reactivity (Cooper et al. 1995). Owing to their lability as autacoids, PGI2 and NO are not therapeutically viable in their natural forms. In contrast, studies in animal models have shown that a recombinant soluble form of human CD39 (solCD39) has platelet-inhibitory properties (Pinsky et al. 2002). Given the progress of solCD39 research as a potential antithrombotic agent, this review focuses on solCD39 in itself, although its use in combination with a synthetic CD73 might offer additional benefit.
Figure 1. Purine metabolism by CD39 and CD73.
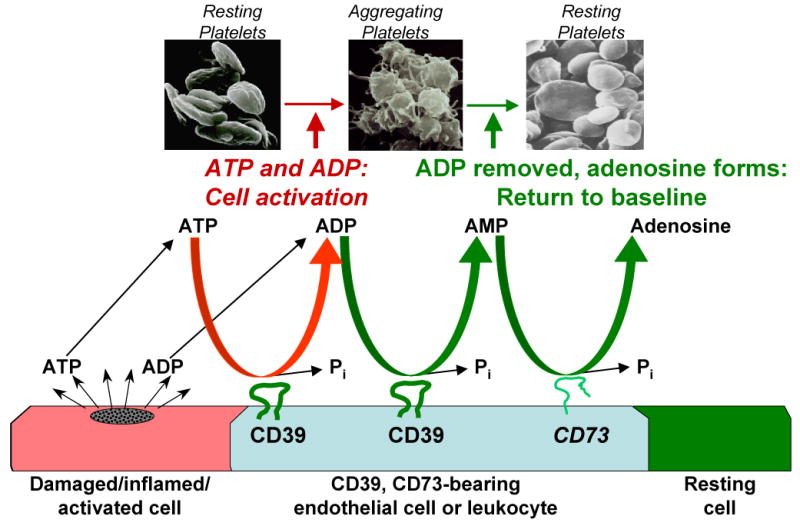
ATP and ADP released from damaged, inflamed, or activated cells are sequentially metabolized to AMP by CD39-bearing endothelial cells and/or leukocytes. AMP is then metabolized to platelet inhibitory adenosine by CD73, reverting platelets to the resting baseline state, thereby resulting in abolition of both platelet activation and recruitment, and thrombus formation.
When vascular integrity is breached, disruption of the protective endothelial layer exposes an underlying subendothelial environment rich in adhesive macromolecules, of which collagen represents a major component. Collagen is also exposed upon fissure of atherosclerotic plaques that are deposited during the aging process (Gawaz 2004). Whether collagen is exposed physiologically or pathologically, platelets bind to it via the immunoglobulin superfamily receptor glycoprotein VI (GPVI) and the α2β1 integrin (Farndale 2006). GPVI functions primarily to generate intracellular signals in response to collagen, while the main role of α2β1 is to promote attachment of platelets to the vessel wall (Varga-Szabo et al. 2008). In addition, under high shear conditions, such as in small arteries and arterioles, collagen-bound von Willebrand factor (vWF) plays a key role in initially tethering platelets to the subendothelium through the platelet glycoprotein (GP) Ib-V-IX complex (Varga-Szabo et al. 2008, Ruggeri and Mendolicchio 2007). Other adhesive proteins such as fibronectin and laminin and platelet surface receptors such as α5β1 are also thought to be involved in platelet tethering and activation (Varga-Szabo et al. 2008, Ruggeri and Mendolicchio 2007). Under conditions of rapid flow in the circulation, there is a narrow “time-window” during which platelets must become sufficiently activated to allow firm adhesion to the vessel wall via activated integrins; otherwise they detach and are swept away. The initial platelet response at the site of injury or ruptured plaque is therefore a key event that determines whether an occlusive thrombus may subsequently form. Alongside receptors for macromolecules exposed upon damage to a vessel wall, emerging evidence now suggests that the ATP-gated P2X1 receptor plays a unique role in the early stages of platelet activation, particularly for collagen (Fung et al. 2005, Hechler et al. 2003, Mahaut-Smith et al. 2004, Oury et al. 2001).
Whether initially activated physiologically or pathologically, platelets that have formed stable attachments to the underlying subendothelium generate an activated monolayer. These activated platelets lead to the local build-up of diffusible agonists that are secreted from dense granules (ADP, ATP and serotonin), synthesised within the platelet (thromboxane A2 (TXA2)) or formed by the coagulation cascade (thrombin). Secondary activation by these agonists is important for amplification of platelet recruitment, especially when the initial stimulus is modest (Jackson 2007). A variety of receptors are involved, but of particular importance in the context of this review is the ADP-dependent Gi-coupled platelet P2Y12 receptor, which synergises with P2Y1 and other Gq-coupled receptors (Gachet 2008) to induce inside-out activation of the major platelet integrin αIIbβ3. Activated αIIbβ3 is the main receptor responsible for attaching platelets to each other in a developing thrombus via its main ligand fibrinogen, and probably also vWF (Ruggeri and Mendolicchio 2007). Concerted platelet activation, in combination with fibrin formation as a result of the coagulation cascade, leads to a hemostatically stable thrombus. However, these thrombi also have the propensity to grow, eventually leading to physical occlusion of the vessel lumen, ischemia and tissue death (Nesbitt et al. 2006).
CD39/NTPDase1 can Transform a Local Prothrombotic Environment into an Antithrombotic Milieu
CD39 belongs to the E-NTPDase family of ectonucleotidases (Robson et al. 2006) and is localized mainly in endothelium and leukocytes; other E-NTPDase family members are absent from vascular cells and endothelium (Kaczmarek et al. 1996). In addition to CD39, CD73 (5′-nucleotidase) is present on vascular cells and converts the AMP generated from CD39 metabolism to adenosine. Thus, in contrast to all other known platelet inhibitors, CD39 acting in concert with CD73 can convert the local environment from a prothrombotic ADP/ATP-rich entity to an antithrombotic adenosine-rich environment (Pinsky et al. 2002) [Figure 1]. This was evident from observations that platelets were unresponsive to all agonists when in motion and in proximity to endothelial cells, even when eicosanoid and NO production were blocked (Marcus et al. 2003). It is important to emphasize that CD39 and CD73 do not act on the platelet per se but act in series to metabolize ATP and ADP secreted from activated platelets to AMP and hence to adenosine (Marcus et al. 1997). This lock-step mechanism enables the two ectonucleotidases to ubiquitously attenuate or abolish platelet activation and recruitment regardless of the original stimulus, without a direct effect on platelets themselves.
Since collagen stimulation initiates dense granule release at the early stages of platelet activation (delay of 15-20 seconds) (Atkinson et al. 2003), the enhanced metabolism of ATP and ADP by therapeutically administered solCD39 would reduce secondary autoamplification and recruitment and therefore, thrombus formation (Marcus et al. 2005). Acting together with endogenous CD73, CD39 will also theoretically raise levels of endogenous adenosine to elevate the basal platelet activation threshold in the local microenvironment (as mentioned above). Additionally, anti-aggregatory and anti-inflammatory effects of adenosine reduce thrombus size and promote tissue repair, respectively (Deaglio et al. 2007, Kim and Liao 2008).
SolCD39 administration in vivo was shown to ameliorate the sequelae of stroke and reverse excessive platelet reactivity in a murine model, even when administered 3 h after stroke induction. This was not associated with bleeding complications such as intracerebral hemorrhage, an inherent complication observed with currently available therapeutic modalities (Pinsky et al. 2002). This also suggests that solCD39 does not interfere with the major signaling pathways involved in hemostasis. Therapeutic benefit of solCD39 has also been demonstrated in animal models of cardiac ischemia (Marcus et al. 2003), development of atherosclerosis (Koziak et al. 2008), regulation of leukocyte activity (Deaglio et al. 2007), inhibition of metastasis (Uluckan et al. 2008), and for therapy in transplantation (Dwyer et al. 2004). Thus, the therapeutic window for optimal concentrations of solCD39 which could abrogate thrombosis without inducing hemorrhage is likely much wider than with existing antiplatelet therapies (Serebruany et al. 2008). Together with the in vivo stability benefits of solCD39 (half-life of 7 days in porcine species) (Buergler et al. 2005), it suggests that solCD39 could represent significant benefit to patients with a low threshold for platelet activation, or who are resistant to existing treatment paradigms (Marcus et al. 2005). Such benefit might be further enhanced by development of a solCD39 and synthetic CD73 analogue combination.
P2X1 Receptors represent a Unique Pathway during Early Platelet Activation
The P2X1 receptor is unique amongst the array of proteins that mediate platelet activation, since it is the only ligand-gated Ca2+-permeable ion channel reported to date on the platelet plasma membrane (Mahaut-Smith et al. 2004). With an activation time course in the order of a few milliseconds, ATP-gated P2X1 receptors provide the most rapid pathway by which an agonist released during vascular injury can elevate cytosolic Ca2+ in the platelet (Sage and Rink 1987, Mahaut-Smith et al. 2000). Intracellular Ca2+ is a key second messenger during platelet activation, able to stimulate most platelet functional events on its own, although it will normally act in synergy with other signaling pathways to promote platelet responses (Jackson and Schoenwaelder 2003). The rapid activation response of P2X1 may be particularly important in faster flowing regions of the circulation. Indeed, studies to date indicate that the greatest contribution of P2X1 receptors to platelet activation occurs in vitro under conditions of high shear and in vivo in thrombosis within small arteries and arterioles (Hechler et al. 2003).
In contrast to the millimolar concentrations of ATP found in the cytosol or the near-molar concentrations stored in platelet granules (Holmsen and Weiss 1979), P2X1 receptors only require micromolar extracellular concentrations of this nucleotide for activation (Valera et al. 1994). Therefore, the non-selective cation channel will be efficiently stimulated by ATP leaked from the cytoplasm of damaged vascular cells or released from platelet dense granules upon agonist stimulation [Figure 2]. Selective activation of P2X1 receptors induces weak platelet activation, including reversible shape change, granule centralization and microaggregate formation (reviewed in (Mahaut-Smith et al. 2004)), but its main effects in vitro are the ability to synergistically enhance platelet activation by low concentrations of other agonists, particularly collagen (Hechler et al. 2003, Oury et al. 2001) and thrombin (Erhardt et al. 2006). The exact mechanism by which P2X1 achieves this effect is unclear, but recent evidence suggests that the ligand-gated ion channel can contribute substantially to the [Ca2+]i increase following stimulation of a variety of other receptors, including those for collagen, thrombin, TXA2 and even ADP (Fung et al. 2007). As with functional responses, the overall contribution of P2X1 is more important at lower concentrations of the primary agonist, at least for collagen and thrombin (Fung et al. 2007). For instance, P2X1 receptor inhibition leads to a >90% reduction of the peak [Ca2+]i increase stimulated by a low dose of collagen (0.25 μg ml-1) (Fung et al. 2005). We have provided evidence that this contribution by P2X1 occurs via a mainly autocrine (as opposed to paracrine) mechanism following release of ATP from dense granules (Fung et al. 2007). This could be particularly important in amplifying activation of individual platelets as they initially tether to the damaged vessel wall, when they are acting relatively independently of their neighbors.
Figure 2. Unique role for P2X1 in platelet activation.

At sites of vascular damage, ATP is released into the circulation from the cytoplasm of endothelial or other injured cells in the vessel wall. In addition, platelet activation by major agonists (e.g. collagen via GPVI receptors, thrombin via PAR receptors and TXA2 via TPα receptors) initiates ATP secretion from the dense granules. ATP activation of P2X1 leads to rapid Ca2+ influx and also potentiates intracellular Ca2+ release via P2Y receptors (Mahaut-Smith et al. 2004). Possible mechanisms for this synergy include 1) upregulating production of IP3 via phospholipase C and 2) directly potentiating IP3 receptor-mediated Ca2+-store mobilization. The increase of [Ca2+]i activates downstream Ca2+-dependent signaling pathways to accelerate platelet activation events, reducing the time window for platelet activation/adhesion.
A further property that distinguishes the P2X1 receptor from other pathways of Ca2+ mobilization is its insensitivity to cAMP and cGMP (Sage et al. 2000). This renders P2X1 receptor-mediated activation insensitive to two of the mechanisms whereby endothelium inhibits platelet activation, namely NO and PGI2, ((Sage et al. 2000); CYE Fung and MP Mahaut-Smith, unpublished observations). In addition, although CD39 is able to metabolize ATP, the rate at which this occurs is likely to be insufficient to prevent local P2X1 activation by released ATP. Consequently, the relative contribution of P2X1 may be more substantial in the very initial stages of platelet activation when residual effects of NO and PGI2 lead to depressed signaling through other receptors.
From a therapeutic standpoint, the above unique properties of the P2X1 receptor suggest that this channel be considered as a novel antithrombotic target. This concept is further strengthened when one considers the minimal effect on the bleeding time in the setting of a substantial reduction in arteriolar thrombosis in mice lacking the P2X1 receptor (Hechler et al. 2003). Since this important role of P2X1 in vivo has been proposed to result from an increased shear rate in small vessels, inhibition of the ligand-gated channel may lead to marked reduction in thrombus formation at sites of atherosclerotic plaques where some of the highest shear rates occur (Strony et al. 1993).
Conclusion
Given the high costs of medical care, diagnostic procedures and therapy associated with cardiovascular disease and its complications, prevention or reversal of early events in thrombosis would provide significant benefits. A number of anti-platelet antithrombotic therapies exist, such as acetylation of cyclooxygenase by aspirin (Jackson and Schoenwaelder 2003) and inhibition of P2Y12 receptors with irreversible, metabolism-requiring compounds such as prasugrel, clopidogrel and ticlopidine, or with reversible direct-acting inhibitors such as cangrelor and AZD6140 (Michelson 2008). While these compounds have beneficial effects for patients, their antithrombotic benefit is complicated by an elevated bleeding risk in some (Buresly et al. 2005), and “resistance” in others (Cerbone et al. 2008). Combination therapy can increase the hemorrhagic risk even further (Serebruany et al. 2008). αIIbβ3 inhibitors, such as abciximab, have also been developed, and can substantially inhibit platelet activation; however, their use is complicated by the associated risk of increased hemorrhage (Serebruany et al. 2008). There are similar concerns for several new anticoagulant compounds in development, such as heparins, Factor Xa (FXa) and thrombin inhibitors that neutralize upstream elements of the coagulation cascade (Greinacher and Warkentin 2008).
We propose that solCD39 or a P2X1 antagonist provide advantages over existing treatment paradigms, since they target early events within the platelet activation pathway in a manner that influences thrombotic events with little effect on hemostasis. This could be of particular benefit to certain patient subsets, for example patients with lower platelet activation thresholds (Buresly et al. 2005), aspirin or clopidogrel low/non-responders (Cerbone et al. 2008), or those where long-term administration leads to bleeding complications. Additional research is necessary to establish the effects of P2X1 inhibition and solCD39 treatment in control of downstream platelet function in patients, their amelioration of clinical cardiovascular events and possible side-effects associated with therapeusis. Nevertheless, these two approaches merit further investigation in the search for safe and effective antiplatelet drugs.
Acknowledgments
Grant support:
CYEF and MPM-S: British Heart Foundation
AJM and MJB: NIH 5R37HL047073 and 5P01HL046403, and a Merit Review Grant from the Department of Veterans Affairs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Atkinson BT, Jarvis GE, Watson SP. Activation of GPVI by collagen is regulated by α2β1 and secondary mediators. J Thromb Haemost. 2003;1:1278–1287. doi: 10.1046/j.1538-7836.2003.00245.x. [DOI] [PubMed] [Google Scholar]
- Buergler JM, Maliszewski CR, Broekman MJ, Kaluza GL, Schulz DG, Marcus AJ, et al. Effects of SolCD39, a novel inhibitor of platelet aggregation, on platelet deposition and aggregation after PTCA in a porcine model. J Thromb Thrombolysis. 2005;19:115–122. doi: 10.1007/s11239-005-1381-y. [DOI] [PubMed] [Google Scholar]
- Buresly K, Eisenberg MJ, Zhang X, Pilote L. Bleeding complications associated with combinations of aspirin, thienopyridine derivatives, and warfarin in elderly patients following acute myocardial infarction. Arch Intern Med. 2005;165:784–789. doi: 10.1001/archinte.165.7.784. [DOI] [PubMed] [Google Scholar]
- Cerbone AM, Macarone-Palmieri N, Saldalamacchia G, Coppola A, Di Minno G, Rivellese AA. Diabetes, vascular complications and antiplatelet therapy: open problems. Acta Diabetol. 2008 doi: 10.1007/s00592-008-0079-y. in press. [DOI] [PubMed] [Google Scholar]
- Cooper JA, Hill SJ, Alexander SP, Rubin PC, Horn EH. Adenosine receptor-induced cyclic AMP generation and inhibition of 5-hydroxytryptamine release in human platelets. Br J Clin Pharmacol. 1995;40:43–50. doi: 10.1111/j.1365-2125.1995.tb04533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaglio S, Dwyer KM, Gao W, Friedman D, Usheva A, Erat A, et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J Exp Med. 2007;204:1257–1265. doi: 10.1084/jem.20062512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer KM, Robson SC, Nandurkar HH, Campbell DJ, Gock H, Murray-Segal LJ, et al. Thromboregulatory manifestations in human CD39 transgenic mice and the implications for thrombotic disease and transplantation. J Clin Invest. 2004;113:1440–1446. doi: 10.1172/JCI19560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt JA, Toomey JR, Douglas SA, Johns DG. P2X1 stimulation promotes thrombin receptor-mediated platelet aggregation. J Thromb Haemost. 2006;4:882–890. doi: 10.1111/j.1538-7836.2006.01849.x. [DOI] [PubMed] [Google Scholar]
- Farndale RW. Collagen-induced platelet activation. Blood Cells Mol Dis. 2006;36:162–165. doi: 10.1016/j.bcmd.2005.12.016. [DOI] [PubMed] [Google Scholar]
- Fung CY, Brearley CA, Farndale RW, Mahaut-Smith MP. A major role for P2X1 receptors in the early collagen-evoked intracellular Ca2+ responses of human platelets. Thromb Haemost. 2005;94:37–40. doi: 10.1160/TH04-11-0732. [DOI] [PubMed] [Google Scholar]
- Fung CY, Cendana C, Farndale RW, Mahaut-Smith MP. Primary and secondary agonists can use P2X1 receptors as a major pathway to increase intracellular Ca2+ in the human platelet. J Thromb Haemost. 2007;5:910–917. doi: 10.1111/j.1538-7836.2007.02525.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gachet C. P2 receptors, platelet function and pharmacological implications. Thromb Haemost. 2008;99:466–472. doi: 10.1160/TH07-11-0673. [DOI] [PubMed] [Google Scholar]
- Gawaz M. Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc Res. 2004;61:498–511. doi: 10.1016/j.cardiores.2003.11.036. [DOI] [PubMed] [Google Scholar]
- Greinacher A, Warkentin TE. The direct thrombin inhibitor hirudin. Thromb Haemost. 2008;99:819–829. doi: 10.1160/TH07-11-0693. [DOI] [PubMed] [Google Scholar]
- Hechler B, Lenain N, Marchese P, Vial C, Heim V, Freund M, et al. A role of the fast ATP-gated P2X1 cation channel in thrombosis of small arteries in vivo. J Exp Med. 2003;198:661–667. doi: 10.1084/jem.20030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmsen H, Weiss HJ. Secretable storage pools in platelets. Annu Rev Med. 1979;30:119–134. doi: 10.1146/annurev.me.30.020179.001003. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Schoenwaelder SM. Antiplatelet therapy: in search of the ‘magic bullet’. Nat Rev Drug Discov. 2003;2:775–789. doi: 10.1038/nrd1198. [DOI] [PubMed] [Google Scholar]
- Jackson SP. The growing complexity of platelet aggregation. Blood. 2007;109:5087–5095. doi: 10.1182/blood-2006-12-027698. [DOI] [PubMed] [Google Scholar]
- Kaczmarek E, Koziak K, Sévigny J, Siegel JB, Anrather J, Beaudoin AR, et al. Identification and characterization of CD39 vascular ATP diphosphohydrolase. J Biol Chem. 1996;271:33116–33122. doi: 10.1074/jbc.271.51.33116. [DOI] [PubMed] [Google Scholar]
- Kim HH, Liao JK. Translational therapeutics of dipyridamole. Arterioscler Thromb Vasc Biol. 2008;28:s39–s42. doi: 10.1161/ATVBAHA.107.160226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koziak K, Bojakowska M, Robson SC, Bojakowski K, Soin J, Csizmadia E, et al. Overexpression of CD39/nucleoside triphosphate diphosphohydrolase-1 decreases smooth muscle cell proliferation and prevents neointima formation after angioplasty1. J Thromb Haemost. 2008;6:1191–1197. doi: 10.1111/j.1538-7836.2008.03019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahaut-Smith MP, Ennion SJ, Rolf MG, Evans RJ. ADP is not an agonist at P2X1 receptors: evidence for separate receptors stimulated by ATP and ADP on human platelets. Br J Pharmacol. 2000;131:108–114. doi: 10.1038/sj.bjp.0703517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahaut-Smith MP, Tolhurst G, Evans RJ. Emerging roles for P2X1 receptors in platelet activation. Platelets. 2004;15:131–144. doi: 10.1080/09537100410001682788. [DOI] [PubMed] [Google Scholar]
- Marcus AJ, Broekman MJ, Drosopoulos JH, Islam N, Pinsky DJ, Sesti C, et al. Heterologous cell-cell interactions: thromboregulation, cerebroprotection and cardioprotection by CD39 (NTPDase-1) J Thromb Haemost. 2003;1:2497–2509. doi: 10.1111/j.1538-7836.2003.00479.x. [DOI] [PubMed] [Google Scholar]
- Marcus AJ, Broekman MJ, Drosopoulos JH, Olson KE, Islam N, Pinsky DJ, et al. Role of CD39 (NTPDase-1) in thromboregulation, cerebroprotection, and cardioprotection. Semin Thromb Hemost. 2005;31:234–246. doi: 10.1055/s-2005-869528. [DOI] [PubMed] [Google Scholar]
- Marcus AJ, Broekman MJ, Drosopoulos JHF, Islam N, Alyonycheva TN, Safier LB, et al. The endothelial cell ecto-ADPase responsible for inhibition of platelet function is CD39. J Clin Invest. 1997;99:1351–1360. doi: 10.1172/JCI119294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelson AD. P2Y12 antagonism: promises and challenges. Arterioscler Thromb Vasc Biol. 2008;28:s33–s38. doi: 10.1161/ATVBAHA.107.160689. [DOI] [PubMed] [Google Scholar]
- Nesbitt WS, Mangin P, Salem HH, Jackson SP. The impact of blood rheology on the molecular and cellular events underlying arterial thrombosis. J Mol Med. 2006;84:989–995. doi: 10.1007/s00109-006-0101-1. [DOI] [PubMed] [Google Scholar]
- Oury C, Toth-Zsamboki E, Thys C, Tytgat J, Vermylen J, Hoylaerts MF. The ATP-gated P2X1 ion channel acts as a positive regulator of platelet responses to collagen. Thromb Haemost. 2001;86:1264–1271. [PubMed] [Google Scholar]
- Pinsky DJ, Broekman MJ, Peschon JJ, Stocking KL, Fujita T, Ramasamy R, et al. Elucidation of the thromboregulatory role of CD39/ectoapyrase in the ischemic brain. J Clin Invest. 2002;109:1031–1040. doi: 10.1172/JCI10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson SC, Sévigny J, Zimmermann H. The E-NTPDase family of ectonucleotidases: Structure function relationships and pathophysiological significance. Purinergic Signal. 2006;2:409–430. doi: 10.1007/s11302-006-9003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res. 2007;100:1673–1685. doi: 10.1161/01.RES.0000267878.97021.ab. [DOI] [PubMed] [Google Scholar]
- Sage SO, Rink TJ. The kinetics of changes in intracellular calcium concentration in fura-2-loaded human platelets. J Biol Chem. 1987;262:16364–16369. [PubMed] [Google Scholar]
- Sage SO, Yamoah EH, Heemskerk JW. The roles of P2X1 and P2T AC receptors in ADP-evoked calcium signalling in human platelets. Cell Calcium. 2000;28:119–126. doi: 10.1054/ceca.2000.0139. [DOI] [PubMed] [Google Scholar]
- Schwarz UR, Walter U, Eigenthaler M. Taming platelets with cyclic nucleotides. Biochem Pharmacol. 2001;62:1153–1161. doi: 10.1016/s0006-2952(01)00760-2. [DOI] [PubMed] [Google Scholar]
- Serebruany VL, Malinin AI, Ferguson JJ, Vahabi J, Atar D, Hennekens CH. Bleeding risks of combination vs. single antiplatelet therapy: a meta-analysis of 18 randomized trials comprising 129 314 patients. Fundamental & Clinical Pharmacology. 2008;22:315–321. doi: 10.1111/j.1472-8206.2008.00582.x. [DOI] [PubMed] [Google Scholar]
- Strony J, Beaudoin A, Brands D, Adelman B. Analysis of shear stress and hemodynamic factors in a model of coronary artery stenosis and thrombosis. Am J Physiol. 1993;265:H1787–H1796. doi: 10.1152/ajpheart.1993.265.5.H1787. [DOI] [PubMed] [Google Scholar]
- Uluckan O, Eagleton MC, Floyd DH, Morgan EA, Hirbe AC, Kramer M, et al. APT102, a novel ADPase, cooperates with aspirin to disrupt bone metastasis in mice. J Cell Biochem. 2008;104:1311–1323. doi: 10.1002/jcb.21709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valera S, Hussy N, Evans RJ, Adami N, North RA, Surprenant A, et al. A new class of ligand-gated ion channel defined by P2× receptor for extracellular ATP. Nature. 1994;371:516–519. doi: 10.1038/371516a0. [DOI] [PubMed] [Google Scholar]
- Varga-Szabo D, Pleines I, Nieswandt B. Cell Adhesion Mechanisms in Platelets. Arterioscler Thromb Vasc Biol. 2008;28:403–412. doi: 10.1161/ATVBAHA.107.150474. [DOI] [PubMed] [Google Scholar]