

Abstract
Cystathionine β-lyase (CBL) catalyzes the hydrolysis of l-cystathionine (l-Cth) to produce l-homocysteine, pyruvate, and ammonia. A series of active-site mutants of Escherichia coli CBL (eCBL) was constructed to investigate the roles of residues R58, R59, D116, W340, and R372 in catalysis and inhibition by aminoethoxyvinylglycine (AVG). The effects of these mutations on the 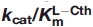 for the β-elimination reaction range from a reduction of only 3-fold for D116A and D116N to 6 orders of magnitude for the R372L and R372A mutants. The order of importance of these residues for the hydrolysis of l-Cth is: R372 >> R58 > W340 ≈ R59 > D116. Comparison of the kinetic parameters for l-Cth hydrolysis with those for inhibition of eCBL by AVG demonstrates that residue R58 tethers the distal carboxylate group of the substrate and confirms that residues W340 and R372 interact with the α-carboxylate moiety. The increase in the pKa of the acidic limb and decrease in the pKa of the basic limb of the versus pH profiles of the R58K and R58A mutants, respectively, support a role for this residue in modulating the pKa of an active-site residue.
for the β-elimination reaction range from a reduction of only 3-fold for D116A and D116N to 6 orders of magnitude for the R372L and R372A mutants. The order of importance of these residues for the hydrolysis of l-Cth is: R372 >> R58 > W340 ≈ R59 > D116. Comparison of the kinetic parameters for l-Cth hydrolysis with those for inhibition of eCBL by AVG demonstrates that residue R58 tethers the distal carboxylate group of the substrate and confirms that residues W340 and R372 interact with the α-carboxylate moiety. The increase in the pKa of the acidic limb and decrease in the pKa of the basic limb of the versus pH profiles of the R58K and R58A mutants, respectively, support a role for this residue in modulating the pKa of an active-site residue.
Keywords: cystathionine, pyridoxal 5′-phosphate, site-directed mutagenesis, transsulfuration, aminoethoxyvinylglycine, enzyme kinetics
Introduction
The enzyme cystathionine β-lyase is a target for the development of new antimicrobial compounds to address the challenge of increasing antibiotic resistance. The design of novel inhibitors of this enzyme will be facilitated by the information presented in this manuscript on the roles of specific amino acids in the active site and their interaction with substrates and inhibitors.
Cystathionine β-lyase (CBL) catalyses the hydrolysis of l-cystathionine (l-Cth) to produce l-homocysteine (l-Hcys), pyruvate, and ammonia. This reaction is the second step in the transsulfuration pathway, which converts l-cysteine (l-Cys) to l-Hcys, the immediate precursor of l-methionine (Fig. 1). The two enzymes of the transsulfuration pathway, cystathionine γ-synthase (CGS) and CBL, are unique to plants and bacteria.1 These enzymes are attractive targets for the development of novel antimicrobial compounds, to address the growing challenge of antibiotic resistance by microbial pathogens, because methionine is a precursor for protein biosynthesis as well as thiamine biosynthesis, via the ubiquitous methyl donor S-adenosylmethionine, thereby providing a link to DNA replication.2 A detailed study of the mechanism of slow-binding inhibition of Escherichia coli CBL (eCBL) by the β,γ-unsaturated amino acid aminoethoxyvinylglycine (AVG) and the structure of the eCBL-AVG complex were reported by Clausen et al.3 This work provides a solid foundation for investigation of the roles of active-site residues and the design of inhibitors, as exemplified by the novel, slow-binding inhibitors of eCBL, with IC50 values in the μM and sub-μM range, recently developed by Ejim et al.2
Figure 1.
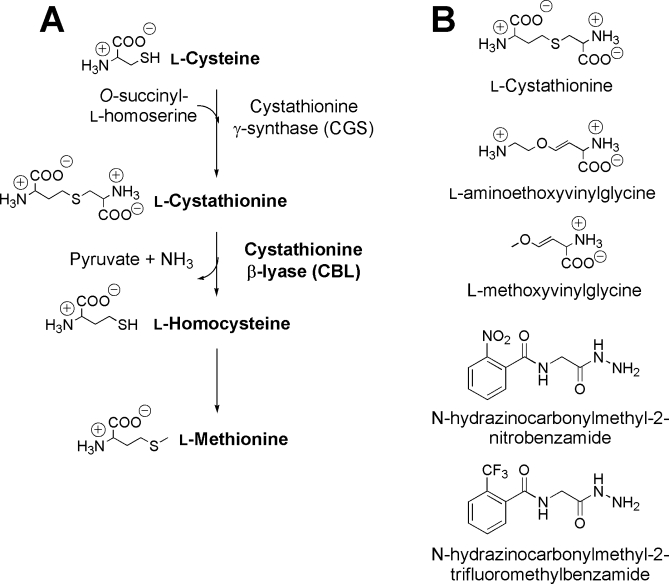
A: The enzymes and metabolites of the transsulfuration pathway. B: The structures of the l-Cth substrate and slow-binding inhibitors of eCBL.2,3
The structures of eCBL, yeast cystathionine γ-lyase (yCGL), the second enzyme of the reverse transsulfuration pathway of mammals and yeast, and E. coli CGS (eCGS) are so similar that the r.m.s. deviation in their least squares superposition is only ∼1.5 Å between ∼350 Cα atoms of the protein backbone.4–6 Therefore, these enzymes provide a useful model system to investigate determinants of specificity, as differences in their substrate and reaction specificity are likely the result of differences in the placement and mobility of active-site residues.4–6
The active site of the homotetrameric eCBL enzyme is situated at the subunit interface of the catalytic dimer and is comprised of residues from each subunit.4 The crystal structures of eCBL in complex with AVG, N-hydrazinocarbonylmethyl-2-nitrobenzamide, and N-hydrazinocarbonylmethyl-2-trifluoromethylbenzamide provide valuable insight into the active site and mechanism of this enzyme (Fig. 1).2–4 The α-carboxylate group of AVG interacts with the side chains of W340 and R372 (Fig. 2).3 Although AVG lacks the distal carboxylate group of l-Cth, an interaction is observed between R58 and the trifluoromethyl moiety of the N-hydrazinocarbonylmethyl-2-trifluoromethylbenzamide inhibitor designed by Ejim et al.2 The side chains of R58 and R59, which approach the bound inhibitor in the eCBL-AVG complex to within 3.6 and 6.7 Å, respectively, have both been proposed to interact with the distal carboxylate group of the l-Cth substrate.3,4,6 A series of site-directed mutants of the eCBL active-site residues R58, R59, D116, W340, and R372 was constructed to probe the specific roles of these residues (Fig. 2). The observed reduction in the activity of the R58A,K, W340F, and R372A,K,L site-directed mutants and the differences in their kinetic parameters for hydrolysis of l-Cth and inhibition by AVG identify R58, W340, and R372 as residues interacting with the distal and α-carboxylate groups of the l-Cth substrate.
Figure 2.
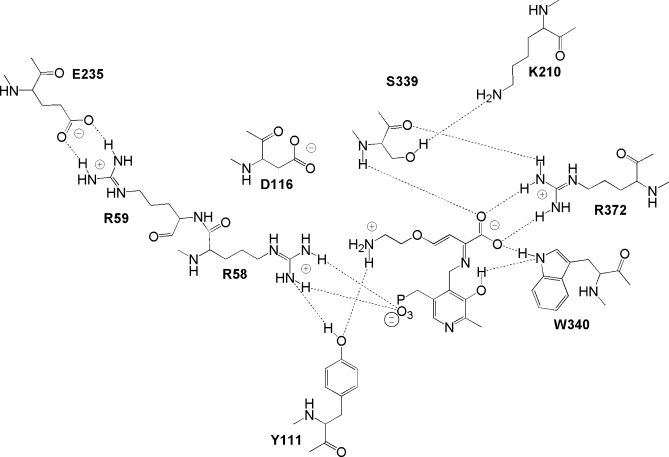
Observed contacts of AVG in the active-site of eCBL.3 The dotted lines represent putative hydrogen bond distances of ≤ 3.3 Å between heteroatoms. The image was constructed using ChemDraw and PDB entry 1CL2.
RESULTS
All of the site-directed mutants were soluble and yields were between 15–41 mg/L, which is comparable to the 56 mg/L reported for the wild-type enzyme.7 The data for the R58K, R372A, and R372L mutants could not be fit to the Michaelis–Menten equation as saturation kinetics were not observed within the solubility limit of the l-Cth substrate. Therefore, with the assumption that 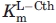 >> [l-Cth], the Michaelis–Menten equation was modified to obtain
>> [l-Cth], the Michaelis–Menten equation was modified to obtain 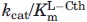 values, but not independent estimates of kcat and
values, but not independent estimates of kcat and 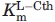 , for these enzymes (Table I). The specific activity versus pH profile of eCBL, and all of the site-directed mutants investigated, is bell-shaped. Upon reaction of eCBL with AVG, the 424-nm peak of the internal aldimine form of the PLP cofactor shifts to 341 nm and an isobestic point at 386 nm is observed, as described by Clausen et al.3
, for these enzymes (Table I). The specific activity versus pH profile of eCBL, and all of the site-directed mutants investigated, is bell-shaped. Upon reaction of eCBL with AVG, the 424-nm peak of the internal aldimine form of the PLP cofactor shifts to 341 nm and an isobestic point at 386 nm is observed, as described by Clausen et al.3
Table I.
Kinetic parameters of eCBL and site-directed mutantsa
| Enzyme | kcat (s−1) |
(mM) |
(M−1s−1) |
|---|---|---|---|
| eCBL | 34.1 ± 0.6 | 0.18 ± 0.01 | (1.9 ± 0.1) × 105 |
| R58A | 9.7 ± 0.3 | 5.1 ± 0.3 | (1.92 ± 0.05) × 103 |
| R58Kb | n.s. | n.s. | 385 ± 1 |
| R59A | 45.8 ± 0.4 | 1.02 ± 0.03 | (4.5 ± 0.1) × 104 |
| R59K | 36.7 ± 0.8 | 1.51 ± 0.09 | (2.4 ± 0.1) × 104 |
| D116A | 41.9 ± 0.7 | 0.67 ± 0.04 | (6.2 ± 0.3) × 104 |
| D116N | 37.9 ± 0.4 | 0.55 ± 0.02 | (6.9 ± 0.2) × 104 |
| W340F | 79 ± 1 | 1.35 ± 0.05 | (5.9 ± 0.2) × 104 |
| R372Ab | n.s. | n.s. | 0.09 ± 0.04 |
| R372K | 8.1 ± 0.8 | 15 ± 2 | 550 ± 20 |
| R372Lb | n.s. | n.s | 0.23 ± 0.03 |
Kinetic parameters reported are for hydrolysis of l-Cth. Reaction conditions: 2 mM DTNB, 0.01–6.4 mM l-Cth and 0.068–6.6 μM wild-type or mutant eCBL, depending on the activity of the enzyme, in assay buffer at 25°C. The data were fit to the Michaelis-Menten equation to obtain kcat and and equation 1 to obtain .
n.s. indicates that exceeds the solubility limit of the l-Cth, such that was determined via linear regression.
The D116A,N and R59A,K mutants
The pH optima of eCBL-D116A and D116N are unchanged and the IC50 and Ki values, for inhibition by AVG, and the kcat and values, for l-Cth hydrolysis, of these mutants are within ∼2-fold and 3-4-fold, respectively, of the wild-type enzyme (Tables I and II). Similarly, the kcat, IC50, and Ki values of R59A are within ∼2-fold of the wild-type enzyme, the pH optima is unchanged and the of this mutant is increased by only 5.7-fold. In contrast, while the kcat of R59K is within ∼2-fold of wild-type eCBL and the pH profile is unchanged, the , IC50 and Ki values of R59K are increased by 8.4, 29, and 6.3-fold, respectively (Tables I and II; Fig. 3).
Table II.
Kinetic parameters for inhibition of eCBL and site-directed mutants by AVG
| Enzyme | IC50 (μM)a | Ki (μM)b | k2 (s−1)b | k1 (s−1)c |
|---|---|---|---|---|
| eCBL | 1.7 ± 0.2 | 1.9 ± 0.6 | (5 ± 1) × 10-4 | 263 |
| R58A | 1.74 ± 0.08 | 6 ± 1 | (2.0 ± 0.4) × 10-3 | 333 |
| R58K | 23 ± 1 | 55 ± 7 | (3.5 ± 0.3) × 10-3 | 64 |
| R59A | 1.34 ± 0.05 | 2.7 ± 0.9 | (1.3 ± 0.4) × 10-3 | 481 |
| R59K | 4.6 ± 0.3 | 12 ± 3 | (6 ± 1) × 10-4 | 50 |
| D116A | 2.6 ± 0.4 | 3.9 ± 0.9 | (4.2 ± 0.9) × 10-4 | 108 |
| D116N | 3.3 ± 0.6 | 2.7 ± 0.8 | (2.6 ± 0.8) × 10-4 | 92 |
| W340F | 43 ± 6 | 40 ± 10 | (7 ± 2) × 10-4 | 17.5 |
| R372A | n.d. | n.d. | n.d. | |
| R372K | 3500 ± 1400 | 9100 ± 400 | (2.30 ± 0.02) × 10-3 | 0.25 |
| R372L | n.d. | n.d. | n.d. |
Reaction conditions for IC50 measurements: Enzyme (0.024–4.3 μM, depending on the activity of the mutant) was incubated with 0.05–104 μM AVG in assay buffer at 25°C for 10 min. Activity was subsequently measured (n = 4) at a l-Cth substrate concentration of 0.1 mM and the data were fit to equation 3 to obtain the IC50 value for each enzyme.
Reaction conditions for measurement of Ki and k2: Wild-type eCBL and site-directed mutants were incubated with 1.5 mM l-Cth and 0.005–7.5 mM AVG in assay buffer and the progress of the reactions was monitored for 30 min. The progress curves were fit to equation 5 to obtain kobs values, which were plotted versus inhibitor concentration and fit to equation 6 to obtain the values of k2 and Ki.
Values of the rate constant k1, for the association of eCBL and AVG were calculated using the equation Ki = k2/k1.
Figure 3.
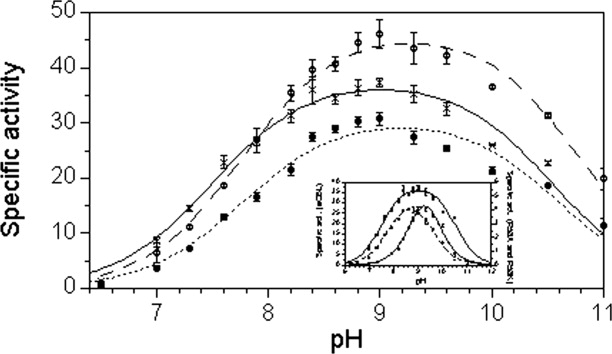
The pH dependence of specific activity for the hydrolysis of l-Cth catalyzed by eCBL (×) and the R59A (○) and R59K (•) mutants site-directed mutants. Inset: Comparison of the pH dependence of the specific activity of eCBL (×), R58A (○) and R58K (•). Reaction conditions: MBP buffer (50 mM MOPS, 50 mM Bicine and 50 mM proline), 20 μM PLP, 2 mM DTNB, 6.25 mM l-Cth and 68 nM eCBL, 0.96 μM R58A, 0.56 μM R58K, 58 nM R59A or 0.13 μM R59K. Measurements were performed in quadruplicate, as represented by the error bars.
The R58A and R58K mutants
The kcat and of R58A are decreased 3.5-fold and increased 28-fold, respectively, and the kcat/ of R58K, which could not be saturated within the solubility limit of l-Cth, is increased ∼500-fold (Table I). In contrast, the IC50 and Ki values of R58A are within 3-fold of those of the wild-type enzyme, while those of R58K are increased by 14 and 29-fold, respectively (Table II, Fig. 5). Mutation of R58 results in a shift in the basic and the acidic limbs of the specific activity versus pH profiles of R58A and R58K, respectively. The pH optima of the R58A and R58K mutants are correspondingly shifted to ∼8.5–9 and 9–9.5, respectively, compared with 8.5–9.5 for the wild-type enzyme (Fig. 3). Therefore, the effect of pH on the kcat/ of R58A and R58K, as well as R372K, which has an 345-fold reduction in kcat/ at pH 8.5, but an unaltered pH optimum, was determined (Fig. 4). The values of pKa1 and pKa2, corresponding to the acidic and basic limbs of the kcat/ versus pH profile, respectively, are presented in Table III. The pKa1 values of R58A and R372K and the pKa2 values of R58K and R372K are similar to the wild-type enzyme, differing by only 0.01–0.24 pH units beyond the experimental error, confirming the trend observed in the specific activity versus pH profiles of these enzymes. In contrast, the pKa1 of R58K is increased 0.66 pH units and pKa2 of R58A is decreased by 0.67 pH units, a change of approximately 0.5 pH units beyond the experimental error in both cases (Table III, Fig. 4).
Figure 5.
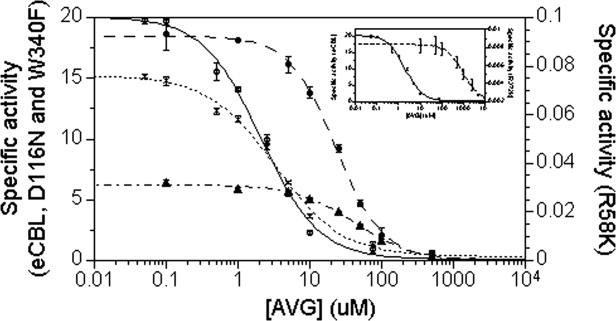
The dependence of enzyme activity on AVG concentration for wild-type eCBL (○) and the R58K (•), D116N (×) and W340 (▴) site-directed mutants. Inset: Comparison of the effect of AVG inhibitor concentration on the activity of eCBL (○) and the R372K (◊) mutant. Reaction conditions: The enzyme (32 nM eCBL, 4.3 μM R58K, 24 nM D116N, 57 nM W340 and 1.9 μM R372K) was mixed with 0.05–104 μM AVG in 50 mM Tris, pH 8.5, containing 20 μM PLP, and incubated at 25 °C for 10 min. Activity was subsequently measured in quadruplicate, as represented by the error bars, at a l-Cth substrate concentration of 0.1 mM and the data was fit to equation 3 to obtain the IC50 value for each enzyme.
Figure 4.
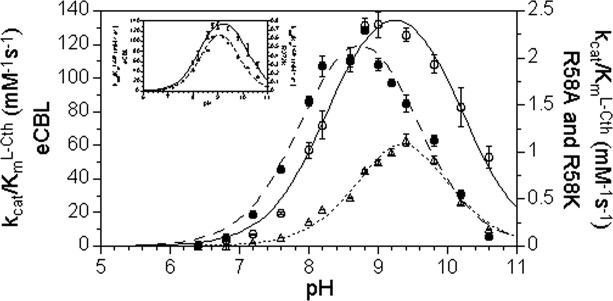
The pH dependence of kcat/ (mM−1s−1) for the hydrolysis of l-Cth catalyzed by eCBL (○) and the R58A (•) R58K (Δ) site-directed mutants. Inset: Comparison of the pH dependence of kcat/ (mM−1s−1) for eCBL (○) and the R372K (♦) mutant. Reaction conditions: MBP buffer (50 mM MOPS, 50 mM Bicine and 50 mM proline), 20 μM PLP, 2 mM DTNB, 0.01–6.25 mM l-Cth and 0.068–4.5 μM enzyme. The data were fit to equation 2.
Table III.
Parameters determined from the versus pH profiles of wild-type eCBL and the R58A, R58K and R372K site-directed mutantsa
| Enzyme | pKa1 | pKa2 |
|---|---|---|
| eCBL | 8.28 ± 0.06 | 10.20 ± 0.06 |
| R58A | 7.93 ± 0.08 | 9.53 ± 0.09 |
| R58K | 8.94 ± 0.09 | 9.8 ± 0.1 |
| R372K | 8.44 ± 0.09 | 9.8 ± 0.1 |
Kinetic measurements for the eCBL-catalyzed hydrolysis of l-Cth were carried out from pH 6.4–10.6 in MBP buffer containing 0.01–6.25 mM l-Cth, 20 μM PLP, 2 mM DTNB and 0.068–4.5 μM enzyme at 25°C. The kcat/ versus pH data were fitted to equation 2 to obtain the values for pKa1 and pKa2.
The W340, R372A, R372L and R372K mutants
The kcat of W340F is within ∼2-fold of the wild-type enzyme and the pH profile of this mutant is unchanged. In contrast, the , for hydrolysis of l-Cth, and the IC50 and Ki values, for inhibition of W340F by AVG, are increased by 10, 25, and 21-fold, respectively (Tables I and II, Fig. 5). The kcat and of R372K are decreased 4.2-fold and increased 83-fold, respectively, and the kcat/ of R372A and R372L, which could not be saturated within the solubility limit of l-Cth, are reduced by 2.1 × 106 and 8.3 × 105-fold, respectively (Table I). The optimum pH of ∼8–9.5 of the R372K mutant is similar to that of the wild-type enzyme. The low activity of R372A and R372L precluded the investigation of their activity as a function of pH as well as the reliable determination of inhibition parameters for these enzymes. The increases of 2100 and 4800-fold in IC50 and Ki, respectively, observed for R372K are the most drastic of the 10 mutants investigated (Table II, Fig. 5). Values of the rate constant k1 for association of eCBL and AVG, calculated using the equation Ki = k2/k1, are within 5-fold of the wild-type enzyme, with the exception of the 15 and ∼1000-fold increases in this parameter observed for the W340F and R372K mutants (Table II). Both of these mutations modify residues observed to interact with the α-carboxylate moiety of the inhibitor in the eCBL-AVG complex.3
DISCUSSION
The identity, position, and flexibility of active-site residues are key determinants of both the substrate and reaction specificity of enzymes. This is particularly true of enzymes dependent on the catalytically versatile PLP cofactor. For example, the effect of a single conservative modification on reaction specificity is demonstrated by the β-elimination activity, not observed for the wild-type enzyme, of the T81A, S82A, T85A, Q157A,E,H, Y158F, and S289A mutants of yeast cystathionine β-synthase and the increased racemase and β-decarboxylase activities of the R292K mutant of E. coli aspartate aminotransferase (eAATase), the archetypical PLP-dependent enzyme.8–10 The γ-subfamily of fold-type I of PLP-dependent enzymes, including eCBL, eCGS, and yCGL, provides a useful model system for the investigation of substrate and reaction specificity, as the overall structures and many active-site residues are conserved in these enzymes.4–6 The characterization of a series of site-directed mutants of five eCBL active-site residues (R58, R59, D116, W340, and R372) is the focus of the current study.
Clausen et al. have suggested, based on the structure of the eCBL-AVG complex, that the partitioning of bound substrate, or reactive intermediates, underlying reaction specificity, is related to the freedom of rotation about the Cα-Cβ bond of the substrate in the enzymes of the γ-subfamily.3 For example, methoxyvinylglycine (MVG) acts as either a substrate or an irreversible inhibitor of PLP-dependent enzymes, depending on the nature of the active site (Fig. 1).3,11–13 Comparison of the reaction of eCBL with l-Cth, MVG, and AVG demonstrates that the differing ability of active-site residues to form hydrogen bonds, tethering the distal portion of these molecules, has an important effect on the ability of the enzyme to catalyze their transformation. The distal methoxy group of MVG lacks the hydrogen bonding ability of AVG and l-Cth (Fig. 1) and is predicted to have a correspondingly greater degree of mobility and rotational freedom within the active site. As a result, MVG is deaminated by eCBL.3 In contrast, AVG is a slow-binding inhibitor of eCBL because the distal amino moiety of AVG enables it to hydrogen bond to the side chain hydroxyl group of Y111, thereby adopting a conformation which results in the slowly-reversible formation of a ketimine intermediate (Figs. 1 and 2).3 The l-Cth substrate of eCBL possesses both distal amino and carboxylate groups (Fig. 1) and is efficiently hydrolyzed to l-homocysteine and iminopropionate, via a β-elimination reaction, which does not include the ketimine intermediate observed upon reaction of eCBL with AVG.
Residue D116
The structure of the eCBL-AVG complex provides a useful guide for the selection of residues to probe their role in binding of the l-Cth substrate (Fig. 2).3 However, the double bond of the β,γ-unsaturated inhibitor is not present in l-Cth and AVG does not possess the sulfur atom at the γ-position or the distal carboxylate group of the substrate (Fig. 1). The ∼2-fold greater B-factor of the distal portion of AVG, compared to the Cα region, demonstrates the relative mobility of this part of the inhibitor in the active site.3 Therefore, the binding mode observed for the distal portion of AVG may not be representative of the eCBL substrate (Fig. 2).3 Residue D116 is conserved as an aspartate in bacterial CBL sequences and as an arginine residue in bacterial CGS and eukaryotic CGL. An interaction between the corresponding R106 of eCGS and its O-succinyl-l-homoserine (OSHS) substrate was observed in docking studies, before energy minimization.5 The side chain of D116 is 8.3 Å from the distal amino group of the inhibitor in the eCBL-AVG complex (Fig. 2). This residue was mutated to alanine and asparagine to investigate the possibility that a conformational change in the active site, upon binding of l-Cth, could bring D116 into contact with the distal portion of the substrate. However, in agreement with the structure of the eCBL-AVG complex, the 3-4-fold increases in the and the ≤2-fold increases in the IC50 and Ki values of the D116A,N mutants demonstrates that this residue is not involved in binding AVG or the distal amino group of l-Cth (Tables I and II).3
Residue W340
The side chain nitrogen of W340 forms a hydrogen bond with the α-carboxylate group of both the trifluoroalanine and AVG inhibitors, but does not interact with the corresponding carbonyl group of N-hydrazinocarbonylmethyl-2-trifluoromethylbenzamide (Figs. 1 and 2).2–4 Residue W340 is conserved in γ-proteobacterial CBL, but is replaced by a leucine residue in CGL from fungi and higher eurkaryotes. The 10-fold increase in and the 25 and 21-fold increases in IC50 and Ki for inhibition of eCBL-W340F by AVG support the proposed role for this residue in binding to the α-carboxylate groups of l-Cth and AVG (Tables I and II).3
The role of arginine residues in the active-site of eCBL
The three arginine residues in the eCBL active site were mutated in this study with the goal of probing the role of each in the binding of l-Cth. The α-carboxylate groups of AVG and trifluoroalanine, in the active sites of the eCBL-inhibitor complexes, both form a pair of hydrogen bonds to the side chain of R372 (Fig. 2).3,4 The low B-factor (∼15 Å2) of the α-carboxylate group of the inhibitor in the eCBL-AVG complex is a result of the interaction with R372, as well as with the backbone amide nitrogen of S339 and the side chain of W340.3 The 83-fold increase in the of R372K, the 2.1 × 106 and 8.3 × 105-fold increases in the kcat/ of the alanine and leucine substitution mutants of R372 (Table I), respectively, and the three order of magnitude increases in the IC50 and Ki values for inhibition of eCBL-R372K by AVG (Table II) demonstrate the importance of this residue (Fig. 2). The additional hydrogen bonds, and ∼1 Å greater length, of the native arginine residue at position R372 of eCBL is expected to contribute to the two and three order of magnitude lower and Ki for AVG, respectively, of the native enzyme, compared with the R372K mutant. The order of magnitude difference between these parameters is likely due to the distal carboxylate moiety of the substrate, which is lacking in AVG. The tyrosine, phenylalanine, alanine, and lysine substitution mutants of the corresponding R386 of eAATase also cause a reduction of at least three orders of magnitude in kcat/Km, demonstrating that replacement with lysine resulted in a similar effect as other nonconservative replacements.10,14,15 Despite the lack of sequence similarity between AATase and CBL, the two enzymes share the common catalytic core of fold-type I PLP-dependent enzymes as well as several active-site features.4 The 4-fold decrease in the kcat of CBL-R372K contrasts with the 55-fold decrease in this parameter reported for AATase-R386K.10,16 Vacca et al. proposed that the observed loss of activity of the R386K mutant reflects differences in the binding orientation and conformation of the substrates, such that they are not optimally positioned for catalysis.10 Therefore, the 14-fold smaller decrease in the kcat of eCBL-R372K likely reflects differences in the nature of the reactions catalyzed by these enzymes, as the hydrolysis of l-Cth is a facile reaction that, in contrast with the transamination of eAATase, does not require the generation of a ketimine intermediate or the binding of a second substrate.
Residues R58 and R59 have been alternatively proposed to interact with the distal carboxylate moiety of l-Cth by Messerschmidt et al. and Clausen et al., respectively.4,6 The corresponding residues of eCGS (OSHS + l-Cys → l-Cth) and yCGL (l-Cth → l-Cys + α-ketobutyrate + NH3) are R48/R49 and R52/S53, respectively. Based on docking studies in the eCGS active site, the distal carboxylate group of OSHS and the α-carboxylate of l-Cys are proposed to interact with R48 and R49, respectively.5 Clausen et al. observed that the salt bridge between E235 and R59 of eCBL is similar to that between eAATase residues D15 and R292, which also interacts with the distal carboxylate of anionic substrates, and proposed that eCBL-R59 could alternatively form an ion pair with E235 in the free enzyme and with the distal carboxylate group of l-Cth, upon substrate binding.4,17 However, docking of l-Cth to the active site of eCBL has indicated that the distal carboxylate group of the substrate interacts with R58.6 Comparison of the kinetic parameters obtained in this study for hydrolysis of the l-Cth substrate and inhibition by the AVG inhibitor, which lacks the distal carboxylate moiety of l-Cth (Fig. 1), provides insight into the roles of R58 and R59. While the is increased 5.7 and 8.4-fold by mutation of R59 to alanine or lysine, a 28-fold increase in this parameter is observed for R58A and R58K could not be saturated within the solubility limit of l-Cth. In contrast, the Ki values for inhibition of R58A, R58K, R59A, and R59K by AVG, were increased by only 3.2, 29, 1.4, and 6.3-fold, respectively (Table II). The similar changes, of less than one order of magnitude, in the and Ki values of R59A and R59K demonstrate that this amino acid is not directly involved in binding l-Cth and AVG. The observed minor effects on the kinetic parameters of these mutants are likely due to loss of the R59-E235 interaction and the resulting alteration in active-site conformation. In contrast, the effect of the alanine and lysine substitutions of R58 on the hydrolysis of l-Cth is much greater than their effect on inhibition by AVG, demonstrating that it is R58 that interacts with the distal carboxylate of the substrate. Residue R58 also interacts with both the phosphate group of the PLP cofactor and the side chain hydroxyl of Y111 in the eCBL active site (Fig. 2).4 In the structure of the eAATase-methylaspartate complex the corresponding R292 forms two hydrogen bonds each with the side chain of D15 and the distal carboxylate group of the substrate analog. Tethering of the eCBL-R58 side chain by the phosphate group of PLP, similar to the R292-D15 interaction of eAATase, may provide rigidity to eCBL-R58 to enable the precise positioning of the distal portion of substrate. The role of arginine residues in the binding of anionic substrates, particularly carboxylate groups, has been observed in a wide range of enzymes.10,17–20 As a result of the ∼1-Å difference in the length of the lysine side chain and its inability to fulfill the number of hydrogen-bonding interactions formed by arginine, the ɛ-amino group of the R58K side chain cannot interact with both the phosphate moiety of the cofactor and the distal carboxylate group of the substrate in the same manner as R58. The 29-fold increase in the Ki for inhibition of R58K by AVG, which lacks a distal carboxylate group, may reflect a weakening in the interaction between Y111 and the distal amino group of AVG in the context of the R58K mutation.
Residue R58 may also be involved in modulating the nucleophilic character of an active-site residue, such as Y111, which forms a π-stacking interaction with the pyridine ring of the cofactor and has been proposed to abstract a proton from the α-amino group of l-Cth and donate a proton to aminoacrylate.4 The observed interaction between R58 and Y111 (Fig. 2) may lower the pKa of the latter, such that the side chain of Y111 exists as a phenolate ion in the pH 8.5–9.5 range, which is pH optimum of eCBL (Fig. 3).7,21 Clausen et al. have suggested that conformational changes in the active site, resulting from substrate binding, would be expected to increase the hydrophobicity of the active-site environment and weaken the R58-Y111 interaction.4 This would result in an increase in the pKa of Y111, thereby facilitating a role for Y111 in proton transfer from the α-amino group of the l-Cth substrate, during transaldimination, to aminoacrylate to facilitate the release of the iminopropionate product. Correspondingly, a shift in the specific activity versus pH profiles was observed only for the R58A and R58K mutants (Fig. 3). Vacca et al. reported a similar narrowing of the specific activity versus pH profile for the corresponding R292K mutant of eAATase, but not for R386K (eCBL-R372K), and proposed that the change may be due to deprotonation of K292, as the pKa of lysine is lower than that of arginine.10 However, a similar mechanism is unlikely in the case of the eCBL-R58K mutant, unless the interaction of R58 with the phosphate moiety of the PLP cofactor, is weakened or not formed in the R58K enzyme (Fig. 2). The kcat/ versus pH profiles of R58A and R58K are also both narrower than that of the wild-type enzyme. The value of pKa2, of the basic limb, of R58A is decreased by 0.66 pH units and pKa1, of the acidic limb, of R58K is increased by 0.67 pH units (Fig. 4, Table III). The pKa of the basic limb of the kcat/ versus pH profile of eCBL may correspond to the substrate, as it is within 0.6 pH units of the pKa of 9.63 determined for one of the amino groups of l-Cth.22 The observed increase in the pKa of the acidic limb of the R58K mutant may be due to the decreased ability of a lysine in this position to modulate the pKa of Y111 to the same extent as R58 in the wild-type enzyme, as the ɛ-amino group of this residue cannot fulfill all of the hydrogen bonding interactions observed (phosphate moiety of the cofactor and side chain of Y111) and proposed (distal carboxylate group of l-Cth) for residue R58. However, the increase in pKa1 and decrease pKa2 observed for R58K and R58A, respectively, are distinct to the each mutant, suggesting that the observed pKa shifts may be an indirect effect resulting from a change in the conformation or charge distribution of the active site.
The CBL enzyme, which is unique to plants and bacteria, is an attractive target for the development of novel antimicrobial compounds because it is linked to a variety of cellular processes, including DNA replication, via the ubiquitous methyl donor S-adenosylmethionine. This study has identified R58, W340, and R372 as residues interacting with the distal and α-carboxylate groups, respectively, of the l-Cth substrate, information that will guide the design of inhibitors of this enzyme. For example, the addition of a distal carboxylate moiety to AVG would allow this compound to form a pair of hydrogen bonds with residue R58 in the active site of eCBL.
Materials and Methods
Reagents
l-Cth and l-aminoethoxyvinylglycine (AVG) were purchased from Sigma. Protease inhibitor (Complete, EDTA-free) tablets were obtained from Roche. Ni-NTA resin was a Qiagen product. 5,5′-Dithiobis(2-nitrobenzoic acid) (DTNB) was from Pierce. Oligonucleotide primers were synthesized by Integrated DNA Technologies and mutants were sequenced by DNA Landmarks before expression and purification.
Construction, expression and purification of site-directed mutants
Site-directed mutants were constructed by overlap-extension polymerase chain reaction and inserted into the pTrc-99aAF plasmid, which encodes an N-terminal, 6-His tag and linker.7 Farsi et al. demonstrated that the presence of this affinity tag does not alter the kinetic properties of eCBL.7 The E. coli KS1000 metC:cat strain, in which the gene encoding eCBL is replaced by that of chloramphenicol acetyltransferase, was employed for expression of the site-directed mutants to avoid contamination with the wild-type E. coli enzyme.7 The wild-type and site-directed mutants of eCBL were expressed and purified as described by Farsi et al.7
Determination of steady-state kinetic parameters
Enzyme activity was measured in a total volume of 100 μL at 25°C using a Spectramax 340 microtiter plate spectrophotometer (Molecular Devices). The assay buffer was comprised of 50 mM Tris, pH 8.5, containing 20 μM PLP, and the hydrolysis of l-Cth (0.01–6.25 mM) was detected (ɛ412 = 13,600 M−1s−1) via the reaction of 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) with the free thiol of the l-Hcys product.7,23,24 A background reading was recorded before initiation of the reaction by the addition of eCBL (0.068–6.6 μM, depending on the activity of the particular site-directed mutant) for all assays. Values of kcat and for the hydrolysis of l-Cth were obtained by fitting of the data to the Michaelis-Menten equation and kcat/ was obtained independently from Eq. (1). Data were fit by nonlinear regression with the SAS software package (SAS Institute, Cary, NC).
| (1) |
Evaluation of the pH dependence of wild-type and site-directed mutants of eCBL
The pH dependence of l-Cth hydrolysis by eCBL was determined using the continuous DTNB assay in a three-component buffer, comprised of 50 mM MOPS (pKa = 7.2), 50 mM Bicine (pKa = 8.3) and 50 mM proline (pKa = 10.7).25,26 The kinetic measurements were carried out at pH 6.4–10.6 in the presence of 20 μM PLP, 2 mM DTNB, 0.06–3.0 μM eCBL (depending on the activity of the site-directed mutant) and 6.25 mM l-Cth, for specific activity measurements, or 0.01–6.25 mM
l-Cth, for the determination of kcat/. Specific activity versus pH measurements were performed in quadruplicate to provide an estimate of the experimental error associated with each point in the pH profile. The kcat/ versus pH data were fit to the bell-shaped curve described by Eq. (2), where 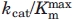 is the upper limit for kcat/ at the pH optimum.24
is the upper limit for kcat/ at the pH optimum.24
| (2) |
Inhibition of wild-type and site-directed mutants of eCBL by AVG
The IC50 values for inhibition of the eCBL enzymes by AVG were determined by measuring enzyme activity between 0.05 μM and 10 mM AVG. The enzyme and inhibitor were mixed and incubated at 25°C for 10 min in assay buffer. Activity was subsequently measured at a l-Cth substrate concentration of 0.1 mM and the data were fit to Eq. (3).2 Measurements were performed in quadruplicate for each enzyme. The parameters Actmax, Actmin, S and IC50 of Eq. (3) correspond to the maximal enzyme activity, the minimal enzyme activity, the slope of the transition between the maximal and minimal activity plateaus and the midpoint of the transition, respectively.
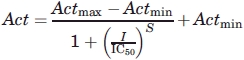 |
(3) |
Values for the dissociation constant Ki and the rate constant k2 for the inhibition of eCBL by AVG were determined as described by Clausen et al.3 In this model the enzyme and inhibitor do not form an initial, rapidly reversible enzyme-inhibitor complex [Eq. (4)].3
| (4) |
The wild-type eCBL and site-directed mutant enzymes were incubated with 1.5 mM l-Cth and 0.005–7.5 mM AVG in assay buffer and the progress of the reactions was monitored for 30 min. Values of kobs were determined from the fit of Eq. (5) to the progress curves. The resulting kobs values were plotted versus inhibitor concentration and values of k2 and Ki were obtained from fitting of the data to Eq. (6).
| (5) |
| (6) |
Acknowledgments
The authors thank Heidi Los and Navya Kalidindi for their technical assistance in protein purification and the reviewers of this paper for their insightful comments and suggestions. This work was supported by a grant from the Natural Sciences and Engineering Research Council of Canada.
Glossary
Abbreviations:
- AVG
aminoethoxyvinylglycine
- CBL
cystathionine β-lyase
- CGL
cystathionine γ-lyase
- CGS
cystathionine γ-synthase
- DTNB
5,5′-dithio-bis-(2-nitrobenzoic acid)
- l-Cth
l-cystathionine
- l-Hcys
l-homocysteine
- IPTG
Isopropyl-β-D-thiogalactopyranoside
- Ni-NTA
Ni-nitrilo triacetic acid
- PLP
pyridoxal 5′-phosphate.
References
- 1.Aitken SM, Kirsch JF. The enzymology of cystathionine biosynthesis: strategies for the control of substrate and reaction specificity. Arch Biochem Biophys. 2005;433:166–175. doi: 10.1016/j.abb.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 2.Ejim LJ, Blanchard JE, Koteva KP, Sumerfield R, Elowe NH, Chechetto JD, Brown ED, Junop MS, Wright GD. Inhibitors of bacterial cystathionine β-lyase: leads for new antimicrobial agents and probes of enzyme structure and function. J Med Chem. 2007;50:755–764. doi: 10.1021/jm061132r. [DOI] [PubMed] [Google Scholar]
- 3.Clausen T, Huber R, Messerschmidt A, Pohlenz HD, Laber B. Slow-binding inhibition of Escherichia coli cystathionine beta-lyase by L-aminoethoxyvinylglycine: a kinetic and X-ray study. Biochemistry. 1997;36:12633–12643. doi: 10.1021/bi970630m. [DOI] [PubMed] [Google Scholar]
- 4.Clausen T, Huber R, Laber R, Pohlenz HD, Messerschmidt A. Crystal structure of the pyridoxal-5′-phosphate dependent cystathionine beta-lyase from Escherichia coli at 1.83 Å. J Mol Biol. 1996;262:202–224. doi: 10.1006/jmbi.1996.0508. [DOI] [PubMed] [Google Scholar]
- 5.Clausen T, Huber R, Prade L, Wahl MC, Messerschmidt A. Crystal structure of Escherichia coli cystathionine gamma-synthase at 1.5 A resolution. EMBO J. 1998;17:6827–6838. doi: 10.1093/emboj/17.23.6827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Messerschmidt A, Worbs M, Steegborn C, Wahl MC, Huber R, Laber B, Clausen T. Determinants of enzymatic specificity in the Cys-Met-metabolism PLP-dependent enzymes family: crystal structure of cystathionine gamma-lyase from yeast and intrafamiliar structure comparison. Biol Chem. 2003;384:373–386. doi: 10.1515/BC.2003.043. [DOI] [PubMed] [Google Scholar]
- 7.Farsi A, Lodha PH, Skanes JE, Los H, Kalidindi N, Aitken SM. Inter-conversion of a pair of active-site residues in E. coli cystathionine γ-synthase, E. coli cystathionine β-Lyase and S. cerevisiae cystathionine γ-Lyase and development of tools for the investigation of mechanism and reaction specificity. Biochem Cell Biol. 2009;87:445–457. doi: 10.1139/o08-144. [DOI] [PubMed] [Google Scholar]
- 8.Aitken SM, Kirsch JF. Role of active-site residues Thr81, Ser82, Thr85, Gln157, and Tyr158 in yeast cystathionine β-synthase catalysis and reaction specificity. Biochemistry. 2004;43:1963–1971. doi: 10.1021/bi035496m. [DOI] [PubMed] [Google Scholar]
- 9.Quazi F, Aitken SM. Characterization of the S289A,D mutants of yeast cystathionine β-synthase. Biochim Biophys Acta. 2009;1794:892–899. doi: 10.1016/j.bbapap.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 10.Vacca RA, Giannattasio S, Graber R, Sandmeier E, Marra E, Christen P. Active-site Arg-Lys substitutions alter reaction and substrate specificity of aspartate aminotransferase. J Biol Chem. 1997;272:21932–21937. doi: 10.1074/jbc.272.35.21932. [DOI] [PubMed] [Google Scholar]
- 11.Rando RR. β,γ unsaturated amino acids as irreversible enzyme inhibitors. Nature. 1974;250:586–587. doi: 10.1038/250586a0. [DOI] [PubMed] [Google Scholar]
- 12.Rando RR, Relyea N, Chong L. Mechanism of irreversible inhibition of aspartate-aminotransferase by bacterial toxin l-2-amino-4-methoxy-trans-3-butenoic acid. J Biol Chem. 1976;251:3306–3312. [PubMed] [Google Scholar]
- 13.Miles EW. New type of pyridoxal-p enzyme catalyzed reaction–conversion of β,γ-unsaturated amino acids to saturated α-keto acids by tryptophan synthase. Biochem Biophy Res Commun. 1975;66:94–102. doi: 10.1016/s0006-291x(75)80299-3. [DOI] [PubMed] [Google Scholar]
- 14.Danishefsky AT, Onuffer JJ, Petsko GA, Ringe D. Activity and structure of the active-site mutants R386Y and R386F of Escherichia coli aspartate-aminotransferase. Biochemistry. 1991;30:1980–1985. doi: 10.1021/bi00221a035. [DOI] [PubMed] [Google Scholar]
- 15.Graber R, Kasper P, Malashkevich VN, Sandmeier E, Berger P, Gehring H, Jansonius JN, Christen P. Changing the reaction specificity of a pyridoxal-5′-phosphate dependent enzyme. Eur J Biochem. 1995;232:686–690. doi: 10.1111/j.1432-1033.1995.tb20861.x. [DOI] [PubMed] [Google Scholar]
- 16.Inoue Y, Kuramitsu S, Inoue K, Kagamiyama H, Hiromi K, Tanase S, Morino Y. Substitution of a lysyl residue for arginine-386 of Escherichia-coli aspartate-aminotransferase. J Biol Chem. 1989;264:9673–9681. [PubMed] [Google Scholar]
- 17.Kirsch JF, Eichele G, Ford GC, Vincent MG, Jansonius JN, Gehring H, Christen P. Mechanism of action of aspartate aminotransferase proposed on the basis of its spatial structure. J Mol Biol. 1984;174:497–525. doi: 10.1016/0022-2836(84)90333-4. [DOI] [PubMed] [Google Scholar]
- 18.Riordan JF, McElvany KD, Borders CL. Arginyl residues–anion recognition in enzymes. Science. 1977;195:884–886. doi: 10.1126/science.190679. [DOI] [PubMed] [Google Scholar]
- 19.Hwang JK, Warshel A. Why ion-pair reversal by protein engineering is unlikely to succeed. Nature. 1988;334:270–272. doi: 10.1038/334270a0. [DOI] [PubMed] [Google Scholar]
- 20.Mitchell JBO, Thornton JM, Singh J, Price SL. Toward an understanding of the arginine aspartate interaction. J Mol Biol. 1992;226:251–262. doi: 10.1016/0022-2836(92)90137-9. [DOI] [PubMed] [Google Scholar]
- 21.Dwivedi CM, Ragin RC, Uren JR. Cloning, purification, and characterization of β-cystathionase from Escherichia coli. Biochemistry. 1982;21:3064–3069. doi: 10.1021/bi00256a005. [DOI] [PubMed] [Google Scholar]
- 22.Aitken SM, Kirsch JF. Kinetics of the yeast cystathionine β-synthase forward and reverse reactions: continuous assays and the equilibrium constant for the reaction. Biochemistry. 2003;42:571–578. doi: 10.1021/bi026681n. [DOI] [PubMed] [Google Scholar]
- 23.Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70–77. doi: 10.1016/0003-9861(59)90090-6. [DOI] [PubMed] [Google Scholar]
- 24.Aitken SM, Kim DH, Kirsch JF. Escherichia coli cystathionine gamma-synthase does not obey ping-pong kinetics. Novel continuous assays for the elimination and substitution reactions. Biochemistry. 2003;42:11297–11306. doi: 10.1021/bi035107o. [DOI] [PubMed] [Google Scholar]
- 25.Peracchi A, Bettati S, Mozzarelli A, Rossi GL, Miles EW, Dunn MF. Allosteric regulation of tryptophan synthase: effects of pH, temperature, and alpha-subunit ligands on the equilibrium distribution of pyridoxal 5′-phosphate-L-serine intermediates. Biochemistry. 1996;35:1872–1880. doi: 10.1021/bi951889c. [DOI] [PubMed] [Google Scholar]
- 26.Jhee KH, McPhie P, Miles EW. Domain architecture of the heme-independent yeast cystathionine β-synthase provides insights into mechanisms of catalysis and regulation. Biochemistry. 2000;39:10548–10556. doi: 10.1021/bi001020g. [DOI] [PubMed] [Google Scholar]