

Abstract
Single-domain allostery has been postulated to occur through intramolecular pathways of signaling within a protein structure. We had previously investigated these pathways by introducing a local thermal perturbation and analyzed the anisotropic propagation of structural changes throughout the protein. Here, we develop an improved approach, the Rotamerically Induced Perturbation (RIP), that identifies strong couplings between residues by analyzing the pathways of heat-flow resulting from thermal excitation of rotameric rotations at individual residues. To explore the nature of these couplings, we calculate the complete coupling maps of 5 different PDZ domains. Although the PDZ domain is a well conserved structural fold that serves as a scaffold in many protein–protein complexes, different PDZ domains display unique patterns of conformational flexibility in response to ligand binding: some show a significant shift in a set of α-helices, while others do not. Analysis of the coupling maps suggests a simple relationship between the computed couplings and observed conformational flexibility. In domains where the α-helices are rigid, we find couplings of the α-helices to the body of the protein, whereas in domains having ligand-responsive α-helices, no couplings are found. This leads to a model where the α-helices are intrinsically dynamic but can be damped if sidechains interact at key tertiary contacts. These tertiary contacts correlate to high covariation contacts as identified by the statistical coupling analysis method. As these dynamic modules are exploited by various allosteric mechanisms, these tertiary contacts have been conserved by evolution.
Keywords: allostery, conformational flexibility, molecular dynamics, PDZ, sidechain
Introduction
Proteins relay signals for biological networks through allosteric regulation.1,2 An event at one site on a protein, such as ligand binding, can induce changes at a distant site on the protein allowing modulation of some functional behavior. Although the original definition of allosteric regulation focused on the behavior of multimeric proteins,3 it is now accepted that even single domains can undergo allosteric regulation.4–6 In single domain proteins, the allosteric interaction must be transmitted through the body of the protein instead of through domain quaternary interactions. For some domains, although changes at the allosteric sites are observed in the crystal structures,7 the structural rearrangements in regions that connect the allosteric sites are subtle. This has led to the idea of allostery without a well-defined conformational change8 where ligand binding results in a general rigidification of the protein that in turn effects function at a distant site.9 One attractive hypothesis is that allosteric interactions are transmitted through the interior of the protein along a specific signaling pathway as suggested by sequence analysis of the PDZ family where a sparse cluster of interconnected residues appear to be coconserved.10
The PDZ domain has been a popular target for the study of single-domain allostery. It is a small protein of ∼100 amino acids with a well-defined carboxylate binding site (Fig. 1) that can bind peptides or protein C-termini. The PDZ domain was originally thought to act solely as a passive scaffold11 but it is increasingly evident that it exhibits a diverse range of dynamical behavior. In this study, we look at 5 PDZ domains and seek structural origins for their differential behavior. The domains are: (1) PSD95-PDZ3: the third domain of Postsynaptic Density 95 Protein12 [Fig. 1(B)]; (2) PTP-PDZ2: the second domain of Tyrosine Phosphatase13 [Fig. 1(C)]; (3) PAR6-PDZ: the PDZ domain of Partition Defective Protein 614 [Fig. 1(D)]; (4) INAD-PDZ5: The 5th domain of Inactivation/No-after-potential D Protein15 [Fig. 1(E)]; (5) GRIP1-PDZ7: the 7th domain of Glutamate Receptor Interacting Protein 116 [Fig. 1(F)].
Figure 1.
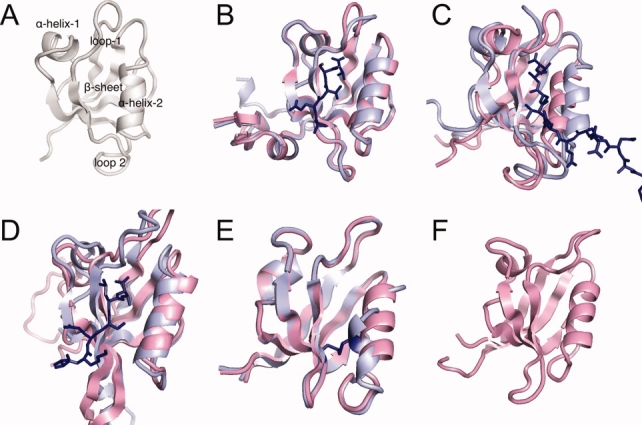
The PDZ domains. (A) Schematic of the structural elements of the PDZ domain. (B) PSD95-PDZ3: the third domain of Postsynaptic Density 95 Protein [1BFE,1BE9]; (C) PTP-PDZ2: the second domain of tyrosine phosphatase [3PDZ,1D5G]; (D) PAR6-PDZ: the binding domain of Partition Defective Protein 6 [1RY4,1RZX]; (E) INAD-PDZ5: The 5th domain of Inactivation/No-after-potential D Protein [2QKT]; and (F) GRIP1-PDZ7: the 7th domain of Glutamate Receptor Interacting Protein 1 [1M5Z]. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Of these domains, PTP-PDZ2 and PAR6-PDZ exhibit large conformational changes upon ligand binding [Fig. 1(C,D)], which has been shown to be allosteric.17,18 The binding of Cdc42 to PAR6 induces a conformational change in the PAR6-PDZ domain that promotes the binding of ligand. In contrast, the structure of PSD95-PDZ3 does not significantly change upon ligand binding [Fig. 1(B)], but induces allostery through incompatible binding surfaces with the ligand.19 INAD-PDZ5 forms an intramolecular disulfide bond depending on the redox potential, which disrupts the carboxylate-binding site15 [Fig. 1(E)]. GRIP1-PDZ7 has no known ligands.16 The origin of these differences in conformational flexibility is not understood but various experiments have studied specific residue-residue interactions through double-mutant experiments.10,20–22 Unfortunately no clear general picture has arisen as the results differ depending on the PDZ domain.
To analyze sidechain–sidechain interactions, computer simulations can be used to analyze dynamics at an atomic level of detail. In our lab, we had previously developed a molecular dynamics method, the anistropic thermal diffusion method (ATD),23 which was designed to probe intramolecular signaling within a protein by simulating flow patterns of kinetic energy. In ATD, the protein is first equilibrated at a low temperature then, a 300 K temperature bath is applied to a single residue. The resultant flow of kinetic energy as monitored by RMSD, was highly nonisotropic and suggested a defined pathway for particular residues. However, there were three practical problems with ATD: to minimize thermal conduction through solvent, the simulations were performed in vacuo using united atom models; positional restraints on backbone and surface atoms were required to reduce spurious fluctuations; and a significant portion of the perturbation energy went into local backbone distortions resulting in only weak coupling to the pathway.
To address these issues, we adapt a method that we have recently developed in our lab, the rotamerically induced perturbation (RIP) method, which was designed to generate large conformational changes through local perturbation of the rotamer degrees of freedom in a sidechain.24 This method is capable of generating large motions of several Ångstroms without disturbing the backbone of the perturbing residue in picoseconds of simulation time. To adapt RIP to identify sidechain–sidechain couplings, we use a much weaker local perturbation that is capable of generating clean signals of heat-flow without significantly disturbing the protein backbone structure. Analyzing the computed couplings across the 5 PDZ domains, we identify a simple relationship between the computed couplings and the observed conformational flexibility of the PDZ domains. If these couplings affect the structural dynamics of the proteins, then they are also likely to probe allosteric couplings. Consequently, we analyze the couplings with respect to the coconservation of paired positions in the PDZ domain as provided by the statistical coupling analysis (SCA).10
Results
Calibrating the rotamerically induced perturbations
The rotamerically induced perturbation (RIP) is designed to generate sidechain perturbations without distorting backbone secondary structure.24 In this method, sidechains are thermally driven to rotate back-and-forth around the initial χ angle at a rotational velocity dependent on a predefined rotational temperature TRIP. Heat is transferred only if the sidechain collides with another residue. As we are interested in using RIP to identify couplings between sidechains, rather than generating large conformational changes, we need to find a TRIP that best reproduces the rotational velocities of sidechains in MD simulations performed in standard conditions of TSTD = 300 K.
To identify a useful TRIP, we use simulations of single amino acids capped with methyl groups (see Ref for more details). As was performed in the original ATD analysis of intramolecular signaling,23 the protein is first equilibrated to 10 K to depress background fluctuations, which allows nonequilibrium heat-flow to be generated from the perturbed residue. We find that TRIP = 26 K generates rotational velocities that best reproduce the rotational velocities of amino acids simulated in a standard MD simulation at TSTD = 300 K [Fig. 2(B)]. The rotational temperature of TRIP = 26 K was deduced from a regression analysis where for each set of rotational velocities simulated at a particular TRIP, we identified the closest set of rotational velocities simulated in standard MD at different temperatures TSTD [Fig. 2(C)]. This gives a linear relationship of TRIP = 0.058 × TSTD + 10 K, resulting in a best fit of TRIP = 26 K to the standard temperature TSTD = 300 K. Another way to think about this is that the rotational energy of the χ angles constitutes ∼10% of the kinetic energy in a normal MD simulation.
Figure 2.

The properties of the Rotamerically Induced Perturbation (RIP) applied to the 18 amino acids with χ angles. (A) Schematic of the χ angles in Phe, which are perturbed in the RIP method. (B) The averaged rotational velocities of the χ angles from standard MD simulations at TSTD=300 K versus simulations of RIP at TRIP=26 K. The box shows the 75% percentile used to calculate the fit, giving a correlation of 0.85. The distribution is fairly close to linear as indicated by the slope of 1 shown in the box. (C) Given a particular set of χ angle rotational velocities measured from simulating the amino acids at a standard simulation at temperature TSTD, the TRIP is identified that generates the closest set of velocities using RIP. From these pairs of TSTD and TRIP, a linear relationship is determined of TRIP = 0.058 × TSTD + 10. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Generating intramolecular signaling pathways
In the original ATD study,23 an RMSD metric was used to detect the response to the perturbations. This gave a clean signal mainly due to the strong positional restraints applied to the backbone atoms, which damped the background drift of backbone motions. However, in the absence of restraints, even at 10 K backbone drift can reduce the signal-to-noise ratio. Instead, we use kinetic energy to identify the response of residues to a RIP perturbation. To demonstrate the properties of the kinetic energy response, we first study in detail two RIP perturbations in the PDZ domain PSD95-PDZ3; a surface residue and a buried residue. To measure the response of the perturbation of the surface residue Lys75 (Lys380 in 1BFE), we plot the kinetic-energy profile of the residues in the protein at the 5th ps [Fig. 3(A)]. To evaluate the profile, we define a background threshold of of 0.042 kcal/mol (mean ± 3σ for the entire set of RIP perturbations; see below). As none of the responding residues has a kinetic energy higher than the background, the protein shows no response to the perturbation. In contrast, perturbing the buried residue Phe20 (Phe325 in 1BFE) induces a large response in disparate regions of the protein [Fig. 3(B)]. This shows that the RIP perturbation can generate a strong signal for a buried residue (Phe20) but no response for a surface residue (Lys75).
Figure 3.
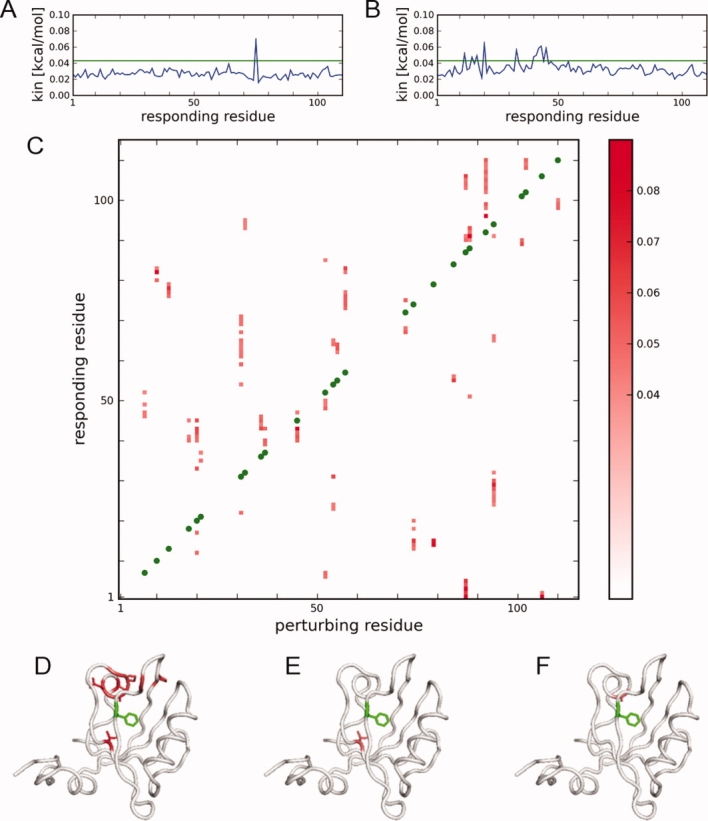
Pathways of heat-flow induced by RIP in PSD95-PDZ3. (A) Average kinetic energy response in the 5th ps of simulation of applying RIP to Lys75; (B) RIP applied to Phe20. (C) Pathways of heat-flow of the RIP perturbations. For each column, the perturbation is applied to the green position, and the colored regions identify the residues that respond with values of kinetic energy in the 5th ps above the background cutoff. (D) Mapping of the responding residues (red) of RIP on Phe20 (green) to the structure. Of these, two responding residues are in spatial proximity to Phe20: (E) Leu44 and (F) Ala42, which define RIP couplings from the Phe20 pathway of heat-flow. These couplings are used to generate the coupling map in Figure 4(A). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
We apply RIP to all 83 residues possessing χ angles in PSD95-PDZ3 to generate a complete map of heat flow pathways. Responding residues with a final kinetic energy in the 5th ps of simulation greater than the background are plotted as a two-dimensional intensity map [Fig. 3(C)]. Thus, a vertical column represents a pathway of heat flow (many residues responding) resulting from the perturbation on the residue denoted on the X-axis. Only responses above a background cut-off of 0.042 kcal/mol (mean + 3σ of the kinetic energy of every responding residue) are shown. The first point to note is that there is very little response near the main diagonal, meaning that RIP does not propagate energy automatically along the backbone from the site of the perturbed residue. This is a desirable behavior as it minimizes spurious coupling to the backbone.
The many vertical continuous segments shown in Figure 3(C) indicate that many residues are strongly coupled to sidechain perturbation of residues distant in sequence. For instance, Phe20 generates a large pathway of heat-flow [Fig. 3(B)], which, when mapped onto the structure [Fig. 3(D)], shows that the responding residues are spatially close to the perturbing residue. The perturbing residue heats physically connected residues [Fig. 3(E,F)], where this transferred kinetic energy can then propagate along the backbone, producing a correlated response along the protein chain [Fig. 3(B)].
RIP-couplings identify sidechain–sidechain interactions
What is the nature of sidechain–sidechain interactions in the interior of the protein? Although it has been assumed that sidechains in close proximity naturally interact by favorable VDW interactions, hydrogen bonds, and hydrophobic clustering, this does not fully take into account the role of sidechain dynamics. By applying a controlled local perturbation to a sidechain in a nonequilibrium environment, we can identify sidechains that interact based on the amount of transferred energy from the perturbed sidechain.
We identify a RIP coupling between two sidechains if a perturbing residue transfers kinetic energy above an energy cutoff to a residue in physical contact (defined by spatial proximity < 3.5 Å). For instance, in the case of the pathway of heat-flow of Phe20 [Fig. 3(D)], we identify two responding residues that are in spatial proximity to the perturbing residue: Leu44 [Fig. 3(E)] and Ala42 [Fig. 3(F)], which results in two couplings: Phe20-Ala42 and Phe20-Leu44. By focusing on pairs of responding residues in spatial contact to a perturbing residue, we generate a coupling map of PSD95-PDZ3 [blue circles Fig. 4(A)]. Similarly, we generate the coupling maps of the apo structures of the other PDZ domains: PTP-PDZ2 [Fig. 4(C)], PAR6-PDZ [Fig. 4(E)], INAD-PDZ5 [Fig. 4(G)], and Grip1-PDZ7 [Fig. 4(I)]. Comparing the coupling to the contact map (red dots; defined by d(Cβ-Cβ) < 6.5 Å), we find that not all residues in contact are coupled and that the contacts differ amongst the PDZ domains. To understand these differences, we analyze the coupling maps with respect to the structure of the proteins.
Figure 4.
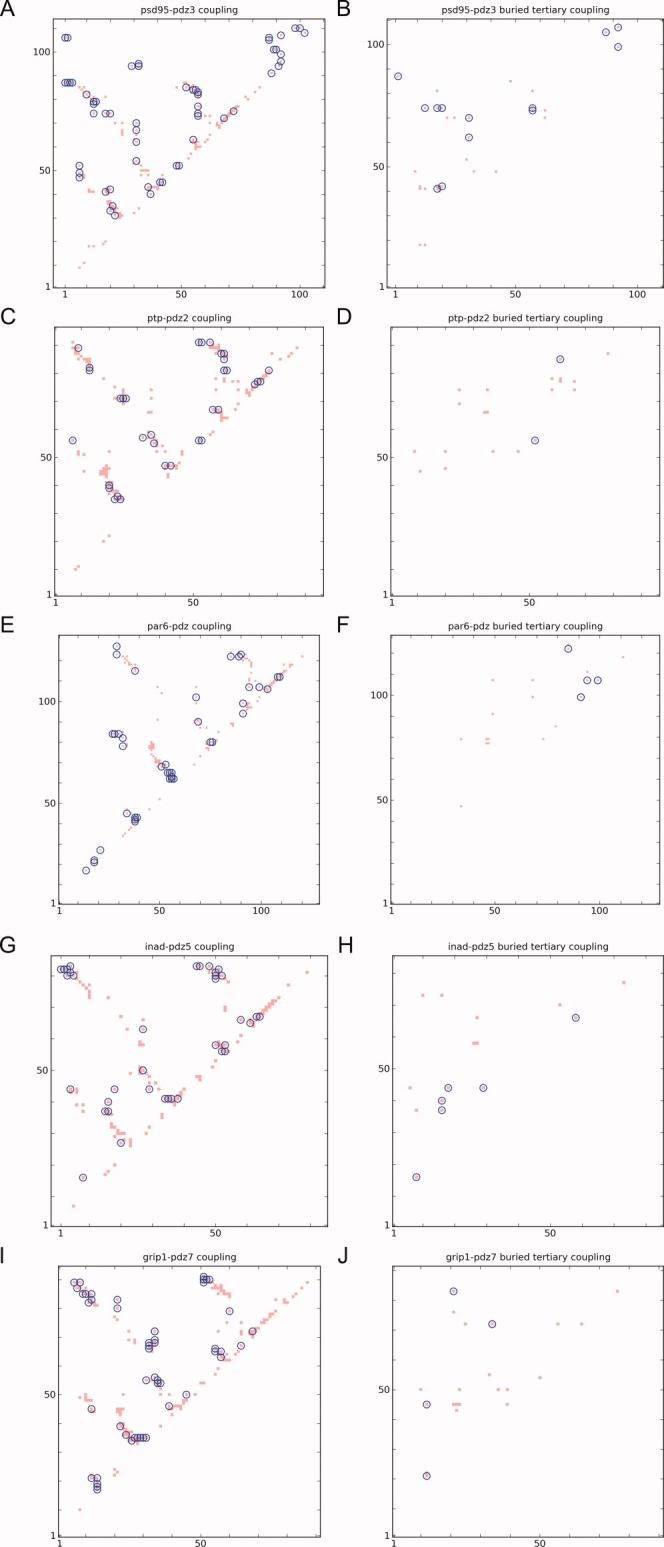
The RIP couplings of the PDZ domains superimposed over the contact maps. Left column: couplings overlaid over the contact map. Right column: buried tertiary couplings. (A,B) PSD95-PDZ3; (C,D) PTP-PDZ2; (E,F) PAR6-PDZ; (G,H) INAD-PDZ5; (I,J) GRIP1-PDZ7. The buried tertiary couplings are significantly different across the 5 domains. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Conformational flexibility and buried tertiary couplings
Although PDZ domains have a well-conserved structure with a well-defined ligand-binding groove, they have quite distinct responses to ligand-binding, ranging from negligible to large changes (left column of Fig. 5). The largest differences in response to ligand-binding lie in the α-helices (left column of Fig. 5). PSD95-PDZ3 [Fig. 5(A)] does not change much upon ligand-binding, whereas PTP-PDZ2 displays large conformational changes in both α-helices [red regions in Fig. 5(C)]. PAR6-PDZ induces large conformational changes only in α-helix-1 [Fig. 5(E)]. INAD-PDZ5 allows the formation of a disulfide bond that disorders the α-helix, but the overall position of the α-helix does not change [Fig. 5(G)]. GRIP7-PDZ1 is found in a closed conformation that precludes ligand-binding [Fig. 5(I)], and does not bind to any known peptides. As the binding surface of GRIP7-PDZ1 involves α-helix-2 but is located on the opposite face of the canonical binding, the α-helix-2 is unlikely to move. In summary, α-helix-1 moves upon ligand-binding in PTP-PDZ2 and PAR6-PDZ, whereas α-helix-2 only moves in PTP-PDZ2. In contrast, the α-helices do not move in PSD95-PDZ3, INAD-PDZ5, and GRIP7-PDZ1.
Figure 5.
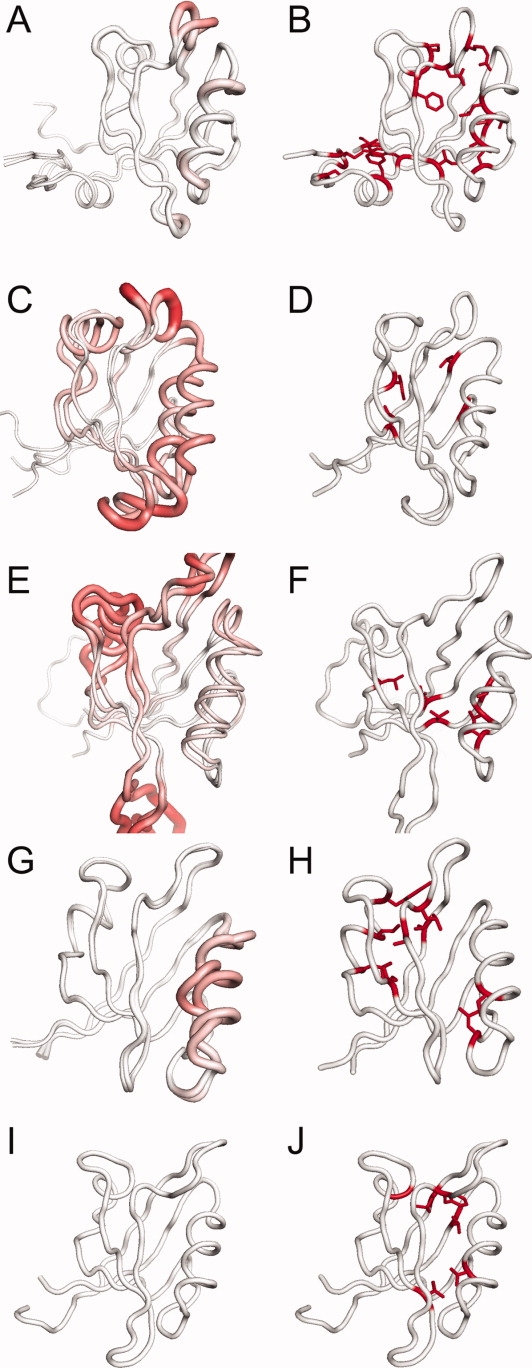
Comparing conformational changes upon ligand-binding (left column) to the buried tertiary RIP couplings (right column). In the left column, bright red is Cα RMSD = 15 Å between the apo and holo structures. In the right column, buried tertiary residues that are involved in RIP couplings are shown in red. (A,B) PSD95-PDZ3; (C,D) PTP-PDZ2; (E,F) PAR6-PDZ; (G,H) INAD-PDZ5; (I,J) GRIP1-PDZ7. The buried tertiary couplings are significantly different across the 5 domains. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
What is the structural origin of the difference in ligand-binding response in the PDZ domains? One possibility is that sidechain-sidechain couplings identified by RIP (left column of Fig. 4) correlate to differences in ligand-response of the PDZ structures (left column of Fig. 5). We analyzed the couplings with respect to the structural features of the PDZ domains (exposed/buried, secondary/tertiary) and could not find any clear pattern except for buried tertiary couplings.
We define a buried tertiary coupling if the two residues are both buried and not in the same secondary structure, as defined by backbone hydrogen-bonding. Two residues are defined to be in the same secondary structure by backbone hydrogen-bonding patterns.25 Residue i and residue j are defined to be in the same segment of secondary structure if 2 hydrogen bonds form between the segment i − 1 to i + 1 and segment j − 1 to j + 1. A hydrogen bond is defined if the backbone O and H atoms are within 2.7 Å. This definition covers the common cases of β-strand pairing and α-helical patterns. A sidechain is defined as buried if the accessible surface area (ASA) of the sidechain in the structure is ≤ 20% of that for the same residue free in solution.26
There are clear differences in the buried tertiary coupling map between the 5 PDZ domains (blue circles in the right column of Fig. 4) where only a fraction of buried tertiary contacts correspond to a coupling. The sparseness of the couplings allows a clear interpretation of the differences when mapped onto the corresponding structure (right column of Fig. 5). In PSD95-PDZ3, there are couplings found throughout the interior of the protein, linking the α-helices to the body of the protein, as well as to the C-terminal domain with the body of the protein [Fig. 5(B)]. In PTP-PDZ2, there are only two couplings, which are both within the body of the protein [Fig. 5(D)]. More importantly, there are no couplings that involve the α-helices. In PAR6-PDZ, there is one coupling between α-helix-2 and the the β-sheet. In both INAD-PDZ5 [Fig. 5(H)] and GRIP1-PDZ7 [Fig. 5(I)], there are several couplings between the small α-helix-1 (towards the back left) and the body of the protein, and one coupling between α-helix-2 on the right and the body (the specific couplings are listed in Table I).
Table I.
Comparison of Coupled Pairs Across the PDZ Domains
| Pair | PSD95-PDZ3 | PTP-PDZ2 | PAR6-PDZ | INAD-PDZ5 | GRIP1-PDZ7 |
|---|---|---|---|---|---|
| [1BFE] | [3PDZ] | [1RY4] | [2QKU-A] | [1M5Z] | |
| 1a | Ile316-Leu323 | Leu11-Leu18 | Leu163-Leu172 | x Leu587-Leu595 | x Leu27-Phe36 |
| 2a | Ile316-Ala347 | Leu11-Ala46 | Leu163-Ala206 | Leu587-Pro617 | x Leu27-Gly60 |
| 3 | x Arg318-Leu379 | Lys13-Leu78 | Lys165-Met238 | Lys589-Phe649 | Lys29-Ile91 |
| 4a | x Leu323-Pro346 | Leu18-Ala45 | Leu172-Leu205 | x Leu595-Tyr616 | Phe36-Pro59 |
| 5a | Leu323-Leu349 | Leu18-Ser48 | Leu172-Ser208 | x Leu595-Ile619 | Phe36-Leu62 |
| 6a | x Phe325-Ala347 | Ile20-Ala46 | Phe174-Ala206 | Leu597-Pro617 | Phe38-Gly60 |
| 7 | Phe325-Leu353 | Ile20-Ile52 | Phe174-Leu212 | x Leu597-Leu623 | Phe38-Leu65 |
| 8a | x Ile336-Leu367 | Ile35-Leu66 | Ile195-Val226 | Cys606-Leu637 | Val49-Thr79 |
| 9b | x Ile336-Ala375 | Ile35-Ala74 | Ile195-Val234 | Cys606-Cys645 | x Val49-Val87 |
| 10b | Leu353-Asp357 | x Ile52-Asp56 | Leu212-Asp216 | Leu623-Asp627 | Leu65-Asp69 |
| 11b | Leu353-Ala390 | Ile52-Leu89 | x Leu212-Val249 | Leu623-Val660 | Leu65-Ile102 |
| 12 | Ile359-Leu367 | Val58-Leu66 | x Val218-Val226 | Ile629-Leu637 | Leu71-Thr79 |
| 13b | Val362-Ala375 | Val61-Ala74 | x Val221-Val234 | Phe632-Cys645 | Val74-Val87 |
| 14b | x Val362-Ala378 | Val61-Thr77 | Val221-Met237 | Phe632-Leu648 | Val74-Leu90 |
| 15b | x Val362-Leu379 | Val61-Leu78 | Val221-Met238 | Phe632-Phe649 | Val74-Ile91 |
| 16 | Val362-Val386 | x Val61-Val85 | Val221-Leu245 | Phe632-Val656 | Val74-Leu98 |
| 17b | Leu367-Ala375 | Leu66-Ala74 | x Val226-Val234 | x Leu637-Cys645 | Thr79-Val87 |
This table allows a comparison of the amino acid identities of the same contact in the PDZ fold across different PDZ domains. Each row refers to a pair of positions in the generic PDZ fold. Each column lists the amino acids of the pairs in a specific PDZ protein [PDB code under the title]. From our simulations, we find that certain pairs in a PDZ domain are energetically coupled, marked with an [x]. We can thus compare, for the same pair position in the PDZ fold, the amino acid identities of coupled pairs to noncoupled pairs across the 5 PDZ domains. In the first column, [a] refers to pair-positions that link the body of the protein to α-helix-1, [b] refers to those that link to α-helix-2 and the rest are pair-positions within the body and loops of the PDZ domain.
The differences in couplings between the PDZ domains can be related to the conformational flexibility of the α-helices upon ligand-binding. In the three PDZ domains, where the overall orientation of the α-helices do not change [Figs. 5(A) and 5(G,I)], we find couplings between both α-helices and the β-sheet [Figs. 5(B) and 5(H,J)]. In PAR6-PDZ where only α-helix-1 undergoes a large conformational change upon ligand-binding [Fig. 5(E)], there are no couplings from the β-sheet to α-helix-1 [Fig. 5(F)] but there is a coupling from the β-sheet to α-helix-2. In contrast, in PTP-PDZ2, where both α-helices move upon ligand-binding [Fig. 5(C)], there are no couplings to the α-helices at all [Fig. 5(D)]. In summary, couplings between the α-helices and the body are only found in domains where the α-helices are rigid. No such couplings are found in domains where the α-helices move upon ligand binding.
Conserved SCA contacts are dominated by buried tertiary couplings
From the previous section, we identified buried tertiary couplings in PDZ domains that correlate with the ligand-binding induced conformational changes. As these conformational changes are likely to be related to intradomain allostery,17,18 these couplings may have been conserved during evolution. To explore possible coconservation of contacts, we use the statistical coupling analysis (SCA) method10 that calculates a statistical free-energy for each pair of positions in the PDZ fold, which measures the amount of covariation in amino-acid identity amongst the PDZ sequences [Fig. 6(A)].
Figure 6.

Analysis of the SCA matrix. (A) the SCA matrix measures the amount of covariation between positions in a multiple sequence alignment of the PDZ family. Higher values gives higher covariation. (B) the SCA contacts is a subset of the SCA matrix that correspond to a Cβ-Cβ contact in PSD95-PDZ3. (C-H) t-test of different categories on the contact SCA matrix. n refers to the number of samples, P refers to the P-values where P < 0.010 indicates that the 2 distributions are statistically different. ΔSCA is the difference between the average SCA free-energies between the distributions. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
The raw information contained in the SCA matrix is not always easy to interpret. Hierarchical clustering was used in the original analysis of the SCA matrix to reduce the SCA matrix into sets of mutually covarying positions. One intriguing result was that a set of positions was found that, when mapped onto a PDZ structure, consisted of an interconnected set of residues spanning one side of the domain to the other. These residues were interpreted as a signaling pathway. Although some double-mutant experiments have found cooperative effects of ligand binding upon along this putative pathway,10 other experiments have not.20–22
There are other ways to reduce the SCA matrix for analysis. Here, we focus on a subset of pair positions of the SCA matrix that map onto the contacts of a representative PDZ domain (PSD95-PDZ3). We call these the contact SCA values [Fig. 6(B)], which measures the amount of covariation of amino acid identity of residues that are in physical contact in the representative PDZ domain. Before we examine the contact SCA values with respect to the RIP-couplings, we can see if there are any particular structural interaction categories (buried, exposed, secondary or tertiary structure) that could provide insights into the high contact SCA values [Fig. 6(C–H)]. For instance, in Figure 6(C), we use a Student t-test to compare the distribution of buried contact SCA values (red) to the distribution of exposed contact SCA values (blue). Given that there are n = 141 samples, we find that these distributions are significantly different (P < 0.001) where the buried contact SCA are typically ΔSCA = 0.19 higher than exposed contact SCA. We also find that tertiary contacts have significantly (P = 0.002) higher SCA values than secondary contacts [Fig. 6(D)]. Buried tertiary contacts covary more than other types of contacts.
This analysis cannot tell us why certain buried tertiary contacts covary more than others. One possibility is that they represent important sidechain-sidechain couplings. If so, then we might expect a correlation of highly covarying contact SCA values with the RIP couplings. Given that buried tertiary RIP-couplings are rather sparse for each individual PDZ domain (right column of Fig. 4), we first map the couplings of all five domains (left column of Fig. 4) onto the contact map of PSD95-PDZ3 [Fig. 7(A)]. Given a particular set of contact SCA values, we divide these into coupling contacts (contact SCA values that coincide with a RIP coupling), and noncoupling contacts (contact SCA values that do not coincide with any couplings). Then we use a Student t-test to calculate the statistical differences between couplings and noncouplings [Fig. 7(C–H)].
Figure 7.

Comparison between the SCA matrix and the RIP couplings.(A) The couplings identified in the 5 PDZ domains and mapped onto the contact map. Only residues that could be aligned across all 5 structures were used. (B) The couplings that were mapped onto the buried tertiary couplings of PSD95-PDZ3. Different domains possess different couplings. (C–H) t-test of couplings vs. noncouplings for different sets of contact SCA values. n refers to the number of samples, P refers to the P-values where P < 0.010 indicates that the two distributions are statistically different. ΔSCA is the difference between the average SCA free-energies between the distributions. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
In the case of the set of all contact SCA values [Fig. 7(C)], there is no statistically significant difference (P = 0.084) of SCA values between coupling contacts and noncoupling contacts. Applying the analysis to different sub-sets of the contact SCA values, we did not find any statistically significant differences amongst the secondary contacts, buried contacts, or exposed contacts. However, we found significant differences amongst tertiary contacts [Fig. 7(F)] where tertiary coupling contacts have significantly higher SCA values (P = 0.001) than tertiary noncoupling contacts. Similarly, buried tertiary couplings have much higher SCA values (ΔSCA = 0.34, P = 0.004) than buried tertiary noncouplings [Fig. 7(H)]. This suggests that highly covarying contacts in the generic PDZ domain identifies buried tertiary contacts where a RIP-coupling can be found in at least one member of the PDZ family.
Discussion
PDZ domains have a conserved fold that binds peptides and the C-termini of proteins in a similar fashion, yet the structural response to ligand-binding varies significantly across family members. Given that the ligand-binding response has been linked to functional allostery, elucidating the structural basis underlying the differential response to ligand binding is important for understanding the evolution of PDZ domain behavior.
As the backbone arrangement is similar across the different PDZ domains, it must be the sidechain–sidechain interactions that dictate the differences in the ligand-binding response. For our purposes here, we consider that two residues are interacting if perturbation by RIP of one residue induces a kinetic energy response in the other. We find that only some of the residues in tertiary contact are coupled; simply being in contact is not evidence of coupling. In previous studies, interactions between sidechains were deduced from the covariation matrix of RMSD in the analysis of equilibrium trajectories.27–29 In the RMSD covariation matrix, residues that undergo collective motion can be identified, such as the residues that constitute the Met20 loop in DHFR.27 However, there is no way to disentangle the effects of collective backbone motions from direct sidechain–sidechain interactions. As the backbone ϕ/ψ angles of consecutive residues are closely coupled by chain connectivity, if a single ϕ angle is perturbed, several ϕ/ψ angles along the chain will also be affected. To identify local interactions, the long-range effects of the ϕ/ψ degrees of freedom need to be avoided, which is obtained in RIP by perturbing only the sidechain χ-angles, which are degrees of freedom orthogonal to the backbone ϕ/ψ angles.
Finally, we focus on buried residues, as buried residues are protected from the solvent, unlike surface residues, buried couplings identified in nonequilibrium conditions would be expected to be also coupled in equilibrium conditions. Table I lists the 17 pair positions in the general PDZ fold where buried tertiary couplings are found in one of the five PDZ domains. Each row refers to a specific pair position where the corresponding amino acid identities are listed for the 5 PDZ domains. The existence of a coupling in a pair position is not related to the physical proximity of the residues. For example, the residues of pair 17 across the PDZ domains are always in close contact (Fig. 8), but they are coupled only in PAR6-PDZ [Fig. 8(C)] and INAD-PDZ5 [Fig. 8(D)].
Figure 8.

The same contact in the PDZ fold across different PDZ domains (corresponds to Pair 17 in Table 1). (A) PSD95-PDZ3, (B) PTP-PDZ2, (C) PAR6-PDZ, (D) INAD-PDZ5, and (E) GRIP1-PDZ7. The contacts in (C) and (D) are RIP-coupled, whilst the other three are not. The coupling depends on the orientation of the χ angles and is not obvious from inspection of the contacts or accessible surface-area. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
We find that PDZ domains with α-helices that are rigid have many couplings between the α-helix and the body of the protein while PDZ domains that have flexible α-helices possess no significant couplings. This coupling varies across the PDZ domains. If we focus on the pair positions that connect α-helix-2 to the body of the protein (denoted by [b] in the first column of Table I), we find that, in different PDZ domains, different couplings bind α-helix-2. For instance, in PSD95-PDZ3, pairs 9, 14, 15, and 17 are coupled. In PAR6-PDZ, pair 11, 13, and 17 provides the coupling. In INAD-PDZ5, pair 17 provides the coupling, whilst in INAD9-PDZ5, pair 9 provides the coupling.
Given that couplings are correlated with conformational flexibility, we may be able to deduce amino acid identities that directly affect the dynamics of a protein. To this end, we can identify pair positions where the couplings are uniquely determined in the amino acid identities, compared to the noncoupling amino-acids. With this criteria, for the pair-positions that connect the body of the protein to α-helix-1, we find uniquely determined couplings in pairs 2, 4, and 5. For the pair positions that connect to α-helix-2, we find uniquely determined couplings in pairs 14 and 17. In other pairs, a more careful analysis is required. Consider pair 1 that connects the body to α-helix-1. In INAD-PDZ5, the residues (Leu587-Leu596) are coupled. However, in PAR6-PDZ, pair 1 has the same residues (Leu163-Leu172) but is not coupled. In contrast, pair 1 in GRIP1-PDZ7 is coupled (Leu27-Phe36) but no other PDZ domain has the same amino acid identities. This suggests that for pair 1, the amino acids of GRIP1-PDZ7 (Leu27-Phe36) uniquely determines a coupling, whilst the amino acids of INAD-PDZ5 do not.
One possible consequence is that uniquely determined amino-acid pairings may be transferrable to other PDZ domains in order to induce couplings. For instance, α-helix-2 is mobile in PTP-PDZ2, whereas α-helix-2 is rigid in the other 4 PDZ domains. From the aforementioned analysis, for pair 17, the amino acids from PAR6-PDZ (Val226-Val234) provide uniquely determined amino acids that induce coupling in α-helix-2. This suggests that the substitutions L66V and A74V in PTP-PDZ2 will induce a coupling that immobilizes α-helix-2. Other possible substitutions are the amino acids of pair 14 in PSD95-PDZ3, and the amino-acids of pair 17 in INAD-PDZ5. The reverse may also work where the amino-acids of PTP-PDZ2 transferred to the other proteins may eliminate the couplings to α-helix-2 in those proteins.
Can we relate these specific pair positions to previous mutation and dynamic studies of PDZ domains? It has been postulated that differences in the mutational experiments between PSD95-PDZ3 and PTP-PDZ2 are due to the rigidity of PSD95-PDZ3 as opposed to a more flexible structure in PTP-PDZ2.22,30 We can identify the specific pairs that determine this difference in rigidity. In PSD95-PDZ3, coupling in pairs 4, 6, 8 fix α-helix-1, whilst couplings in pairs 9, 14, and 15 fix α-helix-2. In PTP-PDZ2, there are no couplings to either of the two α-helices, leading to mobile α-helices in the apo state of the protein.
Another study using NMR measurements of PTP-PDZ2 provide evidence of the direct influence of specific positions on the dynamics of the protein.20 Using the backbone S order parameter, Fuentes and colleagues measured differences between the apo and ligand-bound structures of PTP-PDZ2. Once they established this baseline behavior, they introduced 14 different mutations to PTP-PDZ2. Of these 14 mutants, two induced significant changes in the S order backbone signature in the apo structure, which were strikingly similar to the ligand-bound structure of PTP-PDZ2. The identity of these mutations was I20F, and I35V. We find that I20F is found in pair 6 and 7, whilst I35V is found in pairs 8 and 9 (Table I). From the RIP simulations, we find that these pairs are not coupled in PTP-PDZ2 but are coupled in PSD95-PDZ3. In particular, the I20F mutation is equivalent to bringing the residue from PSD95-PDZ3 to PTP-PDZ2. We can thus interpret these mutations in PTP-PDZ2 as inducing the coupling of the α-helices to the body of the protein, which results in a generally rigid structure similar to PSD95-PDZ3.
This suggests a model of the PDZ domain that intrinsically allows the possibility of mobile α-helices, depending on whether residues at strategic tertiary contacts are coupled. Upon ligand binding, the α-helices rigidify thus transmitting a signal to other parts of the protein, which is consistent with a general proposal for ligand-binding allostery.9 In a recent review, Gunasekaran and Nussinov suggested that all proteins are, to a certain extent, dynamic, and hence potentially allosteric.31 Indeed PDZ dynamics have been directly linked to functional allostery. For example, binding Cdc42 to the PAR6-PDZ fixes α-helix-1 in a new conformation that leads to increased affinity for ligand-binding in the carboxylate binding loop.18 Korkin and coworkers showed that in PSD95-PDZ3, there is no need for flexibility in the α-helix as the two different binding modes of PSD95-PDZ to other domains of PSD95 depends on steric hindrance of the ligand.19 If the function of the protein requires the preservation of the dynamics of the protein, then the identity of the couplings needed to be maintained if rigidity is required (as in PSD95), or couplings need to be avoided if the protein must remain dynamic (as in PTP). This evolutionary pressure leads to the high degree of covariation in the SCA contacts observed for tertiary contacts in the PDZ domain that corresponds to a tertiary couplings in any of the 5 PDZ domains. Of course, this only accounts for a subset of contact pair positions in the SCA matrix. It has been shown that the SCA matrix can also be used to identify protein-protein interaction surfaces32 and structural analysis of allostery.33,34
In the case of the Cooper-Dryden model of single-domain allostery,8 where allosteric interactions are transmitted by the rigidification of the protein upon ligand-binding, the tertiary couplings computed by RIP provides a specific mechanism for identifying allosteric effectors. Using a RIP analysis, we can identify conformational flexibility in structural elements (α-helices, β-sheets, loops) of a protein structure through the absence of tertiary couplings. The flexible structural element will be fixed by the ligand upon ligand-binding, which communicates allostery along the entire surface of the newly fixed element. Mutations to create new couplings that damp the motion of the flexible element, will reduce the allostery of the protein.
Methods
The protocol of the RIP method
The RIP method is implemented as a PYTHON wrapper around the SANDER package of AMBER35 and all analysis code was written in PYTHON. The code is available at http://boscoh.com/rip. The simulations of the RIP method are run in AMBER, using the PARM96 force-field with an GB/SA implicit-solvent term. To prepare for the simulation, ligands and crystallographic waters are removed from the crystal structure. The structure is then minimized. As the bulk of the RIP protocol is performed under constant energy, we first equilibrate the protein to 10 K for 1 ps with a friction constant of 5 ps−1, then equilibrate the protein at constant energy for 10 ps, and then a final equilibration to 10 K for 1 ps. This allows some rearrangements that are favorable only under constant energy but not under a thermostat.
The standard protocol for a RIP method lasts for 10 ps. At the beginning of the RIP method, the equilibrium value of the χ angles of the residue is stored. The run is then broken up into 100 fs intervals where each interval is simulated at constant energy. Between each interval: (1) the direction of the rotational velocity of each χ angle is stored; (2) the atomic velocities of the residue is set to zero; (3) if the value of the χ angle exceeds 60° of the equilibrium χ value, the direction of the rotational velocity is reversed; (4) the magnitude of each χ rotational velocity is calculated from the sidechain conformation; (5) the χ rotational velocities are transformed into atomic velocities and added to each atom; (6) the kinetic energy of the residue is scaled to the rotation temperature. Between the intervals, a Python module translates the AMBER restart files into a Python object, from which the RIP protocol is used to generate new AMBER restart files for the next interval. Finally, the trajectories of all the intervals are spliced into a single trajectory. Since the modifications are made on the velocities, the coordinate trajectories are continuous.
The motion is limited to ± 60° for the χ angles so that each different type of amino acid can fully explore the parameter space of a single rotamer conformation. If it was not restricted to one rotamer in amino acids such as Met with four χ angles, the parameter space would be too large to be fully explored in 10 ps.
Converting rotational velocities into atomic velocities
In the RIP method, a rotational velocity for each χ angle of a sidechain is calculated at the beginning of every interval, from this rotational velocity, the atomic velocities are generated. To generate the rotational velocities of the χ angles, each χ angle is assumed to be an independent degree of freedom. Based on the equipartition theorem, each independent χ angle can be assigned an E derived from the temperature T. This E is drawn randomly from a Gaussian distribution with mean energy ½kT and standard deviation √½kT.
To convert a rotational velocity into an atomic velocity, a frame of reference for the axis of rotation must be chosen. As rotational velocities are only defined relative to the axis of rotation; rotations can occur on either end of the axis, and still give the same rotational velocity. As the purpose of the RIP method is to minimize the motion of the backbone at the perturbing residue, only the sidechain atoms on the side of the rotation axis away from the backbone are rotated. Consequently, the rotational inertia of each χ angle, I = Σ mr2, is calculated as the sum of the moment of inertia of these sidechain atoms.
To convert E into a rotational velocity ω, the equation of rotational energy E = Iω2 is used. This is converted to a tangential velocity v through v = rω, where r is the perpendicular radius of the atom from the χ angle axis. This velocity is applied to the atom along the direction of the tangent to the axis of rotation. The atomic velocities due to each χ angle are then added cumulatively to each atom.
However, the different χ angles of the same sidechain do not represent completely independent degrees of freedom. As such, the final atomic velocities are re-scaled such that the total kinetic energy of the sidechain is E = 3/2 nkT where T = 300 K. This scaling only changes the magnitudes of the rotations and preserves the pure rotation around the χ angles.
Measuring rotational velocities
In the analysis of the RIP simulations, rotational velocities of the χ angles need to be extracted from the trajectories. In the generation of rotational velocities, only atoms that are on the side of the rotation axis of the χ angle away from the backbone contribute to the rotational velocity. Therefore, in the extraction of the rotation velocities, only these atoms are considered. For each atom that fits the criteria, the tangential velocity v to the axis is calculated. This v is converted to a rotational velocity by ω = v/r. However, the contributions of each atom to the total rotational velocity of a χ angle depends on its moment of inertia. A weighting (w) for each atom is calculated from the moment of inertia I = mr2 of the atom where the weighting is given by w = I/Itotal. The overall rotational velocity is then given by ωtotal = Σwω.
Measurable parameters of the simulations
The kinetic energy of a residue is the averaged kinetic energy of the atoms in a residue. The kinetic energy of an atom is simply defined as E = 0.5 mv2. This is proportional to the temperature of that residue.
References
- 1.Swain JF, Gierasch LM. The changing landscape of protein allostery. Curr Opin Struct Biol. 2006;16:102–108. doi: 10.1016/j.sbi.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Kern D, Zuiderweg ER. The role of dynamics in allosteric regulation. Curr Opin Struct Biol. 2003;13:748–757. doi: 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Changeux JP, Edelstein SJ. Allosteric receptors after 30 years. Neuron. 1998;21:959–980. doi: 10.1016/s0896-6273(00)80616-9. [DOI] [PubMed] [Google Scholar]
- 4.Vetter IR, Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 5.Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- 6.Volkman BF, Lipson D, Wemmer DE, Kern D. Two-state allosteric behavior in a single-domain signaling protein. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 7.Daily MD, Gray JJ. Local motions in a benchmark of allosteric proteins. Proteins. 2007;67:385–399. doi: 10.1002/prot.21300. [DOI] [PubMed] [Google Scholar]
- 8.Cooper A, Dryden DTF. Allostery without conformational change—a plausible model. Eur Biophys J Biophys Lett. 1984;11:103–109. doi: 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- 9.Hilser VJ, Thompson EB. Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins. Proc Natl Acad Sci USA. 2007;104:8311–8315. doi: 10.1073/pnas.0700329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 11.Harris BZ, Lim WA. Mechanism and role of PDZ domains in signaling complex assembly. J Cell Sci. 2001;114:3219–3231. doi: 10.1242/jcs.114.18.3219. [DOI] [PubMed] [Google Scholar]
- 12.Doyle DA, Lee A, Lewis J, Kim E, Sheng M, MacKinnon R. Crystal structures of a complexed and peptide-free membrane protein-binding domaMolecular basis of peptide recognition by PDZ. Cell. 1996;85:1067–1076. doi: 10.1016/s0092-8674(00)81307-0. [DOI] [PubMed] [Google Scholar]
- 13.Walma T, Spronk C, Tessari M, Aelen J, Schepens J, Hendriks W, Vuister GW. Structure, dynamics and binding characteristics of the second PDZ domain of PTP-BL. J Mol Biol. 2002;316:1101–1110. doi: 10.1006/jmbi.2002.5402. [DOI] [PubMed] [Google Scholar]
- 14.Garrard SM, Capaldo CT, Gao L, Rosen MK, Macara IG, Tomchick DR. Structure of Cdc42 in a complex with the GTPase-binding domain of the cell polarity protein, Par6. EMBO J. 2003;22:1125–1133. doi: 10.1093/emboj/cdg110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mishra P, Socolich M, Wall MA, Graves J, Wang ZF, Ranganathan R. Dynamic scaffolding in a G protein-coupled signaling system. Cell. 2007;131:80–92. doi: 10.1016/j.cell.2007.07.037. [DOI] [PubMed] [Google Scholar]
- 16.Feng W, Fan JS, Jiang M, Shi YW, Zhang M. PDZ7 of glutamate receptor interacting protein binds to its target via a novel hydrophobic surface area. J Biol Chem. 2002;277:41140–41146. doi: 10.1074/jbc.M207206200. [DOI] [PubMed] [Google Scholar]
- 17.vandenBerk LCJ, Landi E, Walma T, Vuister GW, Dente L, Hendriks WJAJ. An allosteric intramolecular PDZ-PDZ interaction modulates PTP-BL PDZ2 binding specificity. Biochemistry. 2007;46:13629–13637. doi: 10.1021/bi700954e. [DOI] [PubMed] [Google Scholar]
- 18.Peterson FC, Penkert RR, Volkman BF, Prehoda KE. Cdc42 regulates the Par-6 PDZ domain through an allosteric CRIB-PDZ transition. Mol Cell. 2004;13:665–676. doi: 10.1016/s1097-2765(04)00086-3. [DOI] [PubMed] [Google Scholar]
- 19.Korkin D, Davis FP, Alber F, Luong T, Shen MY, Lucic V, Kennedy MB, Sali A. Structural modeling of protein interactions by analogy: application to PSD-95. PLoS Comput Biol. 2006;2:e153. doi: 10.1371/journal.pcbi.0020153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fuentes EJ, Der CJ, Lee AL. Ligand-dependent dynamics and intramolecular signaling in a PDZ domain. J Mol Biol. 2004;335:1105–1115. doi: 10.1016/j.jmb.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 21.Fuentes EJ, Gilmore SA, Mauldin RV, Lee AL. Evaluation of energetic and dynamic coupling networks in a PDZ domain protein. J Mol Biol. 2006;364:337–351. doi: 10.1016/j.jmb.2006.08.076. [DOI] [PubMed] [Google Scholar]
- 22.Chi CN, Elfstrom L, Shi Y, Snall T, Engstrom A, Jemth P. Reassessing a sparse energetic network within a single protein domain. Proc Natl Acad Sci. 2008;105:4679–4684. doi: 10.1073/pnas.0711732105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ota N, Agard DA. Intramolecular signaling pathways revealed by modeling anisotropic thermal diffusion. J Mol Biol. 2005;351:345–354. doi: 10.1016/j.jmb.2005.05.043. [DOI] [PubMed] [Google Scholar]
- 24.Ho BK, Agard DA. Probing the flexibility of large conformational changes in protein structures through local perturbations. PLoS Comput Biol. 2009;5:e1000343. doi: 10.1371/journal.pcbi.1000343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 26.Creamer TP, Srinivasan R, Rose GD. Modeling unfolded states of proteins and peptides. II. Backbone solvent accessibility. Biochemistry. 1997;36:2832–2835. doi: 10.1021/bi962819o. [DOI] [PubMed] [Google Scholar]
- 27.Radkiewicz JL, Brooks CL., III Protein dynamics in enzymatic catalysis: exploration of dihydrofolate reductase. J Am Chem Soc. 2000;122:225–231. [Google Scholar]
- 28.Rod TH, Radkiewicz JL, Brooks CL. Correlated motion and the effect of distal mutations in dihydrofolate reductase. Proc Natl Acad Sci USA. 2003;100:6980–6985. doi: 10.1073/pnas.1230801100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bradley MJ, Chivers PT, Baker NA. Molecular dynamics simulation of the Escherichia coli NikR proteequilibrium conformational fluctuations reveal interdomain allosteric communication pathways. J Mol Biol. 2008;378:1155–1173. doi: 10.1016/j.jmb.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gianni S, Walma T, Arcovito A, Calosci N, Bellelli A, Engström Å, Travaglini-Allocatelli C, Brunori M, Jemth P, Vuister GW. Demonstration of long-range interactions in a PDZ domain by NMR, kinetics, and protein engineering. Structure. 2006;14:1801–1809. doi: 10.1016/j.str.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 31.Gunasekaran K, Ma B, Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 32.Weigt M, White RA, Szurmant H, Hoch JA, Hwa T. Identification of direct residue contacts in protein–protein interaction by message passing. Proc Natl Acad Sci USA. 2009;106:67–72. doi: 10.1073/pnas.0805923106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Süel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nature Struct Biol. 2002;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 34.Shulman AI, Larson C, Mangelsdorf DJ, Ranganathan R. Structural determinants of allosteric ligand activation in RXR heterodimers. Cell. 2004;116:417–429. doi: 10.1016/s0092-8674(04)00119-9. [DOI] [PubMed] [Google Scholar]
- 35.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, Debolt S, Ferguson D, Seibel G, Kollman P. Amber, a package of computer-programs for applying molecular mechanics, normal-mode analysis, molecular-dynamics and free-energy calculations to simulate the structural and energetic properties of molecules. Computer Phys Commun. 1995;91:1–41. [Google Scholar]