

Abstract
The binding of atrial natriuretic peptide (ANP) to its receptor requires chloride, and it is chloride concentration dependent. The extracellular domain (ECD) of the ANP receptor (ANPR) contains a chloride near the ANP-binding site, suggesting a possible regulatory role. The bound chloride, however, is completely buried in the polypeptide fold, and its functional role has remained unclear. Here, we have confirmed that chloride is necessary for ANP binding to the recombinant ECD or the full-length ANPR expressed in CHO cells. ECD without chloride (ECD(−)) did not bind ANP. Its binding activity was fully restored by bromide or chloride addition. A new X-ray structure of the bromide-bound ECD is essentially identical to that of the chloride-bound ECD. Furthermore, bromide atoms are localized at the same positions as chloride atoms both in the apo and in the ANP-bound structures, indicating exchangeable and reversible halide binding. Far-UV CD and thermal unfolding data show that ECD(−) largely retains the native structure. Sedimentation equilibrium in the absence of chloride shows that ECD(−) forms a strongly associated dimer, possibly preventing the structural rearrangement of the two monomers that is necessary for ANP binding. The primary and tertiary structures of the chloride-binding site in ANPR are highly conserved among receptor-guanylate cyclases and metabotropic glutamate receptors. The chloride-dependent ANP binding, reversible chloride binding, and the highly conserved chloride-binding site motif suggest a regulatory role for the receptor bound chloride. Chloride-dependent regulation of ANPR may operate in the kidney, modulating ANP-induced natriuresis.
Keywords: atrial natriuretic peptide, receptor, natriuresis, chloride, allosteric regulation, structural motif, X-ray crystallography, analytical ultracentrifugation, circular dichroism, thermal unfolding
Introduction
Atrial natriuretic peptide (ANP) is a hormone secreted by the heart in response to blood volume expansion. ANP stimulates salt excretion1 and dilates blood vessels.2,3 ANP counterbalances the renin-angiotensin-aldosterone (RAA) system and regulates the blood pressure and blood volume. The hormonal activities of ANP are mediated by the cell-surface ANP receptor (ANPR, also referred to as the A-type natriuretic peptide receptor) coupled to its own guanylate cyclase (GCase) activity in the intracellular domain. ANP binding to the extracellular domain (ECD) stimulates the GCase activity via a novel ligand-induced rotation mechanism,4–6 elevating intracellular cGMP second messenger, which in turn elicits physiologic responses.
The natriuretic activity of ANP has been well documented experimentally. However, there are certain physiologic and pathologic conditions where salt is retained even with high plasma ANP. For example, in transgenic animals overexpressing ANP, salt is retained despite the increased plasma ANP.7–10 In heart failure, plasma levels of ANP are markedly increased, yet sodium is retained.11–13 The mechanism underlying such apparent renal insensitivity to ANP is not understood. Earlier, Misono14 has reported that binding of ANP to the ANPR requires the presence of chloride, and it is chloride concentration dependent, suggesting a possible chloride-dependent regulation of ANPR function. Consistent with this view, we have identified a protein-bound chloride atom in the X-ray structures of ANPR ECD.4,15 On the basis of these findings, we have proposed that chloride may allosterically regulate the ANPR activity and control ANP-natriuresis and further that this control mechanism may explain the apparent renal resistance to ANP observed under certain physiologic and pathologic conditions.14
For chloride to allosterically regulate the ANPR, it needs to reversibly bind. In the X-ray structure of the ANPR ECD,4,15 the bound chloride is completely buried in the polypeptide fold, and it is not immediately clear that it can defuse into or out of the chloride site. The natriuretic peptide clearance receptor (NPCR), whose physiologic role is to bind and remove natriuretic peptides and their fragment from circulation, has a polypeptide fold similar to that of the ANPR and also contains a bound chloride at a corresponding site.16 In contrast to our finding, the ligand-binding activity of the NPCR reportedly does not require the presence of chloride.16 Based on this finding and the fact that chloride is buried, it has been proposed that the bound chloride is a structural element or a remnant of protein evolution but has no functional role.16–18 Thus, the reversibility of chloride binding and its potential role in ANPR regulation have remained controversial.
In this study, we have confirmed the chloride-dependence of ANPR binding activity and obtained physical and structural evidence that chloride indeed binds reversibly and regulates ANP binding. We have found further that the primary and tertiary structures of the chloride binding site in the ANPR is highly conserved among the receptor GCases and the NPCR as well as among the metabotropic glutamate receptors (mGluRs). The mGluRs have the polypeptide folds similar to that of the ANPR and some members of which are known to be regulated by chloride.19 These findings support the proposed chloride-mediate regulation of the ANPR function. The possible role of this ANPR control mechanism in renal salt regulation is discussed.
Results
Chloride-concentration dependence of ANP binding to the full-length ANPR in membranes
We reported earlier that ANP binding to ECD required the presence of chloride and was chloride concentration dependent.14 Here, we confirmed that this chloride dependence of ANP binding also occurs for the full-length ANPR expressed in the cell membrane. The membranes from CHO cells expressing ANPR were suspended and incubated in chloride-free 20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES) buffer, pH 7.0, at 37°C for 1 hr. After this treatment, the membranes showed no detectable ANP-binding activity. The membranes were then incubated with varying concentrations of NaCl for 90 min at room temperature and assayed for ANP binding [Fig. 1(a)]. The ANP-binding activity was restored in a chloride concentration-dependent manner from 0.01 to 10 mM.
Figure 1.
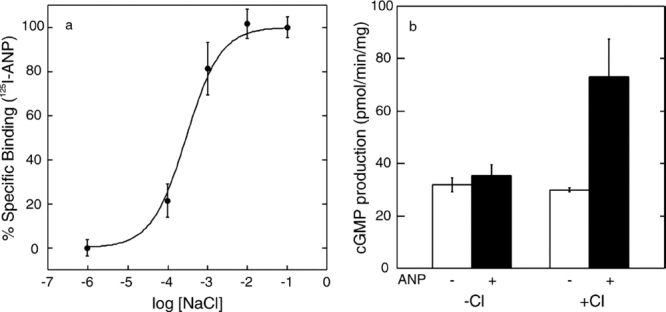
(a) Chloride ion concentration dependence of ANP binding to the full-length ANPR in CHO cell membranes. (b) Stimulation of GCase activity by ANP (1 μM ANP) in the presence and absence of 100 mM NaCl. Bars represent the standard error of triplicate determinations.
Stimulation of GCase activity by ANP requires the presence of chloride
The CHO cell membranes freed from chloride above had a basal GCase activity of 24 to 30 pmol cGMP produced per minute per milligram of protein depending on the preparations. As shown in Fig. 1(b), addition of 1 μM ANP in the absence of chloride had no significant effect on the GCase activity. However, when added with 100 mM NaCl, 1 μM ANP stimulated the GCase activity by 2.5- to 3-fold to approximately 75 pmol/min/mg. Comparable levels of GCase stimulation ranging from 2- to 5-fold have been reported previously with membrane preparations in several reports.20–25
Effectiveness of various halide ions in restoring ANP binding
The purified ECD was freed from chloride by ultrafiltration. Chloride-free ECD (ECD(−)) obtained was incubated with varying concentrations of NaF, NaBr, NaCl, or NaI, and it was assayed for ANP-binding activity [Fig. 2(a)]. NaBr and NaCl both fully restored ANP binding. The concentration of NaCl and NaBr necessary for the half-maximum ANP binding were approximately 1.7 and 0.8 mM, respectively. NaI was less effective, restoring only approximately 50% of binding at 100 mM NaI. NaF at concentrations as high as 100 mM failed to restore ANP binding.
Figure 2.
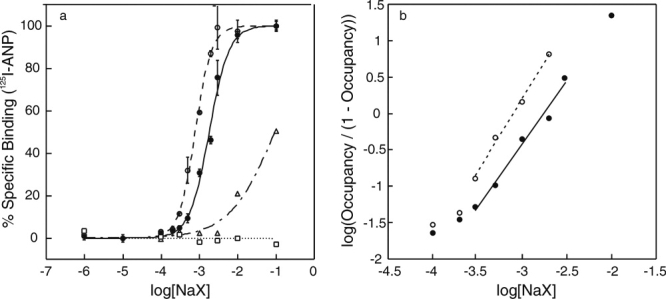
ANP binding to ECD at varying halide concentrations. (a) Binding of 125I-ANP(4-28) to ECD was measured at varying concentrations of NaF (□), NaCl (•), NaBr (○), and NaI (Δ). Bars show the standard error in triplicate determinations. (b) Hill plots for chloride (•) and bromide (○) binding to ECD, in which the occupancy was calculated as the ANP-binding activity divided by the maximum activity at 100 mM halide concentration.
Figure 2(b) shows Hill plots for chloride and bromide binding to ECD(−) using the same data set above. The slopes measured near the half-maximum binding gave estimated Hill coefficients of approximately 1.7 for chloride and 2.0 for bromide.
Crystal structures of ECD(Br)
We previously reported the crystal structures of ECD bound with chloride (ECD(Cl)) with and without bound ANP.4,15 Both structures contained one protein-bound chloride atom in each ECD monomer near the ECD dimerization interface. The loss of ANP binding by removal of chloride and its restoration by bromide addition suggests that the protein-bound chloride can dissociate and be replaced by bromide, and that bromide binding restores the ECD structure and the ANP-binding activity. We prepared ECD bound with bromide (ECD(Br)) by incubating ECD(−) with 10 mM NaBr and crystallized it with and without ANP. Bromide substitution for chloride was then examined by X-ray crystallography.
XAFS scans were taken, and the data were collected above the absorption edge of bromide to maximize the signal. By using the single-wavelength anomalous dispersion technique, the structure of the apo-ECD(Br) dimer was solved (Fig. 3; PDB 3A3K). The structure was essentially identical with that of the apo-ECD(Cl) dimer (root mean square of deviation of Cα, 0.2 Å). The bromide atoms in the apo-ECD(Br) dimer occurred at the same positions as the chloride atoms in the apo-ECD(Cl) dimer structure.
Figure 3.
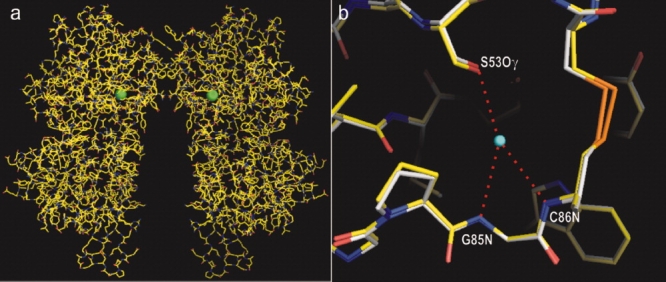
(a) Crystal structure of apo-ECD(Br) dimer determined by single-wavelength anomalous dispersion phasing. Bromide atoms are shown by green balls. (b) Close-up overlay view of the halide binding site in ECD(Br) (carbon atoms shown in white) and that in ECD(Cl) (carbon atoms in yellow). The center of the chloride atom is indicated by a blue dot. Hydrogen bonds are shown by red dotted lines. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
For further confirmation, the anomalous difference Fourier maps were calculated for both apo-ECD(Br) and ANP-ECD(Br) [Fig. 4(a,c)]. The bromide density in the apo-ECD(Br) dimer was localized at positions essentially identical to the chloride atoms in the native apo-ECD(Cl) dimer [Fig. 4(a,c)]. Similarly, the bromide density in ANP-ECD(Br) occurred at the position of chloride atoms in the previously reported ANP-ECD(Cl) structure4 [Fig. 4b,d]. In both cases, the size of the peaks (>45 σ for the apo-ECD(Br) and >28 σ for ANP- ECD(Br)) indicate the presence of bromide (Supporting Information Table II).
Figure 4.
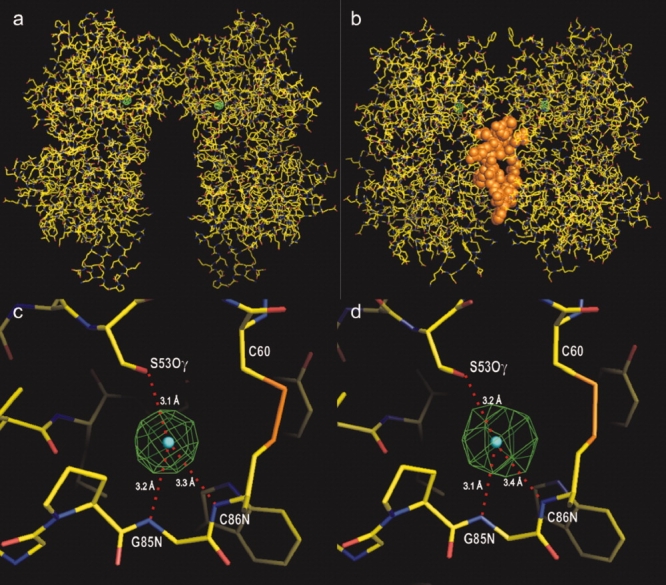
Anomalous difference Fourier map for bromide. (a and b) The electron density maps for bromine (in green) in apo-ECD(Br) and ANP-ECD(Br), respectively, are superimposed onto the apo-ECD(Cl) and ANP-ECD(Cl) structures. ANP is shown in orange. (c and d) Close-up views of the bromide density maps in a and b above, respectively. Bromide densities are contoured at 15 σ. The centers of the chloride atoms in apo-ECD(Cl) and ANP-ECD(Cl) are indicated by blue dots. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Sedimentation equilibrium of ECD in the presence and absence of chloride
To study the effect of chloride on the molecular behavior of ECD in solution, we performed sedimentation equilibrium analysis of ECD in the presence and absence of chloride. The sedimentation equilibrium data in the presence of chloride (Fig. 5, upper panel) showed a weight-average mass Mw well below that of a dimer over the entire concentration range up to 220 μg/mL. The fact that Mw for ECD(Cl) increases systematically with protein concentration, and that the curves for different samples overlap (allowing for experimental noise) are diagnostic of a reversibly self-associating system, and the weak concentration dependence and asymptotic approach toward the expected mass of dimer at higher concentrations are diagnostic of a monomer-dimer equilibrium. These data are fully consistent with reversible monomer-dimer equilibrium with a dissociation constant of approximately 500 nM; a detailed global analysis of these data will be presented elsewhere.
Figure 5.
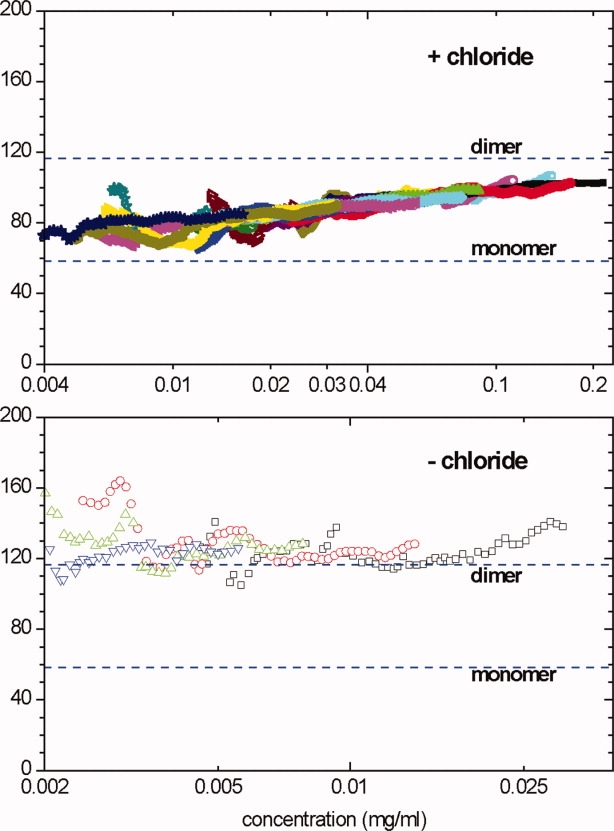
Sedimentation equilibrium data for ECD in the presence (upper) or absence (lower) of 150 mM chloride. The local weight-average molecular mass within regions of the centrifuge cell was calculated from the slope of a ln(concentration) versus (radius)2/2 plot and plotted against the mean concentration for that region. Different data point styles and colors indicate different loading concentrations (see Materials and Methods section for details). The data in the upper include nine different loading concentrations at two rotor speeds; data collected at 9000 rpm are shown as open points, those at 13,000 rpm as filled points. The data in the lower are for four different loading concentrations at 13,000 rpm. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
In the absence of chloride, ECD(−) strongly associates to a dimer, showing an Mw close to that expected for a dimer (Fig. 5, lower panel). No systematic concentration dependence of Mw was found over the concentration range from 30 μg/mL down to 2 μg/mL (as low as we can study by sedimentation equilibrium). Data could not be obtained more than 30 μg/mL for ECD(−) because of irreversible aggregation and precipitation. A global fit of the data for all four loading concentrations as a single ideal species provides a good fit of these data and returns a mass of 126 kDa (95% confidence interval 116–135 kDa), whereas the predicted dimer mass is 116.6 kDa (based on mass spectroscopy). These results show that ECD(−) associates to a tightly bound dimer. These samples also seem to contain a small amount of larger irreversible aggregates. Although those aggregates are mostly pelleted at this rotor speed, they are probably causing the best-fit mass to slightly exceed that for a pure dimer.
Effect of chloride depletion on the structure and stability of ECD
Figure 6(a) shows the far-UV CD spectra of ECD in the presence and absence of chloride. The spectra of ECD(−) and ECD(Cl) are similar, suggesting that the ECD(−) largely retains the native secondary structure. ECD(−) has a CD signal less negative in the 215 to 230 nm wavelength region and more negative at the wavelength shorter than 210 nm compared with ECD(Cl). This spectral change suggests a decrease (∼8%) in the α-helix content and a slight increase in random coil structure.
Figure 6.
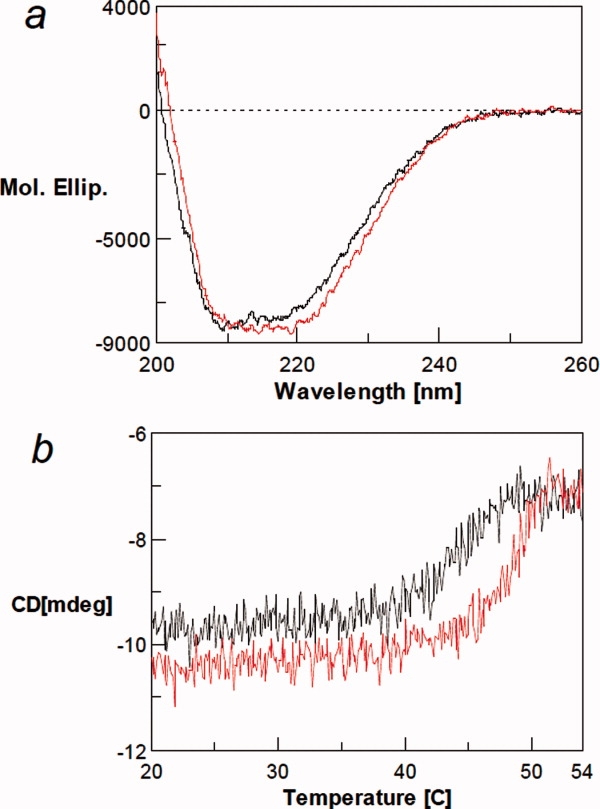
(a) Far-UV CD spectra of ECD in the presence (red line) and absence of 100 mM NaCl (black line). (b) Thermal denaturation of ECD in the presence (red line) and absence of chloride ion (black line) as monitored by CD at 220 nm. Under the conditions used, there is little change in solvent signal with increasing temperature. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Thermal melting of ECD with and without chloride was measured by following the CD at the negative α-helical peak at 220 nm [Fig. 6(b)]. Both ECD(−) and ECD(Cl) show a cooperative structure change over a narrow temperature range, indicating that both states have a distinct, stable tertiary structure and neither form is a molten globule. In the presence of NaCl, the CD intensity was unchanged over the temperature range of 20 to 43°C. The signal magnitude decreased with rising temperature beyond 45°C, with melting complete at 50°C. After the transition, the 220-nm signal is constant at ∼7 milli-degree, indicating that considerable secondary structure remains after the thermal unfolding: note that the CD signal of the solvent is close to 0 independent of the temperature. The photomultiplier voltage changes little with unfolding (data not shown), indicating an absence of light scattering because of aggregation. Although the ECD(Cl) remained soluble and did not aggregate, cooling did not restore the secondary structure content. It is not clear from this experiment whether the observed irreversibility occurred at the end of melting or throughout the transition region. Similar results were obtained in the absence of NaCl, that is, the cooperative transition, retention of postmelted structure, and no aggregation, except that the entire melting curve, including the onset temperature, shifted to a lower temperature by ∼3°C than in the presence of NaCl. This clearly shows that a higher temperature is required to melt the ECD(Cl), although the difference is only 3°C. This small difference may be consistent with the weak binding of chloride−, and therefore, only a small binding energy to stabilize ECD. The observed similar melting behavior of ECD(−) is consistent with the spectral data that the ECD(−) has a distinct fold, resembling the structure of ECD(Cl).
Discussion
We reported earlier that ANP binding to the ANPR requires the presence of chloride, and it is chloride concentration dependent. We have proposed that this chloride-dependent ANP binding may function in the kidney as a switch-off mechanism for the ANPR, thereby allowing sodium reabsorption even in the presence of ANP and preventing the excessive sodium loss.14 Here, we have confirmed the same chloride concentration dependence of ANP binding with the full-length ANPR expressed in CHO cells. When depleted of chloride, the ANPR in membranes show no ANP binding or GCase stimulation. The chloride concentration dependence suggests a dynamic binding equilibrium between the chloride and the receptor, and possible allosteric regulation of the receptor by chloride.
ECD(−) lacking any halide is unable to bind ANP. The binding activity is fully restored by chloride or bromide, partially by iodide, but not by fluoride. This halide selectivity suggests a structure-specific interaction of the halide atom with the binding site rather than a simple charge-to-charge interaction. To confirm that the bound chloride is exchangeable, we crystallized ECD(Br) and ANP-ECD(Br) and determined the location and the identity of the halide atom by crystallographic methods. The bromide density appeared at essentially the same positions as the chloride in ECD(Cl) and ANP-ECD(Cl). The crystal structure of apo-ECD(Br) solved by single-wavelength anomalous dispersion (SAD) phasing is essentially identical with that of ECD(Cl) with the bromide density appearing at corresponding chloride positions. These results provide direct physical evidence for bromide substitution for chloride and restoration of the native receptor structure. Binding of ligands to fully buried sites, with no obvious entry or exit pathway for ligand, has also been observed in other proteins, such as binding of oxygen to heme in myoglobin and hemoglobin.26,27
To examine the mechanism of chloride dependence, we analyzed the molecular characteristics and behavior of ECD(−) in solution by far-UV CD spectroscopy and sedimentation equilibrium analyses. The far-UV CD spectrum of ECD(−) is similar to that of ECD(Cl) with a slight decrease in the estimated α-helix content. Thermal denaturation of ECD(−) monitored by CD at 220 nm shows a cooperative unfolding transition at a temperature lower by 3°C compared with ECD(Cl) [Fig. 6(b)], showing that ECD(−) is slightly less stable but largely retains the native structure. Both the spectra and the melting data clearly indicate that the structure of ECD(−) resembles that of ECD(Cl), and hence, the loss of ANP binding in ECD(−) is not because of protein denaturation.
The sedimentation equilibrium data show that, in the presence of chloride, ECD in solution reversibly dissociates from the dimer seen in crystals to a monomer, and that the monomer is the predominant form at concentrations less than ∼25 μg/mL. In contrast, in the absence of chloride, the equilibrium is strongly shifted to the dimer, with no significant dissociation to monomer detectable down to the lowest concentrations that can be measured (∼2 μg/mL). Thus, chloride affects the ECD dimerization equilibrium, and the loss of chloride shifts the equilibrium strongly to the dimer. In the crystal structure of apo-ECD(Cl), the dimer is formed by association via the membrane-distal subdomain.28,29 To bind ANP, the dimer interface partially opens, and two ECD monomers undergo a twisting motion to capture an ANP molecule. It is possible that the strong dimer association in the absence of chloride may interfere with this opening of the dimer interface, thus preventing the structural change necessary for ANP binding.
The dimer interface involves four strands of α-helix, of which two are provided by each monomer. The decreased α-helix content in ECD(−) indicated by far-UV CD and the strong dimer association observed by sedimentation equilibrium may reflect a change in the dimer interface structure by chloride depletion. We have attempted to crystallize ECD(−) to solve its structure but thus far without success.
The chloride-binding site is located near the dimer interface. Therefore, it is likely that chloride can more readily diffuse into its binding site when ECD is in the monomeric state. The Hill coefficient for chloride binding [Fig. 2(b)] was estimated at approximately 1.7, which suggests a cooperative binding, but the limited titration data presented here are certainly not definitive proof that the binding is cooperative. Note, however, that the sedimentation equilibrium data show that binding of chloride enhances dissociation of the tightly associated ECD dimer to monomers. At the low ECD(−) concentration (0.5 nM) used for the binding assay, it is possible that binding of a single chloride ion to an ECD(−) dimer causes dissociation to monomer, which in turn may facilitate further chloride binding by the other monomer. The fact that chloride binding affects the self-association inevitably leads to a thermodynamic linkage between ligand binding and assembly and the potential for cooperative binding (at least for certain protein concentrations). This linkage between chloride binding and ECD dissociation may in fact be solely responsible for the Hill coefficient apparently being greater than unity, without any direct site-site interactions within the ECD dimer. Because the association of ECD(−) is too strong to measure by sedimentation equilibrium, the strength of the linkage effect on the binding isotherm and Hill plot cannot be estimated. Hence, we cannot say for certain whether binding of one chloride− within a dimer directly influences the chloride-binding affinity of the other monomer. It is of course not necessary that chloride binds cooperatively for it to modulate ANP binding and GCase activity, but cooperativity would enhance the proposed switch-off mechanism by producing switching over a narrower range of chloride concentration.
The reversible dissociation of ECD into monomers we have observed here does not necessarily imply that any significant fraction of ANPR is monomeric in the membranes of physiologically relevant cells at normal chloride concentrations. Association is more strongly favored in the confined 2-dimensional context of a membrane than for ECD free in solution. Even though the self-association of ECD(Cl) is not that strong, it is known that fairly weak interactions with dissociation constants of 1–5 μM are sufficient to dimerize other transmembrane receptors.30,31 Further, it is likely that the transmembrane and/or intracellular portions of the full-length ANPR contribute additional interactions that promote dimerization.
In its binding site, the chloride atom is hydrogen bonded with the hydroxyl group of Ser53 and the backbone NH moieties of Gly85 and Cys86 [Fig. 7(a)]. The binding site also contains a unique cis-peptide bond Gly83—Pro84 and a disulfide bond Cys60—Cys86. An amino acid sequence alignment in the corresponding region [Fig. 7(b)] shows the above features are highly conserved among membrane-bound receptor-GCases and the NPCR. Among them, the crystal structures have been determined, and the presence of a bound chloride has been confirmed only for the ANPR and the NPCR thus far.4,15,16 In the aligned sequences, Ser53 in ANPR (or GCA) is conserved only in GCB and NPCR. It remains to be determined whether other receptors also contain a bound chloride.
Figure 7.
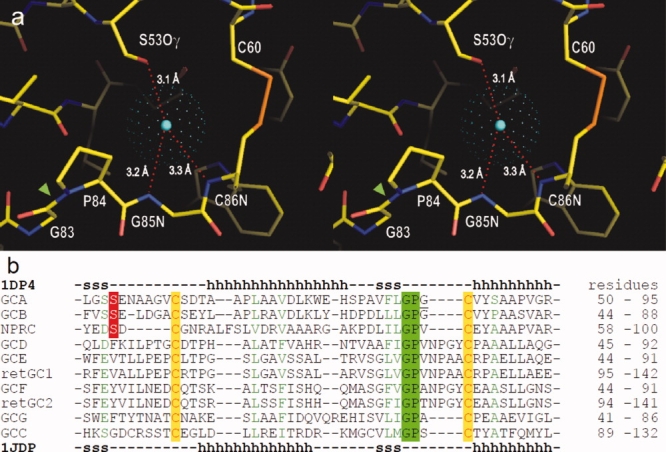
Conserved structural motif for the chloride binding site. (a) Stereo view of the chloride binding site in apo-ECD (PDB 1DP4). The center of chloride atom is shown by a blue dot, and the van der Waals radius is presented by green dots. Chloride is hydrogen bonded to hydroxyl-group of Ser53, and backbone NH moieties of Gly85 and Cys86. The binding site also contains a single cis-peptide bond Gly83-Pro84 (green arrow head) in ECD. (b) Amino acid sequence alignment among natriuretic peptide receptors in the chloride-binding site region: ANPR or GCA75; B-type natriuretic peptide receptor or GCB76; NPCR77; olfactory GCase GCD78; eye retina GCases retGC,79 retGC2,80 GCE,81 and GCF81; GCG82; and guanylin/enterotoxin receptor or GCC.83 The sequences were aligned using the program ClustalW84 and are listed in the order of homology to GCA. The secondary structures in the X-ray structures of ANPR (1DP4) are shown above and below the aligned sequences (h, alpha-helix; s, beta-sheet). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
The NPCR ECD has a polypeptide fold similar to that of the ANPR ECD.16 In contrast to the ANPR, the NPCR reportedly does not show chloride dependence in ligand binding. It has been suggested that the chloride in the NPCR is a structural element and has no functional role.16 The NPCR binds natriuretic peptides and their biologically inactive fragments to clear them from blood circulation. It is not a GCase-linked signaling receptor. The NPCR binds the peptides by a forceps-like clasping motion16 in contrast to a twisting motion by the ANPR.4,5 The twisting motion in the ANPR generates a rotation of the receptor monomers that triggers transmembrane signal transduction.4–6 The difference in the chloride dependence between the ANPR and the NPCR may reflect the difference in their signaling mechanisms and/or physiologic roles.
The mGluRs are seven-transmembrane helix G-protein-coupled receptors that mediate excitatory synaptic transmission in the central nervous system. The extracellular ligand-binding region (LBR) of the mGluR32 also has a polypeptide fold closely resembling that of the ANPR ECD.4,15 The LBR structures with and without bound glutamate has shown that glutamate binding stabilizes both the active dimer and the closed monomer conformation in a dynamic equilibrium and that these movements in the dimer are likely to alter the separation of the transmembrane and intracellular regions and thereby activate the receptor.32 The signaling mechanism of mGluRs hence seems to differ form that of the ANPR. It has been shown that ligand binding by the mGluRs is influenced to different extents by various anions.19,33 In particular, the type-4 mGluR exhibits a strict chloride concentration dependence19 similar to that observed with the ANPR.14 It has been suggested that a transient reduction in the synaptic chloride concentration may reduce agonist binding to mGlu4 and possibly other mGluRs, leading to increased neurotransmitter release.19
Conversely, the ionotropic glutamate receptors (iGluRs) are ligand-gated cation channels with structures distinct from the mGluRs or the ANPR. In the iGluR kainate receptor, chloride ions bind in a cavity formed at the receptor dimer interface. The absence of chloride in this receptor destabilizes the dimer and leads to receptor desensitization.34–36
Alignment of mGluR sequences in the region corresponding to the chloride binding site shows high residue conservation [Fig. 8(a)], including Ser or Thr at the position corresponding to Ser53 in the ANPR and Gly-Pro, Gly-Gly, or Gly-Ala sequence corresponding to Gly83-Pro84 in the ANPR. These peptide linkages likely allow cis-configuration similar to that in the ANPR.
Figure 8.
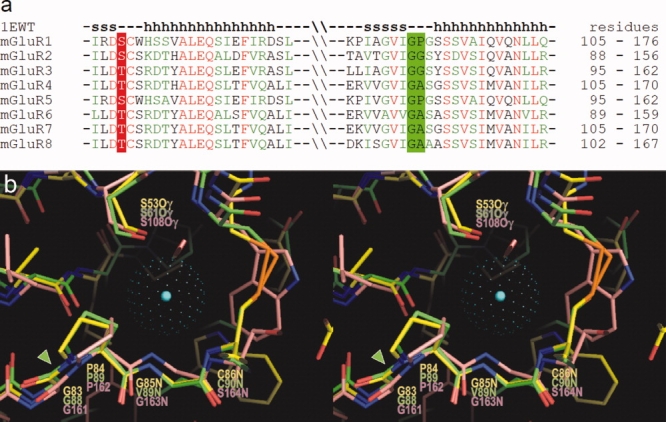
(a) Amino acid sequence alignment among mGluRs in the region homologous to the chloride binding site in ANPR. Residues in mGluRs, corresponding to Ser53 in ANPR, are conserved as either Ser or Thr. The Gly-Pro sequence in ANPR is conserved as Gly-Pro, Gly-Gly, or Gly-Ala. The secondary structures in mGluR1 are shown above the aligned sequences. (b) Overlay of the three-dimensional structures of the chloride binding sites in ANPR (PDB 1DP4)15 and NPCR (1JDP)16 and that of the water bound site in mGluR1 (1EWT)32 in stereo view. The carbon atoms in ANPR, NPCR, and mGluR1 are shown in yellow, green, and pink, respectively. The center of chloride atom in the ANPR is shown by a blue dot, and the van der Waals radius is presented by green dots. The conserved Gly-Pro cis-peptide bonds are highlighted by a green arrowhead. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
The crystal structure of the extracellular LBR of the mGluR1 has been reported (PDB 1EWT).32 The 3D structure of the mGluR1 in the above conserved region is nearly superimposable with the chloride binding site in the ANPR and the NPCR [Fig. 8(b)], including the hydrogen-bonding Ser residue and Gly-Pro cis-peptide bond. In the mGluR1 LBR, a water molecule is bound in this site. It is possible that a chloride or other anion may also bind and regulate ligand binding. Thus, based on the primary and tertiary structure comparison, the unique structural motif of the chloride-binding site in the ANPR is highly conserved among receptor-GCases, NPCR, and mGluRs, all of which have the polypeptide fold similar to that of the ANPR ECD. Binding of chloride or other anion to the site may allosterically regulate ligand binding and, hence, the receptor function. The chloride dependence of ANP binding and conserved chloride-binding site motif strongly suggests a regulatory role for the bound chloride. We have proposed that the chloride-dependent control of ANP binding may occur in the renal collecting tubules to prevent excessive salt loss.14
The kidney rapidly filters the plasma at a rate of roughly 100 mL/min (or ∼150 L/day) and reabsorbs most of the filtered salts and water back into the circulation.37 This rapid filtration-reabsorption process operates in a dynamic equilibrium to maintain the salt–body fluid volume balance. In the kidney, most of the filtered sodium is reabsorbed in the proximal tubule. Further reabsorption occurs in the Loop of Henle and the distal tubule via the Na+-K-2Cl− and Na+-Cl− cotransporters, respectively.37 This suggests that the luminal chloride concentration changes in parallel with the sodium concentration. The final and rate-determining regulation of sodium reabsorption occurs in the collecting tubule, where ANP and counteracting aldosterone exert their hormonal activities. Aldosterone stimulates sodium reabsorption by opening sodium channels, whereas ANP causes natriuresis by decreasing the number of open channels.37 When volume depletion activates the RAA system, the luminary sodium and, hence, chloride concentrations decreases to <1 mEq/L.37–39 At such low chloride concentrations, ANP binding to the receptor is blocked, thus preventing the closure of sodium channels and allowing the sodium reabsorption irrespective of the presence of ANP.
The natriuretic activity of ANP has been well documented experimentally. However, the physiologic role of ANP as a natriuretic hormone continues to be debated,40–42 because there are certain physiologic and pathologic conditions where salt is retained despite increased plasma ANP. For example, in normovolemic animals, infusion of high-dose ANP does not cause a corresponding increase in natriuresis.43 In ANP-overexpressing transgenic animals, plasma ANP levels are markedly increased, yet salt is retained.8,10 An attenuated natriuretic response to ANP is also observed in animals on a low-salt diet.9,44,45 The salt deficit in these animals is associated with avid sodium reabsorption. ANP infusion in these animals does not block sodium reabsorption. This ANP-insensitive sodium retention occurs independent of sympathetic or RAA systems.45 In edematous diseases, such as congestive heart failure, nephrotic syndrome, and hepatic cirrhosis, plasma levels of ANP are markedly increased, yet sodium is retained.11–13 The lack of renal response to ANP in face of increased ANP, which we refer to as natriuretic peptide escape in analogy to aldosterone escape, has suggested an yet undetermined mechanism that directly turns off ANP natriuresis.45 We propose that the chloride-dependent control of the ANPR may be the mechanism that turns off ANP natruresis under these in vivo conditions.
For this mechanism to operate, the ANPR needs to be localized on the luminal side of the collecting tubule. Hirsch et al.46 have reported predominant localization of the ANPR (or GCA) in the luminal membrane of the intercalated cells in the collecting tubule and its involvement in pH regulation by inhibiting Na+/H− exchanger. Luminal localization of GCB47 and GCG46 has also been reported. These findings suggest that natriuretic peptides and other factors filtered into the luminal fluid play regulatory roles in electrolyte-body fluid homeostasis via their receptors. Indeed, small peptides are filtered through the glomerulus, and the presence of ANP and other bioactive peptides in the urine (and hence in the luminal fluid) has been well documented.48–53
Citing from Rose and Rennke,37 the renal filtration (∼150 L/day) “represents a volume that is… approximately 60 times that of the plasma; as a result, survival requires that almost all of the filtered solutes and water be returned to the systemic circulation by tubular reabsorption.” Because survival depends on salt reabsorption, it is conceivable that there exists a mechanism by which the need for sodium reabsorption overrides natriuretic stimuli. The chloride requirement for ANP binding may act as an autonomous switch-off mechanism for the ANPR and ANP natriuresis at low luminal chloride concentrations, thereby enabling sodium reabsorption despite ANP presence. This control mechanism, which is suggested by our molecular studies of the ANPR, may explain the renal resistance ANP observed under certain in vivo conditions. The potential significance of this chloride-dependent control mechanism for the ANPR in renal salt regulation has been noted,54–56 and its clarification awaits further physiologic studies.
Materials and Methods
Expression of the full-length ANPR on CHO cells
CHO cells were transfected with an ANPR expression construct pcDNA3-NPRA encoding the entire receptor sequence (residues 1 through 1029)57 using a Qiagen Effectan Transfection Kit. After 3 days, the cells were treated with G-418 (800 μg/mL) and were cultured through several cycles of trypsinization, dilution, plating, and colony selection steps in the presence of G-418 while monitoring 125I-ANP-binding activity. The clones exhibiting highest binding activity were selected.
Preparation of CHO cell membranes
CHO cells were homogenized in eight volumes of homogenization buffer consisting of Tris-HCl buffer, pH 7.5, 1 mM EGTA, 1 mM EDTA, and 1 mM β glycerol phosphate and protease inhibitors (5 μg/mL leupeptin, 5 μg/mL pepstatin, 5 μg/mL aprotinin, and 1 mM PMSF) using a Polytron at a setting of 7 at 0°C. Membranes were collected by centrifugation at 100,000g for 90 min and resuspended in the same buffer at a protein concentration of 0.3 mg/mL.
Chloride concentration dependence of ANP binding to the ANPR in membranes
The membranes above were freed from chloride as follows. Membranes (30 μg protein) were suspended in 400 μL of 20 mM HEPES buffer (pH 7.4) and incubated at 37°C for 1 hr. The membranes were then spun down and resuspended in 400 μL of the same buffer. To test the effect of chloride on ANP binding, the membranes were incubated with varying concentrations of NaCl in the assay buffer (20 mM HEPES buffer, pH 7.4, 0.05% bovine serum albumin, and 0.01% bacitracin) at 37°C for 90 min. The total salt concentration in the mixture was maintained at 100 mM by supplementing with appropriate concentrations of sodium acetate. ANP-binding assay was performed at room temperature using 125I-ANP (2.4 mCi/nanomole) as the tracer as described.58
Effect of chloride on guanylate cyclase activation by ANP
Chloride-free membranes prepared earlier were incubated in 20 mM HEPES buffer, pH 7.4 containing 100 mM NaCl or 100 mM sodium acetate at 37°C for 90 min. The GCase activation by ANP was measured with the membranes (100 μg) by assaying the activity in the presence and absence of 1 μM ANP by the method of Waldman et al.21
Expression and purification of ECD
ECD consisting of residues 1 through 435 of rat ANPR was expressed and purified as described previously.57
Chloride concentration dependence of ANP binding to ECD
ECD was first freed from chloride by repeating several cycles of ∼20 times dilution with 20 mM sodium phosphate buffer, pH 7.4, and concentration using a Millipore Ultra-free filter tube (10 kDa cutoff). To study the effects of various halides, ECD(−) (5 ng) was incubated in the assay buffer (200 μL) containing varying concentrations of NaF, NaCl, NaBr, or NaI at 37°C for 30 min as previously described.14 The total salt concentration in the mixture was maintained at 100 mM by adding appropriate concentrations of sodium acetate. Binding assay was performed using 125I-ANP (200000 cpm, approximately 50 fmol) as the tracer in the presence and absence of 0.1 μM ANP (approximately 100 times the Kd). ECD-bound 125I-ANP was separated by gel filtration on a Sephadex G-50 column and counted for 125I radioactivity as described.57
Crystallization of ECD(Br) with and without ANP
ECD(−) was incubated with 50 mM NaBr in 50 mM HEPES, pH 7.4 at 15°C for 30 min to obtain ECD(Br), which was then concentrated to 10 mg/mL using a Millipore Ultra-free concentrator (10 kDa cutoff). ANP-ECD(Br) complex was prepared by incubating ECD(Br) with 1.1-fold molar excess of ANP(7-27) (sequence: Cys-Phe-Gly-Gly-Arg-Ile-Asp-Arg-Ile-Gly-Ala-Gln-Ser-Gly-Leu-Gly-Cys-Asn-Ser-Phe-Arg) at room temperature for 1 hr. ANP(7-27) is the smallest size ANP peptide that retains the full biologic activity.59 Crystals of apo-ECD(Br) and ANP-ECD(Br) grew under the conditions similar to those used to grow crystals of apo-ECD(Cl)15 and ANP-ECD(Cl),4,60 respectively.
Collection of X-ray diffraction data
Crystals of apo-ECD(Br) were dialyzed against mother liquor added with 30% ethylene glycol and quickly frozen in liquid nitrogen. ANP-ECD(Br) crystals were dialyzed against mother liquor containing 3.4M ammonium sulfate and were frozen in liquid propane.4
Diffraction data for apo-ECD(Br) were collected at the Advanced Photon Source 19ID beamline at the wavelength 0.92132 Å using the inverse beam method to optimize anomalous signals. Data for ANP-ECD(Br) were collected at the National Synchrotron Light Source X4A beamline at wavelength 0.91973 Å. The diffraction data were integrated and scaled using HKL2000.61 The space group of apo-ECD(Br) crystal was P41212 with the unit cell dimensions, a = b = 120.5 Å, c = 160.6 Å. These parameters were nearly identical to those of the apo-ECD(Cl) crystals.15 The space group of the ANP-ECD(Br) complex crystal was P61 with the unit cell dimensions a = b = 100.2 Å, c = 258.2 Å, which were nearly identical to those of ANP-ECD(Cl) crystals.4,60 Data collection and statistics are summarized in Supporting Information Table I.
Table I.
Refinement Statistics for Apo-ECD(Br) Dimer Structure
| Resolution used for the refinement (Å) | 20–2.5 (2.56–2.50) |
| No. reflections used | 36455 (2266) |
| Rcryst (%) | 20.5 (24.3) |
| Rfree (%) | 24.3 (35.9) |
| Average B-factors (Å2) | |
| ANP receptor | 23.8 |
| Oligosaccharide | 43.5 |
| Solvent | 27.2 |
| Root mean square deviation from ideality | |
| Bond length (Å) | 0.009 |
| Bond angles (o) | 1.2 |
| Bonded B-factors (Å2) (main chain, side chain) | 0.6, 1.3 |
| Ramachandran plot (%) (favored, allowed, generous, disallowed) | 90.6, 8.8, 0.6, 0 |
| Overall figure of merit | |
| After solve | 0.23 (0.11) |
| After resolve | 0.68 (0.36) |
Determination of the ECD(Br) structure
The apo-ECD(Br) structure was determined by the SAD phasing method (PDB 3A3K). The bromide positions were first determined using the program SHELX-D.62 The initial phases were then calculated at 2.5 Å resolution using the program SOLVE63 and improved by density modification using the program RESOLVE.64 The initial figure of merit was 0.21, which was improved to 0.68 at 2.5 Å resolution through repeated cycles of refinement. The program O65 was used for building the structure. The structure was refined with CNS66 by iterative cycles of simulated annealing, imposing noncrystallographic symmetry restraints for the two monomers, which were then released. Further refinement was done using REFMAC5.67 The quality of the structure was assessed with the program PROCHECK.68 The refinement statistics is shown in Table I. Figures were drawn using PYMOL.69 Overlaying of the three-dimensional structures was done using the program O.
Calculation of anomalous difference Fourier maps for confirmation of bromide atoms
An anomalous difference map, using the changes in intensity introduced by the bromide in apo-ECD(Br), was calculated at 2.5 Å resolution with the CNS software package66 using the phases reported for the apo-ECD(Cl) crystal structure (PDB 1DP4).4 The five highest bromide density peaks are shown in Supporting Information Table II.
Sedimentation equilibrium studies
Sedimentation equilibrium runs were performed in a Beckman XL-A or XL-I using six-sector equilibrium cells. The studies of ECD(Cl) were performed in 20 mM sodium phosphate containing 150 mM NaCl, pH 7.4, at loading concentrations of approximately 150, 75, 50, 30, 20, 15, 12, 10, and 8 μg/mL. Scans were recorded at either 278 or 230 nm. Samples were centrifuged at 9000 rpm at 25°C, and scans were recorded after 20 and 24 hr to confirm that equilibrium had been reached. The rotor speed was then increased to 13,000 rpm, and a second set of scans were recorded after 20 and 24 hr. At the end of the run, the rotor speed was increased to 44,000 for 8 hr, and a scan was recorded to establish the baseline offsets.
ECD without chloride was prepared in 50 mM sodium phosphate, pH 7.4, and was loaded at approximately 20, 10, 7.5, and 5 μg/mL. Samples were centrifuged at 13,000 rpm at 25°C, and scans were recorded at 230 nm after 16, 20, and 24 hr to confirm that equilibrium had been reached. At the end of the run, the rotor speed was increased to 48,000 for 8 hr, and a scan was recorded to establish the baseline offsets.
The data were analyzed using the program KDALTON.70,71 Plots of weight-average molecular weight versus concentration were generated by fitting subgroups of 40 adjacent data points within the total radial range for each scan. A partial specific volume of 0.7180 mL/g at 25°C was calculated for ANPR based on a polypeptide partial specific volume calculated from the amino acid composition using the program SEDNTERP by Laue, Hayes, and Philo,72 a carbohydrate content based on the total monomer mass measured by matrix-assisted laser desorption and ionization spectroscopy, and a typical partial specific volume of 0.662 mL/g for carbohydrates in glycoproteins.73
CD spectroscopy
Far-UV CD spectra (between 200 and 260 nm) were recorded on a Jasco J-715 spectrometer at 20°C using a quartz cell with 1 mm optical path length. The temperature was controlled using a Peltier cell holder with a PTC-348WI temperature controller. Measurements were made at a protein concentration of 0.14 mg/mL in 50 mM sodium phosphate buffer in the presence and absence of 100 mM NaCl. CD spectra were analyzed for secondary structures using the program CDNN.74 To study thermal denaturation, the change in secondary structures was monitored by CD at 220 nm under the same conditions as earlier and a temperature scan rate of 30°C/hr.
Acknowledgments
We thank Xiaolun Zhang for able technical assistance. Ogawa, Qiu, and Ogata performed biochemical studies and X-ray crystallography. Philo and Arakawa designed and performed sedimentation equilibrium and circular dichroism studies. Misono designed research and had full access to all of the data. Misono, Philo, and Arakawa drafted the manuscript.
Glossary
Abbreviations:
- ANP
atrial natriuretic peptide
- ANP-ECD
ECD in complex with ANP
- ANPR
ANP receptor (A-type natriuretic peptide receptor)
- ECD
extracellular domain
- ECD(Br)
ECD bound with bromide
- ECD(Cl)
ECD bound with chloride
- ECD(−)
ECD without bound halide
- GCase
guanylate cyclase
- iGluR
ionotropic glutamate receptor
- LBR
ligand-binding region
- mGluR
metabotropic glutamate receptor
- NPCR
natriuretic peptide clearance receptor
- SAD
single-wavelength anomalous dispersion
- RAA
renin-angiotensin-aldosterone.
References
- 1.de Bold AJ, Borenstein HB, Veress AT, Sonnenberg H. A rapid and potent natriuretic response to intravenous injection of atrial myocardial extract in rats. Life Science. 1981;28:89–94. doi: 10.1016/0024-3205(81)90370-2. [DOI] [PubMed] [Google Scholar]
- 2.Grammer RT, Fukumi H, Inagami T, Misono KS. Rat atrial natriuretic factor. Purification and vasorelaxant activity. Biochem Biophys Res Commun. 1983;116:696–703. doi: 10.1016/0006-291x(83)90581-8. [DOI] [PubMed] [Google Scholar]
- 3.Currie MG, Geller DM, Cole BR, Boylan JG, YuSheng W, Holmberg SW, Needleman P. Bioactive cardiac substances: potent vasorelaxant activity in mammalian atria. Science. 1983;221:71–73. doi: 10.1126/science.6857267. [DOI] [PubMed] [Google Scholar]
- 4.Ogawa H, Qiu Y, Ogata CM, Misono KS. Crystal structure of hormone-bound atrial natriuretic peptide receptor extracellular domarotation mechanism for transmembrane signal transduction. J Biol Chem. 2004;279:28625–28631. doi: 10.1074/jbc.M313222200. [DOI] [PubMed] [Google Scholar]
- 5.Misono KS, Ogawa H, Qiu Y, Ogata CM. Structural studies of the natriuretic peptide receptor: a novel hormone-induced rotation mechanism for transmembrane signal transduction. Peptides. 2005;26:957–968. doi: 10.1016/j.peptides.2004.12.021. [DOI] [PubMed] [Google Scholar]
- 6.Garrett RH, Grisham CM. Biochemistry. 4th ed. Boston, MA: Brooks/Cole, Cengage; 2009. pp. 1021–1023. [Google Scholar]
- 7.Sonnenberg H, Honrath U, Chong CK, Field LJ, Veress AT. Proximal tubular function in transgenic mice overexpressing atrial natriuretic factor. Can J Physiol Pharmacol. 1994;72:1168–1170. doi: 10.1139/y94-165. [DOI] [PubMed] [Google Scholar]
- 8.Veress AT, Field LJ, Steinhelper ML, Sonnenberg H. Effect of potassium infusion on renal function in ANF-transgenic mice. Clin Invest Med. 1992;15:483–488. [PubMed] [Google Scholar]
- 9.Veress AT, Chong CK, Field LJ, Sonnenberg H. Blood pressure and fluid-electrolyte balance in ANF-transgenic mice on high- and low-salt diets. Am J Physiol. 1995;269:R186–R192. doi: 10.1152/ajpregu.1995.269.1.R186. [DOI] [PubMed] [Google Scholar]
- 10.Field LJ, Veress AT, Steinhelper ME, Cochrane K, Sonnenberg H. Kidney function in ANF-transgenic mice: effect of blood volume expansion. Am J Physiol. 1991;260 doi: 10.1152/ajpregu.1991.260.1.R1. [DOI] [PubMed] [Google Scholar]
- 11.Warner L, Skorecki K, Blendis LM, Epstein M. Atrial natriuretic factor and liver disease. Hepatology. 1993;17:500–513. erratum in Hepatology 17:1174. [PubMed] [Google Scholar]
- 12.Koepke JP, DiBona GF. Blunted natriuresis to atrial natriuretic peptide in chronic sodium-retaining disorders. Am J Physiol. 1987;252:F865–F871. doi: 10.1152/ajprenal.1987.252.5.F865. [DOI] [PubMed] [Google Scholar]
- 13.Burnett JC, Jr., Kao PC, Hu DC, Heser DW, Heublein D, Granger JP, Opgenorth TJ, Reeder GS. Atrial natriuretic peptide elevation in congestive heart failure in the human. Science. 1986;231:1145–1147. doi: 10.1126/science.2935937. [DOI] [PubMed] [Google Scholar]
- 14.Misono KS. Atrial natriuretic factor binding to its receptor is dependent on chloride concentration: a possible feedback-control mechanism in renal salt regulation. Circ Res. 2000;86:1135–1139. doi: 10.1161/01.res.86.11.1135. [DOI] [PubMed] [Google Scholar]
- 15.van den Akker F, Zhang X, Miyagi M, Huo X, Misono KS, Yee VC. Structure of the dimerized hormone-binding domain of a guanylyl-cyclase-coupled receptor. Nature. 2000;406:101–104. doi: 10.1038/35017602. [DOI] [PubMed] [Google Scholar]
- 16.He X, Chow D, Martick MM, Garcia KC. Allosteric activation of a spring-loaded natriuretic peptide receptor dimer by hormone. Science. 2001;293:1657–1662. doi: 10.1126/science.1062246. [DOI] [PubMed] [Google Scholar]
- 17.Potter LR, Hunter T. Guanylyl cyclase-linked natriuretic peptide receptors: structure and regulation. J Biol Chem. 2001;276:6057–6060. doi: 10.1074/jbc.R000033200. [DOI] [PubMed] [Google Scholar]
- 18.van den Akker F. Structural insights into the ligand binding domains of membrane bound guanylyl cyclases and natriuretic peptide receptors. J Mol Biol. 2001;311:923–937. doi: 10.1006/jmbi.2001.4922. [DOI] [PubMed] [Google Scholar]
- 19.Kuang D, Hampson DR. Ion dependence of ligand binding to metabotropic glutamate receptors. Biochem Biophys Res Commun. 2006;345:1–6. doi: 10.1016/j.bbrc.2006.04.064. [DOI] [PubMed] [Google Scholar]
- 20.Kuno T, Andresen JW, Kamisaki Y, Waldman SA, Chang LY, Saheki S, Leitman DC, Nakane M, Murad F. Co-purification of an atrial natriuretic factor receptor and particulate guanylate cyclase from rat lung. J Biol Chem. 1986;261:5817–5823. [PubMed] [Google Scholar]
- 21.Waldman SA, Rapoport RM, Murad F. Atrial natriuretic factor selectively activates particulate guanylate cyclase and elevates cyclic GMP in rat tissues. J Biol Chem. 1984;259:14332–14334. [PubMed] [Google Scholar]
- 22.Winquist RJ, Faison EP, Waldman SA, Schwartz K, Murad F, Rapoport RM. Atrial natriuretic factor elicits an endothelium-independent relaxation and activates particulate guanylate cyclase in vascular smooth muscle. Proc Natl Acad Sci USA. 1984;81:7661–7664. doi: 10.1073/pnas.81.23.7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishido M, Fujita T, Hagiwara H, Shimonaka M, Saheki T, Hirata Y, Hirose S. Physical and functional association of the atrial natriuretic peptide receptor with particulate guanylate cyclase as demonstrated using detergent extracts of bovine lung membranes. Biochem Biophys Res Commun. 1986;140:101–106. doi: 10.1016/0006-291x(86)91063-6. [DOI] [PubMed] [Google Scholar]
- 24.Abe T, Nishiyama K, Snajdar R, He X, Misono KS. Aortic smooth muscle contains guanylate-cyclase-coupled 130-kDa atrial natriuretic factor receptor as predominant receptor form. Spontaneous switching to 60-kDa C-receptor upon cell culturing. Eur J Biochem. 1993;217:295–304. doi: 10.1111/j.1432-1033.1993.tb18246.x. [DOI] [PubMed] [Google Scholar]
- 25.Woodard GE, Zhao J, Rosado JA. Different effect of ATP on ANP receptor guanylyl cyclase in spontaneously hypertensive and normotensive rats. Acta Physiol (Oxf) 2006;188:195–206. doi: 10.1111/j.1748-1716.2006.01628.x. [DOI] [PubMed] [Google Scholar]
- 26.Srajer V, Ren Z, Teng TY, Schmidt M, Ursby T, Bourgeois D, Pradervand C, Schildkamp W, Wulff M, Moffat K. Protein conformational relaxation and ligand migration in myogloba nanosecond to millisecond molecular movie from time-resolved Laue X-ray diffraction. Biochemistry. 2001;40:13802–13815. doi: 10.1021/bi010715u. [DOI] [PubMed] [Google Scholar]
- 27.Frauenfelder H, McMahon BH, Fenimore PW. Myoglobthe hydrogen atom of biology and a paradigm of complexity. Proc Natl Acad Sci USA. 2003;100:8615–8617. doi: 10.1073/pnas.1633688100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogawa H, Qiu Y, Huang L, Tam-Chang SW, Young HS, Misono KS. Structure of the atrial natriuretic peptide receptor extracellular domain in the unbound and hormone-bound states by single-particle electron microscopy. FEBS J. 2009;276:1347–1355. doi: 10.1111/j.1742-4658.2009.06870.x. [DOI] [PubMed] [Google Scholar]
- 29.Qiu Y, Ogawa H, Miyagi M, Misono KS. Constitutive activation and uncoupling of the atrial natriuretic peptide receptor by mutations at the dimer interface. Role of the dimer structure in signalling. J Biol Chem. 2004;279:6115–6123. doi: 10.1074/jbc.M310225200. [DOI] [PubMed] [Google Scholar]
- 30.Klein P, Mattoon D, Lemmon MA, Schlessinger J. A structure-based model for ligand binding and dimerization of EGF receptors. Proc Natl Acad Sci USA. 2004;101:929–934. doi: 10.1073/pnas.0307285101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Philo JS, Aoki KH, Arakawa T, Narhi LO, Wen J. Dimerization of the extracellular domain of the erythropoietin (EPO) receptor by EPO: one high-affinity and one low-affinity interaction. Biochemistry. 1996;35:1681–1691. doi: 10.1021/bi9524272. [DOI] [PubMed] [Google Scholar]
- 32.Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, Morikawa K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 2000;407:971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- 33.Eriksen L, Thomsen C. [3H]-L-2-amino-4-phosphonobutyrate labels a metabotropic glutamate receptor, mGluR4a. Br J Pharmacol. 1995;116:3279–3287. doi: 10.1111/j.1476-5381.1995.tb15136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Plested AJ, Vijayan R, Biggin PC, Mayer ML. Molecular basis of kainate receptor modulation by sodium. Neuron. 2008;58:720–735. doi: 10.1016/j.neuron.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chaudhry C, Plested AJ, Schuck P, Mayer ML. Energetics of glutamate receptor ligand binding domain dimer assembly are modulated by allosteric ions. Proc Natl Acad Sci USA. 2009;106:12329–12334. doi: 10.1073/pnas.0904175106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Plested AJ, Mayer ML. Structure and mechanism of kainate receptor modulation by anions. Neuron. 2007;53:829–841. doi: 10.1016/j.neuron.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 37.Rose BH, Rennke HG. Renal pathophysiology—the essentials. Baltimore, MD: Williams & Wilkins; 1994. pp. 1–15. [Google Scholar]
- 38.Hanley MJ, Kokko JP. Study of chloride transport across the rabbit cortical collecting tubule. J Clin Invest. 1978;62:39–44. doi: 10.1172/JCI109111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moe OW, Berry CA. Rector FC Renal transport of glucose, amino acid, sodium, chloride, and water. In: Brenner BM, editor. The Kidney. 6th ed. 2000. pp. 375–415. [Google Scholar]
- 40.Drummer C, Franck W, Heer M, Forssmann WG, Gerzer R, Goetz K. Postprandial natriuresis in humans: further evidence that urodilatin, not ANP, modulates sodium excretion. Am J Physiol. 1996;270:F301–F310. doi: 10.1152/ajprenal.1996.270.2.F301. [DOI] [PubMed] [Google Scholar]
- 41.Goetz KL. Evidence that atriopeptin is not a physiological regulator of sodium excretion. Hypertension. 1990;15:9–19. doi: 10.1161/01.hyp.15.1.9. [DOI] [PubMed] [Google Scholar]
- 42.Goetz KL. The tenuous relationship between atriopeptin and sodium excretion. Acta Physiol Scand Suppl. 1990;591:88–96. [PubMed] [Google Scholar]
- 43.Bie P, Wang BC, Leadley RJ, Jr., Goetz KL. Hemodynamic and renal effects of low-dose infusions of atrial peptide in awake dogs. Am J Physiol. 1988;254:R161–R169. doi: 10.1152/ajpregu.1988.254.2.R161. [DOI] [PubMed] [Google Scholar]
- 44.Honrath U, Chong CK, Wilson DR, Sonnenberg H. Dietary salt extremes and renal function in rats: effect of atrial natriuretic factor. Clin Sci (Colch) 1994;87:525–531. doi: 10.1042/cs0870525. [DOI] [PubMed] [Google Scholar]
- 45.Veress AT, Honrath U, Chong CK, Sonnenberg H. Renal resistance to ANF in salt-depleted rats is independent of sympathetic or ANG-aldosterone systems. Am J Physiol. 1997;272:F545–F550. doi: 10.1152/ajprenal.1997.272.4.F545. [DOI] [PubMed] [Google Scholar]
- 46.Hirsch JR, Kruhoffer M, Adermann K, Heitland A, Maronde E, Meyer M, Forssmann WG, Herter P, Plenz G, Schlatter E. Cellular localization, membrane distribution, and possible function of guanylyl cyclases A and 1 in collecting ducts of rat. Cardiovasc Res. 2001;51:553–561. doi: 10.1016/s0008-6363(00)00297-2. [DOI] [PubMed] [Google Scholar]
- 47.Ritter D, Dean AD, Gluck SL, Greenwald JE. Natriuretic peptide receptors A and B have different cellular distributions in rat kidney. Kidney Int. 1995;48:5758–5766. doi: 10.1038/ki.1995.474. [DOI] [PubMed] [Google Scholar]
- 48.Bentzen H, Pedersen RS, Nyvad O, Pedersen EB. Effect of exercise on natriuretic peptides in plasma and urine in chronic heart failure. Int J Cardiol. 2004;93:121–130. doi: 10.1016/S0167-5273(03)00156-6. [DOI] [PubMed] [Google Scholar]
- 49.Totsune K, Takahashi K, Satoh F, Sone M, Ohneda M, Satoh C, Murakami O, Mouri T. Urinary immunoreactive brain natriuretic peptide in patients with renal disease. Regul Pept. 1996;63:141–147. doi: 10.1016/0167-0115(96)00035-3. [DOI] [PubMed] [Google Scholar]
- 50.Mattingly MT, Brandt RR, Heublein DM, Wei CM, Nir A, Burnett JC., Jr. Presence of C-type natriuretic peptide in human kidney and urine. Kidney Int. 1994;46:744–747. doi: 10.1038/ki.1994.329. erratum in Kidney Int 1996;50:1442. [DOI] [PubMed] [Google Scholar]
- 51.Marumo F, Sakamoto H, Ando K, Ishigami T. Concentrations of atrial natriuretic peptide in plasma and urine of kidney disease patients. Clin Chem. 1990;36:1650–1653. [PubMed] [Google Scholar]
- 52.Ando K, Umetani N, Kurosawa T, Takeda S, Katoh Y, Marumo F. Atrial natriuretic peptide in human urine. Klin Wochenschr. 1988;66:768–772. doi: 10.1007/BF01726575. [DOI] [PubMed] [Google Scholar]
- 53.Suzuki Y, Suzuki H, Ohtake R, Tsuchiya T, Muramatsu H, Hashigami Y, Kobori H, Shimoda S. Plasma and urine concentrations of atrial natriuretic peptide in patients with diabetes mellitus. Pancreas. 1988;3:404–408. doi: 10.1097/00006676-198808000-00006. [DOI] [PubMed] [Google Scholar]
- 54.Charloux A, Piquard F, Doutreleau S, Brandenberger G, Geny B. Mechanisms of renal hyporesponsiveness to ANP in heart failure. Eur J Clin Invest. 2003;33:769–778. doi: 10.1046/j.1365-2362.2003.01222.x. [DOI] [PubMed] [Google Scholar]
- 55.Kalra PR, Anker SD, Coats AJ. Water and sodium regulation in chronic heart failure: the role of natriuretic peptides and vasopressin. Cardiovasc Res. 2001;51:495–509. doi: 10.1016/s0008-6363(01)00297-8. [DOI] [PubMed] [Google Scholar]
- 56.Cataliotti A, Boerrigter G, Chen HH, Jougasaki M, Costello LC, Tsuruda T, Lee SC, Malatino LS, Burnett JC., Jr. Differential actions of vasopeptidase inhibition versus angiotensin-converting enzyme inhibition on diuretic therapy in experimental congestive heart failure. Circulation. 2002;105:639–644. doi: 10.1161/hc0502.102962. [DOI] [PubMed] [Google Scholar]
- 57.Misono KS, Sivasubramanian N, Berkner K, Zhang X. Expression and purification of the extracellular ligand-binding domain of the atrial natriuretic peptide (ANP) receptor. Biochemistry. 1999;38:516–523. doi: 10.1021/bi982127v. [DOI] [PubMed] [Google Scholar]
- 58.Misono KS, Grammer RT, Rigby JW, Inagami T. Photoaffinity labeling of atrial natriuretic factor receptor in bovine and rat adrenal cortical membranes. Biochem Biophys Res Commun. 1985;130:994–1001. doi: 10.1016/0006-291x(85)91713-9. [DOI] [PubMed] [Google Scholar]
- 59.Bovy PR. Structure activity in the atrial natriuretic peptide family. Med Res Rev. 1990;10:115–142. doi: 10.1002/med.2610100105. [DOI] [PubMed] [Google Scholar]
- 60.Ogawa H, Zhang X, Qiu Y, Ogata CM, Misono KS. Crystallization and preliminary X-ray analysis of the atrial natriuretic peptide (ANP) receptor extracellular domain complex with ANP: use of ammonium sulfate as the cryosalt. Acta Crystallogr D Biol Crystallogr. 2003;59:1831–1833. doi: 10.1107/s0907444903016445. [DOI] [PubMed] [Google Scholar]
- 61.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:306–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 62.Sheldrick GM. Phase annealing in SHELX-90: direct methods for larger structures. Acta Cryst. 1990;A46:467–473. [Google Scholar]
- 63.Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr D Biol Crystallogr. 1999;55(pt 4):849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr D Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Cryst. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 66.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54(pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 67.Murshudov GN, Vagin AA, Dodson EJ. Refinement of maclomolecular structures by the maximum-likelihood method. Acta Crystallography D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 68.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemistry quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 69.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 70.Philo J, Talvenheimo J, Wen J, Rosenfeld R, Welcher A, Arakawa T. Interactions of neurotrophin-3 (NT-3), brain-derived neurotrophic factor (BDNF), and the NT-3.BDNF heterodimer with the extracellular domains of the TrkB and TrkC receptors. J Biol Chem. 1994;269:27840–27846. [PubMed] [Google Scholar]
- 71.Philo JS. Sedimentation equilibrium analysis of mixed associations using numerical constraints to impose mass or signal conservation. Methods Enzymol. 2000;321:100–120. doi: 10.1016/s0076-6879(00)21189-0. [DOI] [PubMed] [Google Scholar]
- 72.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. Computer-aided interpretation of analytical sedimentation data for proteins. In: Harding SE, Rowe AJ, Horton JC, editors. Analytical ultracentrifugation in biochemistry and polymer science. Cambridge: Royal Society of Chemistry; 1992. pp. 90–125. [Google Scholar]
- 73.Lewis MS, Junghans RP. Ultracentrifugal analysis of molecular mass of glycoproteins of unknown or ill-defined carbohydrate composition. Methods Enzymol. 2000;321:136–49. doi: 10.1016/s0076-6879(00)21191-9. [DOI] [PubMed] [Google Scholar]
- 74.Bohm G, Muhr R, Jaenicke R. Quantitative analysis of protein far UV circular dichroism spectra by neural networks. Protein Eng. 1992;5:191–195. doi: 10.1093/protein/5.3.191. [DOI] [PubMed] [Google Scholar]
- 75.Chinkers M, Garbers DL, Chang MS, Lowe DG, Chin HM, Goeddel DV, Schulz S. A membrane form of guanylate cyclase is an atrial natriuretic peptide receptor. Nature. 1989;338:78–83. doi: 10.1038/338078a0. [DOI] [PubMed] [Google Scholar]
- 76.Schulz S, Singh S, Bellet RA, Singh G, Tubb DJ, Chin H, Garbers DL. The primary structure of a plasma membrane guanylate cyclase demonstrates diversity within this new receptor family. Cell. 1989;58:1155–1162. doi: 10.1016/0092-8674(89)90513-8. [DOI] [PubMed] [Google Scholar]
- 77.Fuller F, Porter JG, Arfsten AE, Miller J, Schilling JW, Scarborough RM, Lewicki JA, Schenk DB. Atrial natriuretic peptide clearance receptor. Complete sequence and functional expression of cDNA clones. J Biol Chem. 1988;263:9395–9401. [PubMed] [Google Scholar]
- 78.Fulle HJ, Vassar R, Foster DC, Yang RB, Axel R, Garbers DL. A receptor guanylyl cyclase expressed specifically in olfactory sensory neurons. Proc Natl Acad Sci USA. 1995;92:3571–3575. doi: 10.1073/pnas.92.8.3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shyjan AW, de Sauvage FJ, Gillett NA, Goeddel DV, Lowe DG. Molecular cloning of a retina-specific membrane guanylyl cyclase. Neuron. 1992;9:727–737. doi: 10.1016/0896-6273(92)90035-c. [DOI] [PubMed] [Google Scholar]
- 80.Lowe DG, Dizhoor AM, Liu K, Gu Q, Spencer M, Laura R, Lu L, Hurley JB. Cloning and expression of a second photoreceptor-specific membrane retina guanylyl cyclase (RetGC), RetGC-2. Proc Natl Acad Sci USA. 1995;92:5535–5539. doi: 10.1073/pnas.92.12.5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang RB, Foster DC, Garbers DL, Fulle HJ. Two membrane forms of guanylyl cyclase found in the eye. Proc Natl Acad Sci USA. 1995;92:602–606. doi: 10.1073/pnas.92.2.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Schulz S, Wedel BJ, Matthews A, Garbers DL. The cloning and expression of a new guanylyl cyclase orphan receptor. J Biol Chem. 1998;273:1032–1037. doi: 10.1074/jbc.273.2.1032. [DOI] [PubMed] [Google Scholar]
- 83.Schulz S, Green CK, Yuen PS, Garbers DL. Guanylyl cyclase is a heat-stable enterotoxin receptor. Cell. 1990;63:941–948. doi: 10.1016/0092-8674(90)90497-3. [DOI] [PubMed] [Google Scholar]
- 84.Higgins DG, Bleasby AJ, Fuchs R. CLUSTAL V: improved software for multiple sequence alignment. Comput Appl Biosci. 1992;8:189–191. doi: 10.1093/bioinformatics/8.2.189. [DOI] [PubMed] [Google Scholar]