

Abstract
Three new kaempferol glycosides, named palmatosides A (1), B (2) and C (3), together with three known kaempferol glycosides, multiflorins A (4) and B (5), and afzelin (6), were isolated from the roots of the fern Neocheiropteris palmatopedata. Palmatosides A (1) and B (2) were determined to be novel kaempferol glycosides, each possessing an unusual sugar moiety containing a 4, 4-dimethyl-3-oxo-butoxy substituent group. The isolated compounds were evaluated for their cancer chemopreventive potential based on their ability to inhibit tumor necrosis factor alpha (TNF-α)-induced NF-κB activity, nitric oxide (NO) production, aromatase, quinone reductase 2 (QR-2) and COX-1/-2 activities. Palmatosides B (2) and C (3) inhibited TNF-α-induced NF-κB activity with IC50 values of 15.7 and 24.1 μM, respectively; multiflorin A (4) inhibited aromatase enzyme with an IC50 value of 15.5 μM; afzelin (6) showed 68.3% inhibition against QR2 at a concentration of 11.5 μg/ml; palmatoside A (1) showed 52 % inhibition against COX-1 enzyme at a concentration of 10 μg/ml; and multiflorin B (5) showed 52 % inhibition against nitric oxide production at a concentration of 20 μg/ml. In addition, compounds 3-6 were shown to bind QR2 enzyme using LC-MS ultrafiltration binding assay.
Keywords: Neocheiropteris palmatopedata, Polypodiaceae, flavone, kaempferol glycosides, palmatosides, cancer chemoprevention, NF-κB, NO, QR-2, aromatase, COX-1, COX-2
1. Introduction
Neocheiropteris palmatopedata (Baker) Christ, belonging to the family Polypodiaceae, is an ornamental fern native only to China. It has been used as a folk medicine to treat gastric obstruction, rheumatism, ovarian cyst, laryngopharyngitis and chronic nutritional disorder (Delectis Florae Reipublicae Popularis Sinicae Agendae, 2000; Wu, 1990). Previous investigation on the constituents of members of the genus Neocheiropteris led to the isolation of one ecdysterone from the species N. ensata (Takemoto et al., 1968). In our search for new bioactive natural products from medicinal plants in Yunnan, China, we investigated the roots of N. palmatopedata, from which three new kaempferol glycosides (1-3) and three known ones [multiflorins A (4) and B (5) and afzelin (6)] were isolated (Fig. 1). Two of the new kaempferol glycosides, palmatosides A (1) and B (2), contain a naturally rare 4, 4-dimethyl-3-oxo-butoxy group substituted in the rhamnose unit of the sugar residue. The third glycoside, palmatoside C (3) was determined to be a conventional kaempferol derivative. The current paper reports the isolation, structure identification, and potential cancer chemopreventive activity of these compounds, based on their ability to inhibit TNF-α-induced NF-κB activity, nitric oxide (NO) production, aromatase, quinone reductase 2 (QR-2) and COX-1/-2 activities.
Fig. 1.

Structures of compounds 1-6.
2. Results and discussion
The 95% EtOH extract of the roots of N. palmatopedata was partitioned with EtOAc and n-BuOH sequentially, and the resulting extracts were subjected separately to chromatographic separation, including silica gel and Sephadex LH-20 gel permeation chromatography to afford palmatosides A (1), B (2) and C (3), multiflorins A (4) and B (5) and afzelin (6).
Palmatoside A (1) was obtained as a yellow amorphous powder. The molecular formula was established as C35H42O17 by HRESIMS, which was supported by 13C NMR and DEPT spectral data. The 1H NMR and 13C NMR spectra (Table 1) showed resonances for two singlet aromatic protons [δH 6.22 (1H, s, H-6), 6.39 (1H, s, H-8)], four ortho-coupled aromatic protons [δH 7.76 (2H, d, J= 8.5 Hz, H-2′, 6′), 6.95 (2H, d, J= 8.5 Hz, H-3′, 5′)], six aromatic methine carbons [δC 100.3 (d, C-6), 95.2 (d, C-8), δC 132.3 (d, C-2′, 6′), 117.0 (d, C-3′, 5′)], eight aromatic quaternary carbons and a carbonyl carbon at δC 180.0 (s, C-4), which characterized 1 as a flavonoid (Bae et al., 2007; Kotsos et al., 2007). The two doublets corresponding to the AA′BB′ system at δH 7.76 and 6.95 indicated a p-substituted ring B. The two singlets at δH 6.22 and 6.39 were assigned respectively to the meta-coupled H-6 and H-8 of ring A, which was confirmed by the presence of the cross peaks between the two protons in 1H-1H COSY spectrum and the long-range correlations of H-6 to C-8 (δC 95.2) and H-8 to C-6 (δC 100.3) in the HMBC spectrum (Fig. 2). The flavonoid moiety of 1 was thus determined as 3, 4′, 5, 7-tetrahydroxyflavone, namely kaempferol.
Table 1.
1H (500 MHz) and 13C NMR (125 MHz) spectroscopic data for 1-3 in CD3OD
| Position | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| 1H | 13C | 1H | 13C | 1H | 13C | |
| 2 | 160.0 | 159.7 | 159.6 | |||
| 3 | 136.5 | 136.5 | 136.4 | |||
| 4 | 180.0 | 180.0 | 180.0 | |||
| 5 | 163.6 | 163.6 | 163.6 | |||
| 6 | 6.22, 1H, br s | 100.3 | 6.22, 1H, br s | 100.4 | 6.40, 1H, d, 1.6 | 100.3 |
| 7 | 166.3 | 166.4 | 166.3 | |||
| 8 | 6.39, 1H, br s | 95.2 | 6.39, 1H, br s | 95.3 | 6.22, 1H, d, 1.6 | 95.2 |
| 9 | 159.0 | 159.0 | 159.0 | |||
| 10 | 106.4 | 106.4 | 106.4 | |||
| 1′ | 123.1 | 123.1 | 123.0 | |||
| 2′ | 7.76, 1H, d, 8.5 | 132.3 | 7.77, 1H, d, 8.5 | 132.3 | 7.78, 1H, d, 8.5 | 132.3 |
| 3′ | 6.95, 1H, d, 8.5 | 117.0 | 6.94, 1H, d, 8.5 | 117.0 | 6.96, 1H, d, 8.5 | 117.1 |
| 4′ | 162.0 | 162.0 | 162.0 | |||
| 5′ | 6.95, 1H, d, 8.5 | 117.0 | 6.94, 1H, d, 8.5 | 117.0 | 6.96, 1H, d, 8.5 | 117.1 |
| 6′ | 7.76, 1H, d, 8.5 | 132.3 | 7.77, 1H, d, 8.5 | 132.3 | 7.78, 1H, d, 8.5 | 132.3 |
| Rha-1″ | 5.37, 1H, br s | 103.7 | 5.39, 1H, br s | 103.7 | 5.43, 1H, d, 1.5 | 103.4 |
| 2″ | 4.28, 1H, m | 72.2 | 4.27, 1H, m | 72.2 | 4.20, 1H, m | 72.6 |
| 3″ | 3.95, 1H, m | 72.5 | 3.96, 1H, m | 72.6 | 3.84, 1H, m | 72.3 |
| 4″ | 3.52, 1H, m | 83.7 | 3.52, 1H, m | 83.5 | 3.55, 1H, m | 80.4 |
| 5″ | 3.39, 1H, m | 70.9 | 3.40, 1H, m | 70.9 | 3.32, 1H, m | 70.6 |
| 6″ | 0.96, 3H, d, 6.2 | 18.2 | 1.03, 3H, d, 6.2 | 18.3 | 0.96, 3H, d, 6.2 | 18.2 |
| Glc-1‴ | 4.49, 1H, d, 8.0 | 106.0 | 4.62, 1H, d, 8.0 | 105.8 | 4.76, 1H, d, 8.0 | 102.1 |
| 2‴ | 3.22, 1H, m | 76.3 | 3.33, 1H, m | 74.6 | 4.69, 1H, m | 76.1 |
| 3‴ | 3.38, 1H, m | 78.5 | 4.90, 1H, m | 79.7 | 3.54, 1H, m | 76.6 |
| 4‴ | 3.24, 1H, m | 72.2 | 3.50, 1H, m | 70.0 | 3.39, 1H, m | 72.2 |
| 5‴ | 3.45, 1H, m | 75.7 | 3.33, 1H, m | 78.2 | 3.28, 1H, m | 78.2 |
| 6‴ | 4.37, 4.21, 2H, m | 65.2 | 3.83, 3.72, 2H, m | 62.7 | 3.86, 3.70, 2H, m | 63.1 |
| CH3CO | 2.00, 3H, s | 21.1 | 2.15, 3H, s | 30.0 | 2.22, 3H, s | 21.6 |
| CH3CO | 173.1 | 173.2 | 172.9 | |||
| 1⁗ | 2.20, 3H, s | 32.5 | 2.20, 3H, s | 32.5 | ||
| 2⁗ | 212.0 | 212.0 | ||||
| 3⁗ | 2.65, 2H, s | 56.5 | 2.65, 2H, s | 56.5 | ||
| 4⁗ | 71.0 | 71.0 | ||||
| 5⁗, 6⁗ | 1.26, 6H, s | 30.0 | 1.27, 6H, s | 30.0 | ||
Fig. 2.
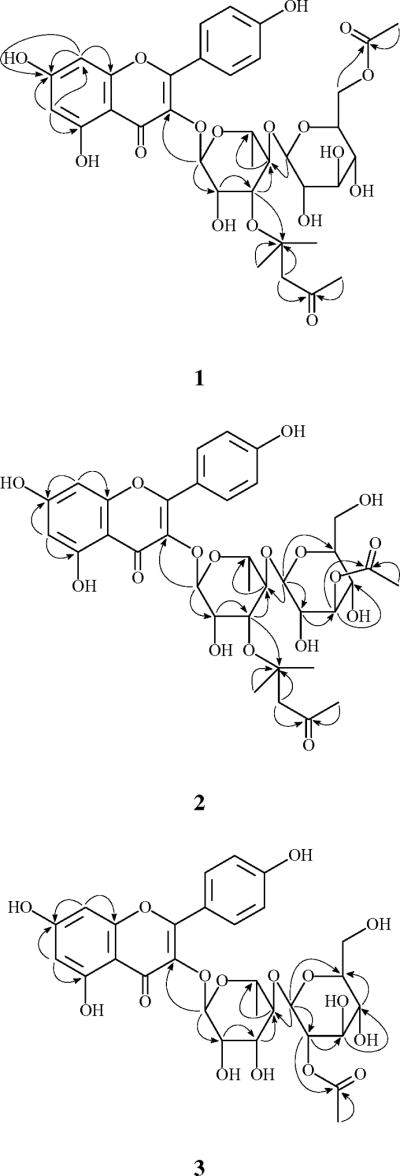
Key HMBC correlations of compounds 1-3.
The 1H NMR spectrum of 1 also exhibited a series of sugar signals at δH 3.22–4.37, with two anomeric protons at δH 5.37 (1H, brs, H-1″) and 4.49 (1H, d, J= 8.0 Hz, H-1‴). By acid hydrolysis, 1 afforded D-glucose and L-rhamnose, which were detected by direct co-TLC comparison with authentic samples. The β-anomeric configuration for the glucose was determined based on the coupling constant (J = 8.0 Hz) of H-1‴, and the α-anomeric configuration for the rhamnose was determined based on the singlet of H-1″ (Agrawal, 1992; Piccinelli et al., 2007; Tian et al., 2006). The presence of the HMBC correlation between the rhamnosyl anomeric proton H-1″ and C-3 [δC 136.5 (s)] suggested glycosidation at C-3, and the interglycosidic linkage between the glucose and the rhamnose units was determined to be glucopyranosyl- (1→4)-rhamnopyranose by the presence of the HMBC correlation between H-1‴ and C-4″ [δC 83.7 (d)].
The remaining sub-structure of 1 was deduced by 1H, 13C NMR and DEPT spectral data to possess an acetoxy group [δH 2.00 (3H, s); δC 173.1 (s), 21.1 (q)] and a six carbon unit composed of three singlet methyls [δH 2.20 (3H, s, H-1⁗), 1.26 (6H, s, 5⁗); δC 32.5 (q, C-1⁗), 30.0 (2C, q, C-5⁗ and C-6⁗)], a methylene (δH 2.65 (2H, s, H-3⁗); δC 56.5 (t, C-3⁗)], a ketone group [δC 212.0 (s, C-2⁗)] and an oxygenated quaternary carbon [δC 71.0 (s, C-4⁗)]. The six carbon unit was determined as 4, 4-dimethyl-3-oxo-butoxy due to the presence of the HMBC correlations from H-5⁗/6⁗ and H-3⁗ to C-4⁗, and from H-1⁗ and H-3⁗ to C-2⁗ as well as the observation of the base peak at m/z 139 ([C6H10O2+Na]+) in the ESIMS. The acetoxy group was determined to be linked at the glucosyl C-6‴ due to the presence of the cross peak between H-6‴ [δH 4.37, 4.21 (each 1H, m)] and the acetyl carbonyl carbon at δC 173.1 in the HMBC spectrum. The presence of the HMBC correlation between H-3″ of the rhamnose and C-4⁗ determined the 4, 4-dimethyl-3-oxo-butoxy unit to be linked to C-3″ of the rhamnose unit through an ether bond between C-3″ and C-4⁗. Accordingly, the structure of 1 was elucidated as kaempferol 3-O-[(6‴-O-acetyl-β-D-glucopyranosyl)-(1→4)-3″-O-(4⁗, 4⁗-dimethyl-3-oxo-butyl)-α-L-rhamnopyranoside], and given the trivial name of palmatoside A. The total assignment of the 1H and 13C NMR spectra of compound 1 was determined by a combination of 1H-1H COSY, HSQC, HMBC and ROESY spectral data.
Palmatoside B (2) was also obtained as a yellow amorphous powder. The molecular formula was established as C35H42O17, which is the same as that of 1 by HRESIMS. The ESIMS, 1H, 13C NMR and DEPT spectra of 2 (Table 1) were very similar to those of 1. Detailed analysis of the NMR spectral data between the two compounds revealed that 2 differs from 1 only by the location of the acetoxy group. The presence of an HMBC correlation between the acetyl carbonyl resonance at δC 173.2 and the glucosyl H-3‴ at δH 4.90 (1H, m) (Fig. 2) determined the acetoxy group at the C-3‴ of the glucose residue. Thus, compound 2 was identified as kaempferol 3-O-[(3‴-O-acetyl-β-D-glucopyranosyl)-(1→4)-3″-O-(4⁗, 4⁗-dimethyl-3-oxo-butyl)-α-L-rhamnopyranoside], and was given trivial name palmatoside B.
Compound 3, obtained as a yellow amorphous powder, had the molecular formula of C29H32O16, as deduced by a negative HRESIMS. The 1H, 13C NMR and DEPT spectra of 3 (Table 1) were very similar to those of 1 and 2 except for the absense of the 4, 4-dimethyl-3-oxo-butoxy moiety, suggesting it to be a conventional kaempferol diglycoside. NMR signals of an acetoxy group [δH 2.22 (3H, s); δC 172.9 (s), 21.6 (q)] were also observed in the1H and 13C NMR spectra of 3. This acetoxy group was determined to be at C-2‴ of the glucose residue due to the presence of the HMBC correlation between the acetyl carbonyl resonance at δC 172.9 and the glucosyl H-2‴ at δH 4.69 (1H, m) (Fig. 2). Thus, compound 3 was identified as kaempferol 3-O-[(2‴-O-acetyl-β-D-glucopyranosyl)-(1→4)-α-L-rhamnopyranoside], and given the trivial name of palmatoside C.
Compounds 4-6 were identified as the known compounds multiflorins A and B (Yamasaki et al., 1977), and afzelin (Jang et al., 2006), respectively, by analysis of their spectroscopic data and a comparison of the data with those reported in the literature.
Although flavonoids are found to exist abundantly in ferns, flavonoid glycosides having a 4, 4-dimethyl-3-oxo-butoxy group in the sugar moiety are unusual. This is the first report of flavonoid glycosides possessing a 4, 4-dimethyl-3-oxo-butoxy group in a sugar moiety from the family Polypodiaceae. The present study also represents the first chemical investigation of the genus Neocheiropteris since the report of the isolation of the steroid ecdysterone from N. ensata by Takemoto et al. in 1968. Hence, additional phytochemical studies of the plants in this genus merit consideration.
Compounds 1-6 were evaluated for their cancer chemopreventive potential based on their ability to inhibit TNF-α-induced NF-κB activity, nitric oxide (NO) production, aromatase, quinone reductase 2 (QR-2) and COX-1/-2 activities. NFκB is a transcription factor that plays roles associated with cell apoptosis, differentiation, and migration. Upon activation, it may promote cell proliferation and prevent cell death through anti-apoptotic factors (Baldwin, 2001). Inhibition of NFκB signaling has the potential application for the treatment or prevention of cancer.
Aromatase is an enzyme that catalyzes the conversion of androgen to estrogen, the female sex hormone that is needed for breast cancer cells to grow in most breast cancer cases, especially those in post-menopausal patients. Aromatase inhibition blocks the production of estrogen, which in turn will slow the growth of breast cancer cells. Aromatase inhibitors have been used in anticancer therapy to treat breast cancer in postmenopausal women. Animal studies have shown that they may also be used as potential chemopreventive agents (Lubet et al., 1994; Gunson et al., 1995).
Nitric oxide (NO) is an ubiquitous signaling molecule that impacts many physiological and pathological processes. It has been shown to be associated with the development of cancers in the early stages with in vivo studies (Crowell et al., 2003). The blocked production of NO is a potential mechanism for chemoprevention.
The enzyme quinone reductase 2 (QR2) is a close homologue of quinone reductase 1 (QR1). Unlike QR1, which is determined as a detoxifying enzyme, QR2 is a multifunctional enzyme. Although the two enzymes are structurally similar, their catalytic properties are very different (Chen et al., 2000). In vivo studies with mice demonstrated that QR1 and QR2 showed opposite properties toward menadione toxicity (Long et al., 2002a, 2002b). Another study showed that QR2 is abundantly expressed in prostate cancer cells (Wang et al., 2004). While the precise function of QR2 remains to be determined, some chemopreventive agents such as resveratrol and melatonin were found to be potent inhibitors of QR2 (Buryanovskyy et al., 2004; Calamini et al., 2008), suggesting that this enzyme may be a new target for the development of new chemopreventive agents.
Finally, inhibitors of cyclooxygenase (COX) activity have been associated with neoplastic transformation, and inhibition of this activity provides a strategy for the prevention of cancer (Cuendet and Pezzuto, 2000).
Although all six compounds are kaempferol glycosides, they showed different inhibition profiles in the aforementioned assays, as shown in Table 2. The chemical structures of compounds 1-3 are very similar, especially for 1 and 2, which differ from each other only by the position of an acetyl group. However, while 2 and 3 exhibited inhibition of TNF-α-induced NF-κB activity with IC50 values of 15.7 and 24.1 μM, respectively, 1 was completely devoid of such activity at a concentration of 20 μg/ml. When examining downstream activities associated with the NF-κB cascade, of the six compounds, only 5 showed more than 50 % inhibition against nitric oxide production at a concentration of 20 μg/ml; 1 was the only compound that showed more than 50 % inhibition against COX enzymes at a concentration of 10 μg/ml.
Table 2.
Cancer chemoprevention potential of compounds 1-6
| Compound | NOa | NFb | NFc | ARd | ARe | CX1f | CX2g | QRh | QRMSi |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 7 % | 0 % | 23 % | 52 % | 15 % | 24 % | |||
| 2 | 9 % | 91 % | 15.7 | 65 % | -3 % | -32 % | 29 % | ||
| 3 | 10 % | 72 % | 24.1 | 58 % | 25 % | 13 % | 4 % | 636 | |
| 4 | 8 % | 0 % | 95 % | 15.5 | -29% | 16 % | 39 % | 594 | |
| 5 | 52 % | 0 % | 38 % | 22 % | -28 % | 9 % | 636 | ||
| 6 | 8 % | 10 % | 23 % | -16 % | -21 % | 68 % | 432 |
Percentage inhibition of NO production at 20 μg/ml.
Percentage inhibition of NFκB at 20 μg/ml.
IC50 value (μM) against NFκB.
Percentage inhibition of aromatase at 20 μg/ml.
IC50 value (μM) against aromatase.
Percentage inhibition of COX-1 at 10 μg/ml.
Percentage inhibition of COX-2 at 10 μg/ml.
Percentage inhibition of QR2 at 11.5 μg/ml.
Molecular weight of compound interacting QR2.
Aromatase inhibitory activity of the six compounds (1-6) is clearly different. Among them, 4 was the most active, with an IC50 value of 15.5 μM. Compound 4 is different from 5 only by lack of an acetyl group at C-6‴, and it is different from 3 only by the position of the acetyl group. In addition, the Glc group at C-4″ is clearly important for activity, since 6 is not active.
As for QR2 activity, we conducted both binding and inhibition activity of the six compounds. The binding assay is a fast tool to identify active compounds even from among a mixture. We could apply the binding assay to speed up our pre-screening process of a large number of samples since we are able to incubate more than 10 samples with the target macromolecule in one experiment. We only carry out inhibition assays on those samples that show binding activity if a large number of samples are involved. For the current study, we applied both binding and inhibition assays to the six compounds. When examining QR2 enzymic activity, afzelin (6) was the only compound that showed more than 50 % inhibition at the standard test concentration of 11.5 μg/ml. When evaluated for ability to interact with QR2 using an LC-MS ultrafiltration binding assay, four of the compounds (3-6) were shown to mediate positive response. That only afzelin (6) showed more than 50 % inhibition activity among the four binding active compounds (3-6) suggests that the other three compounds (3-5) might only interact with a binding site of the enzyme rather than the catalytic site.
Finally, compounds 1-6 were evaluated for cytotoxic potential with Hepa1c1c7 and MCF-7 cells in culture. No significant growth inhibitory effects were observed when tested at a concentration of 20 μg/ml. In the context of cancer chemoprevention, the lack of nonspecific cytotoxicity is generally considered beneficial, since a high therapeutic index is required for disease prevention. Thus, while the activities of these compounds in the chemoprevention assays described herein are not especially potent, the modulation of unique chemopreventive targets in the absence of general toxicity provides a rationale for further consideration.
3. Experimental
3.1. General
MS were determined on a Finnigan MAT 90 instrument and a VG Auto Spec-3000 spectrometer. NMR spectra were measured on a Bruker DRX-500 spectrometer. Silica gel (200-300 mesh, Qingdao Marine Chemical Co., China), MCI GEL® (Mitsubishi Chemical Corp.), and Sephadex LH-20 (25–100 μm, Pharmacia Fine Chemical Co. Ltd.) were used for column chromatography and silica gel GF254 for TLC (Qingdao Marine Chemical Co., China). Solvents were of industrial purity and distilled prior to use.
3.2. Plant materials
The roots of Neocheiropteris palmatopedata were collected in Kunming, Yunnan, China in March, 2006, and identified by Prof. Shugang Lu, School of Life Science, Yunnan University, where a voucher specimen (No.0603014) is deposited.
3.3. Extraction and isolation
The dried roots of N. palmatopedata (0.5 kg) were powered and extracted with 95% EtOH at room temperature (5×, each 5 l). The EtOH extract (85 g) was evaporated in vacuo, suspended in H2O (1.0 l) and then extracted with EtOAc and n-BuOH successively (4×, each 1.5 l). The EtOAc extract (18 g) was subjected to a silica gel column, eluting with petroleum ether containing increasing amounts of EtOAc to obtain 6 fractions (fractions I-VI). Fraction V (3.1 g) was separated using MCI GEL® ion exchange column chromatography eluting with a gradient of 50-90% MeOH in H2O to afford five subfractions V1–V5. Fraction V2 (1.7 g) was purified on a Sephadex LH-20 column eluting with CHCl3/MeOH 5: 1 to yield 1 (1.13 g). Fraction V3 (300 mg) was separated by repeated CC on a Sephadex LH-20 column eluting with MeOH to yield 2 (22 mg). Fraction V5 (250 mg) was purified by repeated CC on Sephadex LH-20 eluting with MeOH to provide 3 (15 mg) and 6 (18 mg), respectively. Fraction VI (5.7 g) was separated via MCI column chromatography eluting with a gradient of 60-90% MeOH in H2O to afford four subfractions VI1–VI4. Fraction VI1 (580 mg) was purified by repeated CC on Sephadex LH-20 eluting with MeOH to afford 5 (389 mg). The n-BuOH extract (18.5 g) was subjected to CC on silica gel eluting with a gradient of 10-100% MeOH in CHCl3 to obtain four fractions (fractions A-D). Fraction A (0.4 g) was purified by CC on silica gel eluting with CHCl3/MeOH 5: 1, and then on Sephadex LH-20 eluting with MeOH to provide 4 (4 mg).
Palmatoside A (1)
Yellow amorphous powder. 1H and 13C NMR data see Table 1. ESIMS m/z (ret. int.): 659 [M+Na-C6H10O]+ (5), 139 [C6H10O2 (4, 4-dimethyl-3-oxo-butoxy)+Na]+(100). HRESIMS m/z: 659.1592 [M+Na-C6H10O]+ (calcd for [C35H42O17+Na-C6H10O]+, 659.1588).
Palmatoside B (2)
Yellow amorphous powder. 1H and 13C NMR data see Table 1. ESIMS m/z (ret. int.): 659 [M+Na-C6H10O]+ (7), 139 [C6H10O2 (4, 4-dimethyl-3-oxo-butoxy)+Na]+ (100). HRESIMS m/z: 659.1585 [M+Na-C6H10O]+ (calcd for [C35H42O17+Na-C6H10O]+, 659.1588).
Palmatoside C (3)
Yellow amorphous powder. 1H and 13C NMR data see Table 1. Negative FABMS: 635 [M-H]- (100). Negative HRESIMS m/z: 635.1629 [M-H]- (calcd for [C29H32O16-H]+, 635.1612).
3.4. NF-κB luciferase assay
Panomic (Fremont, CA) has established a number of stably-transfected NFκB reporter cell lines. We have employed human embryonic kidney cells 293 for monitoring any changes occurring along NFκB pathway. Cells were seeded into sterile 96-well plates at a density of 20 × 103 cells per well. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen Co., Carlsbad, CA), supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine. After a 48 hr incubation, medium was replaced and various concentrations of test compounds were added (dissolved in PBS). Human recombinant TNF-α (2 ng/ml; 0.14 nM) (Calbiochem, Gibbstown, NJ) was used as activator. The plate was incubated for 6 hr. Spent media was discarded and the cells were washed once with PBS. Then, the cells were lysed by adding 50 μl/well of Reporter Lysis Buffer (diluted 5-fold with water) (Promega, Madison, WI) and incubating for 5 min on a shaker. At this point, plates can be stored at -80°C for subsequent analysis. The luciferase assay was performed using the Luc assay system from Promega. The gene product, luciferase enzyme, reacts with luciferase substrate, emitting light which was detected using a luminometer (LUMIstar Galaxy BMG). Dose-response curves were constructed and data were expressed IC50 values (i.e., concentration of tested sample required to inhibit TNF-α–induced NFκB activity by 50%) (Homhual et al., 2006).
3.5. Aromatase assay
Aromatase activity was assayed as previously reported (Lee et al., 2001)., with the necessary modifications to utilize a 384-well format. Briefly, test compounds (3.5 μL) were preincubated with 30 μl of a NADPH-regenerating system (2.6 mM NADP+, 7.6 mM glucose 6-phosphate, 0.8 U/ml glucose 6-phosphate dehydrogenase, 13.9 mM MgCl2, and 1 mg/ml albumin in 50 mM potassium phosphate buffer, pH 7.4) for 10 min at 37 °C. The enzyme and substrate mixture [33 μl of 1 μM CYP19 enzyme (BD Biosciences), 0.4 μM dibenzylfluorescein, 4 mg/ml albumin, in 50 mM potassium phosphate, pH 7.4] was added, and the plate was incubated for 30 min at 37 °C before quenching with 25 μl of 2 N NaOH. After termination of the reaction and shaking for 5 min, the plate was further incubated for 2 hr at 37 °C. This enhances the ratio of signal to background. Fluorescence was measured at 485 nm (excitation) and 530 nm (emission). IC50 values and dose-response curves were based on three independent experiments performed in duplicate using five concentrations of test substance. Naringenin (IC50 = 0.23 μM) was used as a positive control.
3.6. QR2 assay
Quinone reductase 2 (QR2) assay was conducted as previously described (Calamini et al., 2008). Data were expressed as percentage of inhibition or IC50 values (concentration required to inhibit QR2 activity by 50 %). Resveratrol was used as a positive control, which showed 50 % inhibition against QR2 at a concentration of 0.96 μM.
3.7. COX-1 and COX-2 assays
COX-2 (0.2 μg) or COX-1 (0.2 μg) was activated by adding 146 μl Tris-HCl buffer (pH 8.0), 2 μl hematin (1 μM final), 10 μl L-epinephrine (2 mM final) at room temperature for 2 min on ice. Then, 2 μl of each test solution was added and preincubated for 10 min in a water bath at 37°C for 10 min. Negative control incubations were identical except that 2 μl of Tris-HCl buffer was added instead of the test solution. Celecoxib and indomethacin were used as positive controls in the COX-2 and COX-1 inhibition assays, respectively. The reactions were initiated by adding 20 μl arachidonic acid (5 μM, final concentration) and terminated after 2 min by adding 10 μl 2.0 M HCl. 20 μL d4-[PGE2] at 50 ng/ml was then added as internal standard. Both PGE2 and [d4]-PGE2 were extracted from incubates using 800 μl water saturated with EtOAc. The EtOAc phase was then collected, evaporated to dryness, and reconstituted in 100 μl MeOH/H2O (50:50, v/v). The formation of the COX product prostaglandin E2 was measured using the LC-MS-MS method as described previously (Cao et al., 2008). An Applied Biosystems (Foster City, CA) API 4000 triple quadrupole mass spectrometer with negative ion electrospray and a collision energy of 22 eV equipped with a Shimadzu (Columbia, MD) Prominence UFLC system with a Waters (Milford, MA) XTerra MS C18 (2.1 × 50 mm, 3.5 μm) analytical column was used for PGE2 measurement.
3.8. Ultrafiltration LC-MS binding assay for QR2
Ultrafiltration LC-MS assay for QR2 binding was carried out using a modified protocol that was previously published (van Breemen et al., 1997). Briefly, test compounds were incubated with ovine QR2 for 1 hr at 37 °C. The mixture was then filtered through a 30,000 Da molecular weight cut-off ultrafiltration membrane. After washing each sample 3 times with buffer, the ligands were dissociated from QR2 using methanol. The ligand ultrafiltrates were dried under nitrogen and reconstituted in 50 % aqueous methanol prior to LC-MS analysis.
3.9. Cytotoxicity assay
Hepa1c1c7 cells were maintained in AMEM (Alpha Minimum Essential Medium) medium. MCF-7 cells were maintained in MEME (Eagle's Minimum Essential Medium) medium containing 10 mg/l of insulin. In each case, PSF (100 units/ml penicillin G, 100 μg/ml streptomycin sulfate, 250 ng/ml amphotericin B) was added. All media were supplemented with 10 % heat-inactivated FBS. Compounds were tested according to established protocols (Mi et al., 2002).
Acknowledgments
This investigation was supported in part by a program project P01 CA48112 awarded by the National Cancer Institute USA, a grant (2005DFA30670) for international collaborative research from the Chinese Ministry of Science and Technology, a grant (30560178) from Natural Science Foundation of China, a Program for New Century Excellent Talents in University (NCET-05-0824), and the West Light Program from the Chinese Academy of Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agrawal PK. NMR spectroscopy in the structural elucidation of oligosaccharides and glycosides. Phytochemistry. 1992;31:3307–3330. doi: 10.1016/0031-9422(92)83678-r. [DOI] [PubMed] [Google Scholar]
- Bae KH, Jin WY, Thuong PT, Min BS, Na MK, Lee YM, Kang SS. A new flavonoid glycoside from the leaf of Cephalotaxus koreana. Fitoterapia. 2007;78:409–413. doi: 10.1016/j.fitote.2007.02.008. [DOI] [PubMed] [Google Scholar]
- Baldwin AS. Control of oncogenesis and cancer therapy resistance by the transcription factor NF-κB. J Clin Invest. 2001;107:241–246. doi: 10.1172/JCI11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buryanovskyy L, Fu Y, Boyd M, Ma YL, Hsieh TC, Wu JM, Zhang ZT. Crystal structure of quinone reductase 2 in complex with resveratrol. Biochemistry. 2004;43:11417–11426. doi: 10.1021/bi049162o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calamini B, Santarsiero BD, Boutin JA, Mesecar AD. Kinetic, thermodynamic and X-ray structural insights into the interaction of melatonin and analogues with quinone reductase 2. Biochem J. 2008;413:81–91. doi: 10.1042/BJ20071373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Xiao L, Park G, Wang X, Azim AC, Christman JW, van Breemen RB. An improved LC-MS/MS method for the quantification of prostaglandins E(2) and D(2) production in biological fluids. Anal Biochem. 2008;372:41–51. doi: 10.1016/j.ab.2007.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wu K, Knox R. Structure–function studies of DT-diaphorase (NQO1) and NRH: quinone oxidoreductase (NQO2) Free Radic Biol Med. 2000;29:276–284. doi: 10.1016/s0891-5849(00)00308-7. [DOI] [PubMed] [Google Scholar]
- Crowell JA, Steele VE, Sigman CC, Fay JR. Is inducible nitric oxide synthase a target for chemoprevention? Mol Cancer Ther. 2003;2:815–823. [PubMed] [Google Scholar]
- Cuendet M, Pezzuto JM. The role of cyclooxygenase and lipoxygenase in cancer chemoprevention. Drug Metab Drug Interac. 2000;17:109–157. doi: 10.1515/dmdi.2000.17.1-4.109. [DOI] [PubMed] [Google Scholar]
- Delectis Florae Reipublicae Popularis Sinicae Agendae, Academiae Sinicae Edita. Flora Reipublicae Popularis Sinicae. 2. Vol. 6. Science Press; Beijing: 2000. p. 32. [Google Scholar]
- Gunson DE, Steele RE, Chau RY. Prevention of spontaneous tumors in female rats by fadrozole hydrochloride, an aromatase inhibitor. Br J Cancer. 1995;72:72–75. doi: 10.1038/bjc.1995.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homhual S, Zhang HJ, Bunyapraphatsara N, Kondratyuk TP, Santasiero BD, Mesecar AD, Herunsalee A, Chaukul W, Pezzuto JM, Fong HHS. Bruguiesulfurol, a new sulfur compound from Bruguiera gymnorrhiza. Planta Med. 2006;72:255–260. doi: 10.1055/s-2005-873171. [DOI] [PubMed] [Google Scholar]
- Jang DS, Kim JM, Lee YM, Yoo JL, Kim YS, Kim JH, Kim JS. Flavonols from Houttuynia cordata with protein glycation and aldose reductase inhibitory activity. Nat Prod Sci. 2006;12:210–246. [Google Scholar]
- Lee D, Bhat KPL, Fong HHS, Farnsworth NR, Pezzuto JM, Kinghorn AD. Aromatase inhibitors from Broussonetia papyrifera. J Nat Prod. 2001;64:1286–1293. doi: 10.1021/np010288l. [DOI] [PubMed] [Google Scholar]
- Long DJ, 2nd, Gaikwad A, Multani A, Pathak S, Montgomery CA, Gonzalez FJ, Jaiswal AK. Disruption of the NAD(P)H:quinone oxidoreductase 1 (NQO1) gene in mice causes myelogenous hyperplasia. Cancer Res. 2002a;62:3030–3036. [PubMed] [Google Scholar]
- Long DJ, 2nd, Iskander K, Gaikwad A, Arin M, Roop DR, Knox R, Barrios R, Jaiswal AK. Disruption of dihydronicotinamide riboside:quinone oxidoreductase 2 (NQO2) leads to myeloid hyperplasia of bone marrow and decreased sensitivity to menadione toxicity. J Biol Chem. 2002b;277:46131–46139. doi: 10.1074/jbc.M208675200. [DOI] [PubMed] [Google Scholar]
- Lubet RA, Steele VE, Casebolt TL, Eto I, Kelloff GJ, Grubbs CJ. Chemopreventive effects of the aromatase inhibitors vorozole (R-83842) and 4-hydroxyandrostenedione in the methylnitrosourea (MNU)-induced mammary tumor model in Sprague-Dawley rats. Carcinogenesis. 1994;15:2775–2780. doi: 10.1093/carcin/15.12.2775. [DOI] [PubMed] [Google Scholar]
- Kotsos MP, Aligiannis N, Mitakou S. A new flavonoid diglycoside and triterpenoids from Stachys spinosa L. (Lamiaceae) Biochem Syst Ecol. 2007;35:381–385. [Google Scholar]
- Mi QW, Lantvit D, Reyes-Lim E, Chai HB, Zhao WM, Lee IS, Peraza-Sanchez S, Ngassapa O, Kardono LBS, Riswan S, Hollingshead MG, Mayo JG, Farnsworth NR, Cordell GA, Kinghorn AD, Pezzuto JM. Evaluation of the potential cancer chemotherapeutic efficacy of natural product isolates employing in vivo hollow fiber tests. J Nat Prod. 2002;65:842–850. doi: 10.1021/np010322w. [DOI] [PubMed] [Google Scholar]
- Piccinelli AL, Veneziano A, Passi S, Simone FD, Rastrelli L. Flavonol glycosides from whole cottonseed by-product. Food Chem. 2007;100:344–349. [Google Scholar]
- Takemoto T, Hikino Y, Arai T, Knono C, Nabetani S, Hikino H. Isolation of insect moulting substances from Pleopeltis thunbergiana, Neocheiropteirs ensata and Lemmaphyllum microphyllum. Chem Pharm Bull. 1968;16:759–760l. [Google Scholar]
- Tian JM, Hao XJ, He HP. A new lignan and four new lignan glycosides from Mananthes patentiflora. Helv Chim Acta. 2006;89:291–298. [Google Scholar]
- van Breemen RB, Huang CR, Nikolic D, Woodbury CP, Zhao YZ, Venton DL. Pulsed Ultrafiltration mass spectrometry: a new method for screening combinatorial libraries. Anal Chem. 1997;69:2159–2164. doi: 10.1021/ac970132j. [DOI] [PubMed] [Google Scholar]
- Wang Z, Hsieh TC, Zhang Z, Ma Y, Wu JM. Identification and purification of resveratrol targeting proteins using immobilized resveratrol affinity chromatography, Biochem. Biophys Res Commun. 2004;323:743–749. doi: 10.1016/j.bbrc.2004.08.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZY. XinHuaBenCaoGangYao. Vol. 3. Shanghai Press of Science and Technology; Shanghai: 1990. pp. 712–713. [Google Scholar]
- Yamasaki K, Kasai R, Masaki Y, Okihara M, Tanaka O, Oshio H, Takagi S, Yamaki M, Masuda K, Nonaka G, Tsuboi M, Nishioka I. Application of C-13 NMR to the structural elucidation of acylated plant glycosides. Tetrahedron Lett. 1977;18:1231–1234. [Google Scholar]