

Abstract
Activation of the transcription factor Nrf2 in astrocytes coordinates the upregulation of antioxidant defenses and confers protection to neighboring neurons. Dominant mutations in Cu/Zn-superoxide dismutase (SOD1) cause familial forms of amyotrophic lateral sclerosis (ALS), a fatal disorder characterized by the progressive loss of motor neurons. Non-neuronal cells, including astrocytes, shape motor neuron survival in ALS and are a potential target to prevent motor neuron degeneration. The protective effect of Nrf2 activation in astrocytes has never been examined in a chronic model of neurodegeneration. We generated transgenic mice over-expressing Nrf2 selectively in astrocytes using the glial fibrillary acidic protein (GFAP) promoter. The toxicity of astrocytes expressing ALS-linked mutant hSOD1 to cocultured motor neurons was reversed by Nrf2 over-expression. Motor neuron protection depended on increased glutathione secretion from astrocytes. This protective effect was also observed by crossing the GFAP-Nrf2 mice with two ALS-mouse models. Over-expression of Nrf2 in astrocytes significantly delayed onset and extended survival. These findings demonstrate that Nrf2 activation in astrocytes is a viable therapeutic target to prevent chronic neurodegeneration.
Keywords: Nrf2, astrocytes, glutathione, motor neurons, neuronal death, neuroprotection
Introduction
Induction of phase II detoxification and antioxidant enzymes expression is governed by a cis-acting regulatory element termed the antioxidant response element (ARE). ARE-containing genes are regulated by the nuclear factor erythroid 2-related factor 2 (Nrf2), a member of the Cap‘n’Collar/basic-leucine zipper family of transcription factors (Kobayashi and Yamamoto, 2005). ARE-driven genes are preferentially activated in astrocytes (Johnson et al., 2002; Lee et al., 2003; Kraft et al., 2004) and Nrf2/ARE activation in astrocytes confers protection to neighboring neurons (Shih et al., 2003; Kraft et al., 2004).
Increased levels of glutathione [γ-l-glutamyl-l-cysteinyl-glycine (GSH)] seems to be a major component of the protection conferred by Nrf2 activation. Glutathione is synthesized by the consecutive action of two enzymes, glutamate-cysteine ligase (GCL) and glutathione synthetase. Nrf2 regulates both enzymes and GCL is the rate-limiting enzyme for glutathione synthesis. Increased production and secretion of glutathione by astrocytes is known to improve the antioxidant status of cocultured neurons and protect them from oxidative insults (Dringen et al., 2000).
Amyotrophic lateral sclerosis (ALS) is the most common adult-onset motor neuron disease, caused by the progressive degeneration of motor neurons in the spinal cord, brainstem, and motor cortex (Rowland and Shneider, 2001). Approximately 10–20% of familial ALS is caused by a toxic gain-of-function induced by mutations of the Cu/Zn-superoxide dismutase (SOD1) (Rosen et al., 1993). Rodents over-expressing mutated forms of hSOD1 generally develop an ALS-like phenotype (Gurney et al., 1994; Howland et al., 2002). Although the molecular mechanism underlying this toxic gain-of-function remains unknown, toxicity to motor neurons requires mutant SOD1 expression in non-neuronal cells as well as in motor neurons (Clement et al., 2003).
A large proportion of non-neuronal cells in the ventral horn of the spinal cord are astrocytes. In ALS patients and rodent models, a strong glial reaction typically surrounds degenerating motor neurons (Barbeito et al., 2004). Primary spinal cord astrocyte monolayers support the survival of purified embryonic motor neurons in the absence of added trophic factors. However, astrocytes isolated from hSOD1G93A rats (Vargas et al., 2006) or mice (Di Giorgio et al., 2007; Nagai et al., 2007) are toxic to cocultured motor neurons. Therefore, functional alterations in activated astrocytes can shape the interaction with surrounding cells such as damaged neurons, microglia and immune cells, and consequently could modulate motor neuron survival (Barbeito et al., 2004; Pehar et al., 2004; Cassina et al., 2005; Yamanaka et al., 2008). In addition, current evidence (Vargas et al., 2006; Pehar et al., 2007) suggests that Nrf2 activation could be beneficial in the context of hSOD1G93A toxicity.
Despite the fact that Nrf2 activation plays a critical role in protecting neurons in many acute models of neuronal damage, it is currently unknown whether Nrf2 may help counteract the progressive loss of neurons in chronic neurodegeneration. To determine whether Nrf2 activation in astrocytes prevents motor neuron degeneration, we generated mice over-expressing Nrf2 under the control of the hGFAP promoter and showed here that astrocytic Nrf2 over-expression prevented toxicity of hSOD1G93A-astrocytes toward motor neurons and extended survival in ALS-mice models.
Materials and Methods
Construction and mating of transgenic mice.
The hGFAP-Nrf2 transgene was constructed by excising the CMV promoter from pCMVβ (BD Biosciences) using EcoRI and XhoI and replacing it with a custom multiple cloning site (MCS) consisting of 5′-EcoRI, SalI, AvrII, KpnI, Msc I, BglII, XhoI-3′. A BglII-BamHI fragment containing the hGFAP promoter, was excised from pgfa2-lac2 (Brenner et al., 1994) and inserted into the BglII site of the MCS. Beta-galactosidase was excised from hGFAP-βgal sequence using NotI and replaced with mNrf2 (NotI-NotI fragment) from pEF-Nrf2 (received from Dr. Jawed Alam, Alton Ochsner Medical Foundation, New Orleans, LA) (Alam et al., 1999). The fragment for microinjection was excised with SalI, isolated by agarose gel electrophoresis and purified using GeneClean Turbo Kit (MP Biomedicals). Transgenic mice were generated by pronuclear microinjection using fertilized eggs of the FVB/N strain. Founder mice were identified by PCR analysis with primers complementary to hGFAP promoter sequence (5′-TAGCCCACTCCTTCATAAAGCCCT-3′) and mNrf2 (5′-TCAAATCCATGTCCTGCTGGGACT-3′). All experiments reported here used mice from the line denominated Tg173.4. The cycling parameters were as follows: 95°C, 30 s; 56°C, 30 s; 72°C, 30 s. Three different breeding schemes were used. B6SJLTg(SOD1*G93A)1Gur/J males (Gurney et al., 1994) were mated to GFAP-Nrf2 F1 (B6SJLxFVB/N) females. Unless indicated, all experiments were performed with these animals. In this way, although in a mixed background, the contribution of each background was maintained constant throughout the study. In addition, hSOD1H46R/H48Q (C3H/HeJxC57BL/6J) males (obtained from Dr. David Borchelt, University of Florida College of Medicine, Gainesville, FL) (Wang et al; 2002) were mated to GFAP-Nrf2 F1 (B6SJLxFVB/N) females. To rule out mice background effects, hSOD1G93A males in pure FVB/N background (generated by backcrossing B6SJLTg(SOD1*G93A)1Gur/J animals to non-transgenic FVB/N for 10 generations) were mated to GFAP-Nrf2 (FVB/N) females. For survival and onset experiments double-transgenic animals were always compared with their contemporaneously produced hSOD1G93A or hSOD1H46R/H48Q littermates. End-stage was determined by the inability of the animal to right itself within 20 s when placed on its side. All animal procedures comply with Animal Care and Use Committee requirements of the University of Wisconsin-Madison. Disease onset was determined by rotarod performance. Mice were trained for a week to become familiar with the rotarod apparatus (Columbus Instruments). Training started at 60 d of age and followed by assessment of performance twice a week. In each session animals were first allowed to run at 5 r.p.m. for 1 min followed by three trials at 15 r.p.m. for 3 min. The inability of mice to stay on the rod in two consecutive sessions was used as sign of disease onset. Mice were weighed twice per week. For the experiments with animals in pure FVB/N background, mice were weighed three times per week and disease onset was retrospectively determined as the time when mice reached peak body weight (Boillée et al., 2006).
Immunofluorescence and histochemistry.
Mice were transcardially perfused with 0.1 m PBS, followed by 4% paraformaldehyde in PBS, pH 7.4. Spinal cords were removed and cryoprotected in 30% sucrose or dehydrated, and paraffin embedded. Frozen sections (20 μm) were stained with antibodies anti-hPAP (1:250; Abcam) and anti-GFAP (1:500; Millipore). For paraffin embedded tissues antigen retrieval was performed in a microwave oven in 0.01 m sodium citrate, pH 6.0, and serial 5 μm sections were stained with anti-GFAP (1:500; Dako) or anti-Mac2 (1:250; Cedarlane). All sections were permeabilized with 0.1% Triton X-100 in PBS and non-specific binding was blocked with 10% goat serum, 2% bovine serum albumin, 0.1% Triton X-100 diluted in PBS for 1 h at room temperature. Sections were incubated with primary antibodies diluted in blocking solution overnight at 4°C. Secondary antibodies diluted in blocking solution were incubated for 1 h at room temperature. Sections were mounted with Fluoro-Gel (EMS). Controls were performed omitting the primary antibody.
Medial gastrocnemius was cryoprotected in 20% sucrose–PBS overnight and flash-frozen in isopentane. Muscles were sectioned at 20 μm and stained with Alexa Fluor 594-conjugated bungarotoxin (1 μg/ml, Invitrogen) and antibodies to neurofilament (NF160 1:200, Millipore) and to synaptophysin (SYN 1:250; Abcam). For quantitative analyses, end-plate-neuromuscular junctions were scored as innervated in the case of complete overlap between bungarotoxin and NF/SYN staining, intermediate in case of partial overlap and denervated in the absence of NF/SYN labeling at the end-plate.
Secondary antibodies were Alexa Fluor 488-conjugated goat anti-mouse, Alexa Fluor 488-conjugated goat anti-rat, Alexa Fluor 488-conjugated goat anti-rabbit, Alexa Fluor 594-conjugated goat anti-mouse and Alexa Fluor 594-conjugated goat anti-rabbit (5 μg/ml each, Invitrogen). Sections were analyzed in an Axiovert microscope (Zeiss) with Axiovision 4.6 image software.
Motor neuron numbers were determined in 10 μm serial sections across the lumbar spinal cord stained with cresyl violet. Every fifth section was counted and a total of 32 sections per 4-week-old-animals were analyzed.
Cell cultures.
Primary astrocyte cultures were prepared from spinal cords of 1-d-old mice as previously described (Vargas et al., 2006). Astrocytes were plated at a density of 2 × 104 cells/cm2 in 35 mm Petri dishes or 24-well plates and maintained in DMEM supplemented with 10% fetal bovine serum, HEPES (3.6 g/L), penicillin (100 IU/ml) and streptomycin (100 lg/ml). Astrocyte monolayers were >98% pure as determined by GFAP immunoreactivity and devoid of OX42-positive microglial cells. Motor neuron cultures were prepared from 12.5 embryonic day mouse spinal cord by a combination of Optiprep-gradient centrifugation and immunopanning against p75NTR (Millipore). For coculture experiments, motor neurons were plated on mouse astrocyte monolayers at a density of 300 cells/cm2 and maintained in supplemented L15 medium (Vargas et al., 2006). Motor neurons were identified with anti-neurofilament or anti-choline acetyltransferase (Millipore) and survival was determined by counting all cells displaying intact neurites longer than four cells in diameter Counts were performed over an area of 0.90 cm2 in 24-well plates. All cell culture experiments were conducted under normoxic conditions in a tri-gas incubator with 5%CO2 and 5%O2.
Cell treatment and siRNA transfection.
Confluent astrocyte monolayers were changed to L15 supplemented media before treatment. tert-butyl hydroperoxide (t-BOOH) was diluted in Dulbecco's PBS and applied to astrocyte monolayers at the indicated final concentrations. Survival was assessed 24 h later by the reduction of [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS; Promega). MK-571 (20 μm, Alexis) was added to the cocultures 3 h after motor neuron plating. siRNA transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Astrocytes were transfected with 25 nm of a Silencer Predesigned Mrp1-siRNA (ID#155312, Ambion) or Silencer Cy3-labeled negative control siRNA (Ambion) 24 h before motor neuron plating.
Real-time PCR.
Total RNA was isolated using TRIZol reagent (Invitrogen). RNA quality was assessed with the 2100 Bioanalyzer (Agilent Technologies) and 2 μg were randomly reverse transcribed using SuperScript II reverse transcriptase (Invitrogen) according to the manufacturer's protocol. PCRs were performed in a 20 μl reaction with 1X LightCycler480 SYBR Green I Master (Roche) containing 1 μl of cDNA and 20 pmol of each specific primer in a LightCycler480 Real-time PCR System (Roche). The cycling parameters were as follows: 95°C, 10 s; 55°C, 10 s; 72°C, 15 s. Minus reverse transcriptase controls were included in each assay. Specific primers were: Nrf2/5′ (5′-TTCTTTCAGCAGCATCCTCTCCAC-3′), Nrf2/3′ (5′-ACAGCCTTCAATAGTCCCGTCCAG-3′), NQO1/5′ (5′-GCGAGAAGAGCCCTGATTGTACTG-3′), NQO1/3′ (5′-TCTCAAACCAGCCTTTCAGAATGG-3′), HO-1/5′ (5′-CAAGCCGAGAATGCTGAGTTCATG-3′), HO-1/3′ (5′-GCAAGGGATGATTTCCTGCCAG-3′), GCLM/5′ (5′-GCCACCAGATTTGACTGCCTTTG-3′), GCLM/3′ (5′-TGCTCTTCACGATGACCGAGTACC-3′), GCLC/5′ (5′-ACATCTACCACGCAGTCAAGGACC-3′), GCLC/3′ (5′-CTCAAGAACATCGCCTCCATTCAG-3′), GFAPF (5′-CGAGTCCCTAGAGCGGCAAATG-3′), GFAPR (5′-CGGATCTGGAGGTTGGAGAAAGTC-3′). Mrp1/5′ (5′-GATGGCTCCGATCCACTCT-3′), Mrp1/3′ (5′-AGGTAGAAACAAGGCACCCA-3′) ActinF (5′-CATGAAGATCCTGACCGAGCGTG-3′), ActinR (5′-TCTGCTGGAAGGTGGACAGTGAGG-3′)
Glutathione measurement.
Total glutathione levels (GSH and GSSG) were determined using the Tietze method as previously described (Vargas et al., 2006). Cells were lysed with ice-cold 3% perchloric acid while tissues were lysed in 5 volumes of 5% sulfosalicylic acid. Glutathione content was corrected by protein concentration determined by BCA protein assay (Thermo Scientific). For glutathione secretion assays, 1.75 × 105 cells were plated on 35 mm Petri dishes. When confluent, astrocyte monolayers were transfer to supplemented L15 without phenol red and glutathione was determined as described above.
Western blot analysis.
Protein samples (30 μg) were resolved on 12% SDS–polyacrylamide gel and transferred to Hybond-P membrane (Amersham). Membranes were blocked for 1 h in TBS, 0.1% Tween 20 and 5% non-fat dry milk, followed by an overnight incubation with primary antibody (monoclonal anti-SOD1, clon SD-G6, Sigma-Aldrich) diluted in the same buffer. After washing with 0.1% Tween in Tris-buffered saline, the membrane was incubated with peroxidase-conjugated secondary antibody (Amersham) for 1 h, and then washed and developed using the ECL chemiluminescent detection system (Amersham). Densitometric analyses were performed using the NIH ImageJ program and normalized against the signal obtained by reprobing the membranes with anti-actin (Sigma-Aldrich).
Placental alkaline phosphatase assay and histochemistry.
Tissue was immediately frozen in liquid nitrogen, followed by mortar grinding and lysed in TMNC buffer (0.05 m Tris pH 7.5, 0.005 M MgCl2, 0.1 M NaCl, 4% CHAPS) plus 1X Complete protease inhibitor cocktail-EDTA free (Roche). Samples were sonicated, centrifuged at 12,000 × g at 4°C and the supernatant was used to determine hPAP activity as described (Johnson et al., 2002). hPAP activity was corrected by protein content determined by BCA assay. hPAP histochemistry was performed in 20 μm lumbar spinal cord cryostat sections as described (Johnson et al., 2002).
Statistical analysis.
Each experiment was performed by duplicate and repeated at least three times. Groups of at least three animals were used for biochemical analysis and unless indicated, all data are reported as mean ± SD. Survival and onset data were analyzed with Kaplan–Meier curves and log rank test. Multiple group comparison was performed by one-way ANOVA with Bonferroni's post test and differences were declared statistically significant if p < 0.05. All statistical computations were performed using GraphPad Prism 4.0 (GraphPad Software).
Results
Astrocyte-specific Nrf2 over-expression in GFAP-Nrf2 mice
We developed a transgenic mouse that over-expresses Nrf2 under the control of the hGFAP promoter, a cytoskeleton protein selectively expressed in astrocytes. Cell-specificity of Nrf2 over-expression was confirmed by breeding GFAP-Nrf2 animals with mice expressing the human placental alkaline phosphatase (hPAP) driven by a promoter containing the core sequence from the rat NADPH:quinone oxidoreductase 1 (NQO1) ARE (ARE-hPAP) (Johnson et al., 2002). Although the hGFAP promoter occasionally drives expression in some neuronal populations, it does not direct expression in spinal motor neurons (Lee et al., 2008). In GFAP-Nrf2/ARE-hPAP mice, increased hPAP expression was restricted to the CNS and in the spinal cord to GFAP-positive astrocytes (Fig. 1A–C). Nrf2 over-expression in the spinal cord increased transcription of prototypical ARE-driven genes, including NQO1, heme oxygenase-1 (HO-1), GCL modifier (GCLM) and catalytic subunit (GCLC) (Fig. 1D).
Figure 1.

Increased Nrf2 activity is restricted to the CNS in GFAP-Nrf2 mice. A, Spinal cord sections from 60-d-old ARE-hPAP and GFAP-Nrf2/ARE-hPAP animals were stained for hPAP (green) and GFAP (red). The merged image depicts increased hPAP expression restricted to GFAP-positive spinal cord astrocytes. Scale bars: 200 μm (left panels), 40 μm (right panels). B, hPAP histochemistry in lumbar spinal cord sections from 60-d-old ARE-hPAP or ARE-hPAP/GFAP-Nrf2 mice. Scale bar: 200 μm (top), 40 μm (bottom). C, hPAP enzymatic activity in tissues collected from 60-d-old ARE-hPAP or ARE-hPAP/GFAP-Nrf2 mice. Activity is expressed as the logarithm of relative luminescence units (RLU) per mg of protein. CX, Frontal cortex; Crbm, cerebellum; BS, brainstem; SC, spinal cord; Muscle, quadriceps. Each bar represents the mean ± SD (n = 3). *Significantly different from its respective ARE-hPAP tissue (p < 0.05). The fold change in hPAP activity is depicted in parenthesis above the bars. D, Increased mRNA expression of Nrf2 and ARE-driven genes in the spinal cord of 60-d-old GFAP-Nrf2 mice. Nrf2, NQO1, HO-1, GCLM, GCLC and GFAP mRNA levels were determined by real-time PCR and corrected by actin mRNA levels. No difference in GFAP mRNA levels was observed. Data are expressed as mean ± SD (n = 4). *Significantly different from NonTG (p < 0.05).
Primary GFAP-Nrf2 spinal cord astrocytes displayed a 2.5-fold increase in Nrf2 messenger. Increased levels of Nrf2 caused an increase in GCLC and GCLM expression that resulted in a twofold increase in glutathione content/secretion (Fig. 2A,B). Accordingly, GFAP-Nrf2 astrocytes displayed increased resistance to oxidative stress as reflected by reduced vulnerability to tert-butyl hydroperoxide toxicity (Fig. 2C). In astrocytes, glutathione is exported to the extracellular milieu mainly through the multidrug resistance-associated protein 1 (Mrp1), hence glutathione secretion from astrocytes can be reduced by inhibiting Mrp1 (Hirrlinger and Dringen, 2005). A siRNA against Mrp1 mRNA effectively diminished Mrp1 mRNA expression and glutathione secretion from hSOD1G93A/GFAP-Nrf2 astrocytes (Fig. 2D,E). A control non-targeting siRNA had no effect on Mrp1 mRNA 24 h posttransfection (data not shown).
Figure 2.
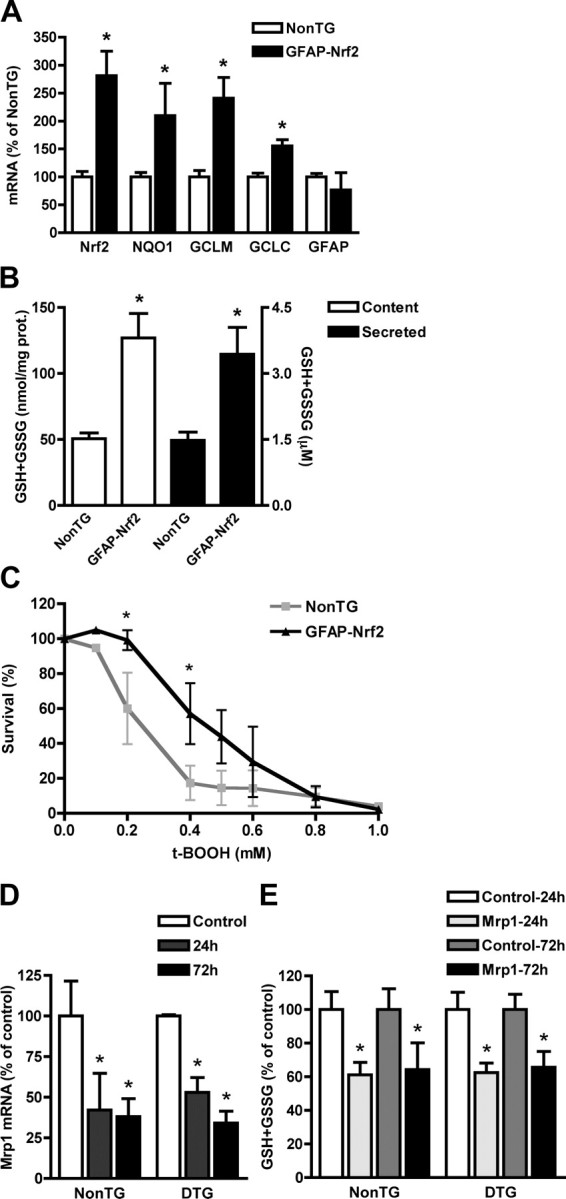
Increased mRNA expression of Nrf2 and ARE-driven genes in GFAP-Nrf2 spinal cord astrocytes increases glutathione secretion and resistance to oxidative stress. A, Total RNA was extracted from purified non-transgenic or GFAP-Nrf2 confluent astrocyte monolayers and Nrf2, NQO1, GCLM, GCLC and GFAP mRNA levels were determined by real-time PCR and corrected by actin mRNA levels. mRNA levels are expressed as percentage of non-transgenic astrocytes (NonTG). B, Increased total glutathione (GSH+GSSG) content (white bars, left axis) and secretion (black bars, right axis) in GFAP-Nrf2 spinal cord astrocyte cultures. C, Astrocyte monolayers were treated with the indicated concentrations of tert-butyl hydroperoxide (t-BOOH), and survival was assessed 24 h later by MTS assay. Data are expressed as percentages of respective controls. Similar results were obtained when astrocyte survival was assayed by LDH release or ethidium homodimer-1 nuclear staining (data not shown). A–C, Each bar represents the mean ± SD (A, n = 4; B, n = 3; C, n = 3; D, n = 3) *Significantly different from NonTG (p < 0.05). D, Non-transgenic (NonTG) and hSOD1G93A/GFAP-Nrf2 (DTG) astrocyte monolayers were transfected with a multidrug resistance-associated protein 1 (Mrp1)-siRNA and 24 or 72 h later Mrp1 mRNA levels were determined by real-time PCR and corrected by Actin mRNA levels. E, Secreted total glutathione (GSH+GSSG) from non-transgenic (NonTG) and hSOD1G93A/GFAP-Nrf2 (DTG) astrocytes 24 and 72 h after Mrp1-siRNA transfection. D, E, Data are expressed as percentages (mean ± SD; n = 3) of respective controls. *Significantly different from its respective control (p < 0.05).
Nrf2 over-expression in astrocytes protects motor neurons from mutant hSOD1 toxicity
In contrast to the trophic support provided by non-transgenic astrocytes, a ∼40% of decrease in motor neuron survival is observed when cocultured on resting astrocytes isolated from hSOD1G93A rats (Vargas et al., 2006) or mice (Nagai et al., 2007). Nrf2 over-expression in hSOD1G93A/GFAP-Nrf2 astrocytes completely reversed the toxicity of hSOD1G93A astrocytes toward cocultured motor neurons (Fig. 3A). When glutathione secretion from astrocytes was prevented by inhibiting Mrp1 expression (Mrp1-siRNA) or activity (MK-571), the protection achieved by Nrf2 over-expression in hSOD1G93A/GFAP-Nrf2 double transgenic astrocytes was lost (Fig. 3B).
Figure 3.

Increased ARE-driven gene expression in GFAP-Nrf2 astrocytes protects cocultured motor neurons from mutant hSOD1 toxicity. A, Purified non-transgenic motor neurons were cocultured on top of confluent non-transgenic (NonTG), wild type hSOD1 (WT), hSOD1G93A (G93A), hSOD1G93A/GFAP-Nrf2 double transgenic (DTG) and GFAP-Nrf2 astrocyte monolayers. Motor neuron survival was determined 72 h later. Motor neuron loss observed in cocultures with G93A astrocytes was prevented by Nrf2 over-expression in DTG astrocytes. B, Motor neuron protection conferred by Nrf2 over-expression in cocultures with DTG astrocytes was lost when glutathione secretion from astrocytes was prevented by inhibiting multidrug resistance-associated protein 1 (Mrp1) by the addition of MK-571 (MK, 20 μm) or a Mrp1-siRNA. Control cocultures with MK-571 and Mrp1-siRNA did not modify motor neuron survival. A control non-targeting siRNA (c-siRNA) did not modify motor neuron survival. Data are expressed as mean ± SD (A, n = 5; B, n = 3). *Significantly different from NonTG control (p < 0.05).
Nrf2 over-expression in astrocytes delayed onset and increased survival in ALS-mice
Over-expression of Nrf2 in astrocytes extended median survival in hSOD1G93A/GFAP-Nrf2 mice by 20.5 d (Fig. 4A). Disease onset, as reflected by rotarod performance and body weight loss, was delayed for 17 d (Fig. 4B,C). The extension observed in survival was due to a delay in median onset since no significant difference was observed in progression from onset to death (G93A, 30.1 ± 10.7 d vs DTG, 29.6 ± 12.3 d). Four weeks-old hSOD1G93A/GFAP-Nrf2 mice had no difference in the number of spinal cord motor neurons or the levels of hSOD1G93A expression compared with hSOD1G93A animals (Fig. 4D,E). Although we use a breeding scheme that minimizes the possible effect of the different transgenic strain backgrounds (see material and methods), we ruled out any possible background contribution by mating GFAP-Nrf2 mice with hSOD1G93A mice in a pure FVB background and founded a similar extension in median onset (19 d) and survival (21.5 d) (Fig. 5A,B). No significant difference was observed in progression from onset to death in a pure FVB background (G93A, 26.5 ± 9.8 d vs DTG, 30.7 ± 10.4 d). We also crossed the GFAP-Nrf2 mice with the hSOD1H46R/H48Q ALS model. The metal-deficient hSOD1H46R/H48Q mutant differs from the hSOD1G93A in that it lacks SOD1 activity (Wang et al., 2002). hSOD1H46R/H48Q/GFAP-Nrf2 animals displayed an extension of 17 d in the median survival (Fig. 5C).
Figure 4.

Nrf2 over-expression in astrocytes extended survival in hSOD1G93A mice. A, Nrf2 over-expression increased the median survival from 128.5 d in G93A animals (n = 16) to 149 d in DTG animals (n = 16). Survival curves are significantly different p < 0.0001 (χ2 = 19.31). B, Extension in the median onset from 96 d in G93A animals (n = 13) to 113 d in DTG animals (n = 9). Onset curves are significantly different p < 0.0001 (χ2 = 17.98). C, Body mass curves from G93A males (n = 6), DTG males (n = 4), G93A females (n = 7), and DTG females (n = 5). Each point represents the mean ± SEM. *Significantly different from G93A (p < 0.05). D, Number of ventral horn motor neurons in 4-week-old non-transgenic (NonTG), hSOD1G93A (G93A), hSOD1G93A/GFAP-Nrf2 (DTG) and GFAP-Nrf2 mice. Data are presented as percentages of NonTG animals (mean ± SD; n = 3). No significant difference was observed among the four groups of mice. E, hSOD1 protein expression in 4-week-old spinal cord extracts from non-transgenic (NonTG), hSOD1G93A transgenic (G93A) and hSOD1G93A/GFAP-Nrf2 double transgenic (DTG) animals. No difference was observed in hSOD1 levels between hSOD1G93A (100 ± 19; n = 3) and hSOD1G93A/GFAP-Nrf2 (106 ± 17; n = 3) mice when quantified and corrected by actin levels.
Figure 5.

Increased Nrf2 activation in astrocytes protects motor neurons from mutant hSOD1 toxicity regardless of the strain background. A, In pure FVB background, the median survival increased from 136 d in G93A animals (n = 14) to 157.5 d in DTG animals (n = 12). Survival curves are significantly different, p < 0.001 (χ2 = 13.65). B, The median disease onset increased from 107 d in G93A animals (n = 14) to 126.5 d in DTG animals (n = 12). Onset curves are significantly different, p < 0.0001 (χ2 = 17.15). C, Increased Nrf2 activation in astrocytes extends median survival in hSOD1H46R/H48Q mice from 213 d (n = 9) to 230 d (n = 8) in DTG animals. Survival curves are significantly different, p < 0.001 (χ2 = 11.25).
Motor neuron survival in hSOD1G93A/GFAP-Nrf2 mice was accompanied by continued innervation of neuromuscular junctions (Fig. 6A) and thus improved motor function and delayed muscular atrophy. Our coculture results indicate that astrocyte-motor neuron interaction can be modified by the amount of glutathione produced/secreted by astrocytes (Fig. 3B). hSOD1G93A/GFAP-Nrf2 mice presented a ∼25% increase of glutathione content in the spinal cord, brainstem and cerebellum (Fig. 6B). Although we did not observe changes in disease progression, delayed astrocyte and microglia activation was observed in hSOD1G93A/GFAP-Nrf2 mice compared with age-matched hSOD1G93A (Fig. 6C).
Figure 6.

Delayed muscle denervation and glial activation in hSOD1G93A/GFAP-Nrf2 animals. A, Quantitative analysis of neuromuscular denervation in medial gastrocnemius sections from 90-d-old hSOD1G93A (G93A) and hSOD1G93A/GFAP-Nrf2 (DTG) mice. Data are expressed as percentage (mean ± SD) of neuromuscular junction (NMJ) from 3 different animals, classified as innervated, intermediate or denervated. *Significantly different from its respective G93A category p < 0.05. B, Total glutathione (GSH+GSSG) content in different regions of the CNS from 4-week-old non-transgenic (NonTG), hSOD1G93A (G93A), hSOD1G93A/GFAP-Nrf2 (DTG) and GFAP-Nrf2 mice. CX, Frontal cortex; Crbm, cerebellum; BS, brainstem; SC, spinal cord. Each bar represents the mean ± SD, n = 3. *Significantly different from its respective NonTG tissue (p < 0.05). C, Immunofluorescence against GFAP (red) and Mac2 (green) in the anterior horn of lumbar spinal cord from 90- and 120-d-old non-transgenic (NonTG), hSOD1G93A (G93A), hSOD1G93A/GFAP-Nrf2 (DTG) and GFAP-Nrf2 mice. Scale bar, 20 μm.
Discussion
It is well documented that Nrf2 activation exerts beneficial effects in many acute models of neuronal damage (Calkins et al. 2005; Shih et al., 2005a,b; Wang et al., 2007; Zhao et al., 2007; Satoh et al., 2008). Here, we provide the first evidence that Nrf2 activation counteracts the progressive loss of neurons in chronic neurodegeneration. ALS-mice with specific astrocyte Nrf2 over-expression developed the disease later, survived longer and had lower glial reactivity. These data not only validate Nrf2 as a viable therapeutic target in ALS but also stress the role of astrocytes in determining motor neuron fate in the disease.
Several hypotheses, including oxidative stress, glutamate excitotoxicity, formation of high molecular weight aggregates and mitochondrial dysfunction have been proposed to explain the toxic effect of mutated hSOD1 (Beckman et al., 2001; Cleveland and Rothstein, 2001; Bruijn et al., 2004; Manfredi and Xu, 2005). The proposed non-cell autonomous mechanism of the disease suggests that, while the expression of the mutant enzyme in motor neurons affects disease onset (Boillée et al., 2006), expression within the glial compartment influences disease progression (Beers et al., 2006 Boillée et al., 2006, Yamanaka et al., 2008). Transgenic mice expressing hSOD1G86R exclusively in astrocytes displayed astrocytosis but fail to develop the disease (Gong et al., 2000). However, in vitro, restricted expression of mutant hSOD1 in astrocytes alone is toxic to cocultured wild-type motor neurons (Vargas et al., 2006; Nagai et al., 2007). Although the nature of the toxicity remains controversial, the over-expression of Nrf2 completely reversed it. Although Nrf2 over-expression directly conferred astrocytes increased resistance to oxidative stress, motor neuron protection depended on glutathione secretion to the extracellular compartment. Therefore, these data suggest that Nrf2 may not directly modify the toxic component of hSOD1G93A expression in astrocytes but the ability of motor neurons to cope with it. Several studies have demonstrated that by releasing glutathione, astrocytes can improve the antioxidant defenses in cocultured neurons. Astrocyte-secreted glutathione can boost glutathione levels in neurons and also act as an antioxidant in the extracellular compartment (Dringen et al. 1999, 2000, 2001). In agreement, glutathione secretion by astrocytes has been demonstrated to protect neurons against oxidative stress (Shih et al., 2003; Kraft et al., 2004), the deleterious effect of nitric oxide (Bolaños et al., 1996; Chen et al., 2001; Gegg et al., 2005) and p75NTR-dependent apoptosis (Vargas et al., 2006).
According to the current notion of non-cell autonomous toxicity in ALS (Clement et al., 2003; Boillée et al., 2006; Yamanaka et al., 2008), a modification that directly affects astrocytes will predict a shift in the disease duration by affecting progression. However Nrf2 over-expression in astrocytes increased the survival in hSOD1G93A mice by delaying onset without affecting the progression of the disease. In principle, transgenic Nrf2 over-expression could incorrectly appear to shift onset in hSOD1G93A mice if during development alters the baseline number of spinal cord motor neurons, however no differences were observed among the four groups of animals studied. Therefore, delayed onset implies that motor neurons were spared for a longer period of time in hSOD1G93A/GFAP-Nrf2 animals. This idea is also supported by the fact that hSOD1G93A/GFAP-Nrf2 animals displayed a larger number of innervated neuromuscular junctions, improved rotarod performance and delayed muscular atrophy, when compared with age-matched hSOD1G93A.
The results obtained from the astrocyte-motor neuron coculture indicate that the amount of glutathione secreted by astrocytes is a main component of the neuroprotection conferred by Nrf2 over-expression. Although it has proven extremely difficult to experimentally increase glutathione content in the CNS, transgenic over-expression of Nrf2 in hSOD1G93A/GFAP-Nrf2 animals resulted in ∼25% increase in glutathione content in the spinal cord. In vivo, increased astrocytic glutathione secretion could modify astrocyte-motor neuron interaction and in part account for the observed motor neuron preservation. In turn, preventing the initial damage caused by mutant hSOD1 within motor neurons might attenuate glial activation (Barbeito et al., 2004; Yamanaka et al., 2008). In addition, increased glutathione secretion from astrocytes might also alter the way in which they interact with other cells and contribute to a decreased microglia response.
Together, these findings demonstrate that it is possible to modify non-neuronal cells-motor neuron interaction in ALS by targeting astrocytes to alter the course of the disease. Our results also validate Nrf2 as a viable therapeutic target in chronic neurodegeneration.
Footnotes
This work was supported by grants from the ALS Association and the Robert Packard Center for ALS Research at Johns Hopkins, the National Institute of Environmental Health Sciences Grants ES08089 and ES10042 (J.A.J.), and NS060120 and HD03352 (A.M.). M.R.V. is a recipient of the Milton Safenowitz Postdoctoral Fellowship for ALS Research. We thank Dr. Luis Barbeito for critical reading of this manuscript.
References
- Alam J, Stewart D, Touchard C, Boinapally S, Choi AM, Cook JL. Nrf2, a Cap‘n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J Biol Chem. 1999;274:26071–26078. doi: 10.1074/jbc.274.37.26071. [DOI] [PubMed] [Google Scholar]
- Barbeito LH, Pehar M, Cassina P, Vargas MR, Peluffo H, Viera L, Estévez AG, Beckman JS. A role for astrocytes in motor neuron loss in amyotrophic lateral sclerosis. Brain Res Brain Res Rev. 2004;47:263–274. doi: 10.1016/j.brainresrev.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Estévez AG, Crow JP, Barbeito L. Superoxide dismutase and the death of motoneurons in ALS. Trends Neurosci. 2001;24:S15–S20. doi: 10.1016/s0166-2236(00)01981-0. [DOI] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel SH. Wild-type microglia extend survival in PU.1 knockout mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Bolaños JP, Heales SJ, Peuchen S, Barker JE, Land JM, Clark JB. Nitric oxide-mediated mitochondrial damage: a potential neuroprotective role for glutathione. Free Radic Biol Med. 1996;21:995–1001. doi: 10.1016/s0891-5849(96)00240-7. [DOI] [PubMed] [Google Scholar]
- Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A. GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci. 1994;14:1030–1037. doi: 10.1523/JNEUROSCI.14-03-01030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- Calkins MJ, Jakel RJ, Johnson DA, Chan K, Kan YW, Johnson JA. Protection from mitochondrial complex II inhibition in vitro and in vivo by Nrf2-mediated transcription. Proc Natl Acad Sci U S A. 2005;102:244–249. doi: 10.1073/pnas.0408487101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassina P, Pehar M, Vargas MR, Castellanos R, Barbeito AG, Estévez AG, Thompson JA, Beckman JS, Barbeito L. Astrocyte activation by fibroblast growth factor-1 and motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. 2005;93:38–46. doi: 10.1111/j.1471-4159.2004.02984.x. [DOI] [PubMed] [Google Scholar]
- Chen Y, Vartiainen NE, Ying W, Chan PH, Koistinaho J, Swanson RA. Astrocytes protect neurons from nitric oxide toxicity by a glutathione-dependent mechanism. J Neurochem. 2001;77:1601–1610. doi: 10.1046/j.1471-4159.2001.00374.x. [DOI] [PubMed] [Google Scholar]
- Clement AM, Nguyen MD, Roberts EA, Garcia ML, Boillée S, Rule M, McMahon AP, Doucette W, Siwek D, Ferrante RJ, Brown RH, Jr, Julien JP, Goldstein LS, Cleveland DW. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non-cell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R, Pfeiffer B, Hamprecht B. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci. 1999;19:562–569. doi: 10.1523/JNEUROSCI.19-02-00562.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain. Metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem. 2000;267:4912–4916. doi: 10.1046/j.1432-1327.2000.01597.x. [DOI] [PubMed] [Google Scholar]
- Dringen R, Gutterer JM, Gros C, Hirrlinger J. Aminopeptidase N mediates the utilization of the GSH precursor CysGly by cultured neurons. J Neurosci Res. 2001;66:1003–1008. doi: 10.1002/jnr.10042. [DOI] [PubMed] [Google Scholar]
- Gegg ME, Clark JB, Heales SJ. Co-culture of neurons with glutathione deficient astrocytes leads to increased neuronal susceptibility to nitric oxide and increased glutamate-cysteine ligase activity. Brain Res. 2005;1036:1–6. doi: 10.1016/j.brainres.2004.11.064. [DOI] [PubMed] [Google Scholar]
- Gong YH, Parsadanian AS, Andreeva A, Snider WD, Elliott JL. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J Neurosci. 2000;20:660–665. doi: 10.1523/JNEUROSCI.20-02-00660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hirrlinger J, Dringen R. Multidrug resistance protein 1-mediated export of glutathione and glutathione disulfide from brain astrocytes. Methods Enzymol. 2005;400:395–409. doi: 10.1016/S0076-6879(05)00023-6. [DOI] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DA, Andrews GK, Xu W, Johnson JA. Activation of the antioxidant response element in primary cortical neuronal cultures derived from transgenic reporter mice. J Neurochem. 2002;81:1233–1241. doi: 10.1046/j.1471-4159.2002.00913.x. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. J Neurosci. 2004;24:1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278:12029–12038. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- Lee Y, Messing A, Su M, Brenner M. GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia. 2008;56:481–493. doi: 10.1002/glia.20622. [DOI] [PubMed] [Google Scholar]
- Manfredi G, Xu Z. Mitochondrial dysfunction and its role in motor neuron degeneration in ALS. Mitochondrion. 2005;5:77–87. doi: 10.1016/j.mito.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehar M, Cassina P, Vargas MR, Castellanos R, Viera L, Beckman JS, Estévez AG, Barbeito L. Astrocytic production of nerve growth factor in motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. 2004;89:464–473. doi: 10.1111/j.1471-4159.2004.02357.x. [DOI] [PubMed] [Google Scholar]
- Pehar M, Vargas MR, Robinson KM, Cassina P, Díaz-Amarilla PJ, Hagen TM, Radi R, Barbeito L, Beckman JS. Mitochondrial superoxide production and nuclear factor erythroid 2-related factor2 activation in p75 neurotrophin receptor-induced motor neuron apoptosis. J Neurosci. 2007;27:7777–7785. doi: 10.1523/JNEUROSCI.0823-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi RE, Halperin JJ, Herzfeldt B, Van den Bergh R, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med. 2001;344:1688–1700. doi: 10.1056/NEJM200105313442207. [DOI] [PubMed] [Google Scholar]
- Satoh T, Kosaka K, Itoh K, Kobayashi A, Yamamoto M, Shimojo Y, Kitajima C, Cui J, Kamins J, Okamoto S, Izumi M, Shirasawa T, Lipton SA. Carnosic acid, a catechol-type electrophilic compound, protects neurons both in vitro and in vivo through activation of the Keap1/Nrf2 pathway via S-alkylation of targeted cysteines on Keap1. J Neurochem. 2008;104:1116–1131. doi: 10.1111/j.1471-4159.2007.05039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Johnson DA, Wong G, Kraft AD, Jiang L, Erb H, Johnson JA, Murphy TH. Coordinate regulation of glutathione biosynthesis and release by Nrf2-expressing glia potently protects neurons from oxidative stress. J Neurosci. 2003;23:3394–3406. doi: 10.1523/JNEUROSCI.23-08-03394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Li P, Murphy TH. A small-molecule-inducible Nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. J Neurosci. 2005a;25:10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AY, Imbeault S, Barakauskas V, Erb H, Jiang L, Li P, Murphy TH. Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo. J Biol Chem. 2005b;280:22925–22936. doi: 10.1074/jbc.M414635200. [DOI] [PubMed] [Google Scholar]
- Vargas MR, Pehar M, Cassina P, Beckman JS, Barbeito L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J Neurochem. 2006;97:687–696. doi: 10.1111/j.1471-4159.2006.03742.x. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Gonzales V, Coonfield M, Fromholt D, Copeland NG, Jenkins NA, Borchelt DR. Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol Dis. 2002;10:128–138. doi: 10.1006/nbdi.2002.0498. [DOI] [PubMed] [Google Scholar]
- Wang J, Fields J, Zhao C, Langer J, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Doré S. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic Biol Med. 2007;43:408–414. doi: 10.1016/j.freeradbiomed.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Moore AN, Redell JB, Dash PK. Enhancing expression of Nrf2-driven genes protects the blood brain barrier after brain injury. J Neurosci. 2007;27:10240–10248. doi: 10.1523/JNEUROSCI.1683-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]