

Abstract
Prostate cancer (CaP) progresses from prostatic intraepithelial neoplasia through locally invasive adenocarcinoma to castration resistant (CR) metastatic carcinoma1. Although radical prostatectomy, radiation and androgen ablation are effective therapies for androgen-dependent (AD) CaP, metastatic CR-CaP is a major complication with high mortality2. Androgens stimulate growth and survival of prostate epithelium and early CaP. Although most patients initially respond to androgen ablation, many develop CR-CaP within 12-18 months2. Despite extensive studies, the mechanisms underlying CR-CaP emergence remain poorly understood and their elucidation is critical for development of improved therapies. Curiously, CR-CaP remains androgen receptor (AR) dependent and potent AR antagonists induce tumor regression in castrated mice3. The role of inflammation in CR-CaP has not been addressed, although it was reported that intrinsic NF-κB activation supports its growth4. Inflammation is a localized protective reaction to injury or infection, but it also has a pathogenic role in many diseases, including cancer5. Whereas acute inflammation is critical for host defense, chronic inflammation contributes to tumorigenesis and metastatic progression. The inflammation-responsive IκB kinase (IKK) β and its target NF-κB have important tumor promoting functions within malignant cells and inflammatory cells6. The latter, including macrophages and lymphocytes, are important elements of the tumor microenvironment7-9, but the mechanisms underlying their recruitment remain obscure, although thought to depend on chemokine and cytokine production10. We found that CaP progression is associated with inflammatory infiltration and activation of IKKα, which stimulates metastasis by an NF-κB-independent, cell autonomous, mechanism11. We now show that androgen ablation causes infiltration of regressing AD tumors with leukocytes, including B cells, in which IKKβ activation results in production of cytokines that activate IKKα and STAT3 in CaP cells to enhance hormone-free survival.
To determine whether IKKβ-driven NF-κB participates in development of CR-CaP, we conditionally deleted the Ikkβ gene in prostate epithelial cells of TRAMP mice, in which CaP is induced by prostate specific expression of SV40 T antigen12. Counter to previous expectations4, IKKβ ablation in prostate epithelial cells had no effect on genesis and progression of AD-CaP (Fig. S1) or development of CR-CaP after castration (Fig. S2). To facilitate mechanistic analysis of CR-CaP development we used subcutaneous (SC) allografts of the mouse AD-CaP cell line myc-CaP, which is derived from the FVB genetic background13. Silencing of IKKβ in myc-CaP cells did not affect primary tumor growth or CR-CaP re-growth in castrated FVB mice (Fig. S3).
By contrast, deletion of IKKβ in interferon-responsive cells upon induction of Mx1-Cre expression prevented castration-induced metastatic spread in TRAMP/IkkβF/F/Mx1-Cre mice (Fig. S4A). To narrow down the role of IKKβ to bone marrow (BM)-derived cells (BMDCs), we reconstituted irradiated FVB mice with BM from IkkβF/F and IkkβΔ/Δ mice [IkkβF/F/Mx1-Cre mice injected with poly(IC) to induce Mx1-Cre] and inoculated the resulting chimeras (Fig. S4B) with myc-CaP cells. Absence of IKKβ in BMDC had no impact on primary tumor growth, but delayed CR-CaP emergence after castration (Fig. S4C). A similar delay in CR-CaP growth was seen in castrated tumor-bearing mice treated with specific IKKβ inhibitors14, 15 (Fig. S4D and data not shown).
Dependence of CR-CaP emergence on IKKβ in BMDC, suggested that androgen deprivation elicits a tumor-associated inflammatory response. Castration of mice bearing myc-CaP tumors resulted in CaP cell death, peaking within one week (Fig. 1A). Concurrently, the regressing tumors were infiltrated with T and B lymphocytes, NK cells and myeloid cell types (Fig. 1B; Fig. S5). Infiltration was transient, declining by 2 weeks after castration. B and T lymphocyte infiltration was also detected in 100% of human CaP samples (untreated patients with Gleason scores of 6-8), but B cells were undetectable in normal prostate or benign prostatic hyperplasia (Fig. 1C). The mRNAs for many inflammatory chemokines were also upregulated in the myc-CaP allografts, but no changes in AR mRNA expression were found (Fig. S6A). These chemokines may recruit lymphoid and myeloid cells into the regressing tumor. Indeed, antibody mediated inhibition of CXCL13, a B cell chemoattractant16, prevented castration-induced B cell recruitment (Fig. S6B,C). Inflammatory cytokine mRNAs, including IL-6, IL-12, TNF-α and lymphotoxin (LT), were also upregulated in the regressing myc-CaP allograft, but only LT expression was reduced upon CXCL13 inhibition (Fig. S7). Castration resulted in nuclear export of AR, but after 3 weeks AR was nuclear again (Fig. S8), suggesting it is activated at late phases of CR-CaP growth despite androgen depletion.
Figure 1. Androgen ablation induces tumor inflammatory infiltration.

Six week old FVB males (n=10) were inoculated with myc-CaP cells. When tumors reached 1000 mm3, mice were left untreated, castrated or sham operated. Tumors were collected when indicated for analysis. A. Paraffin-embedded tumor sections were TUNEL stained to determine apoptotic cell frequency (results are averages, n=3). B. Total RNA was isolated from tumor samples and expression of indicated cell marker mRNAs was quantitated and normalized to that of cyclophilin A (norm= normal, C1,2,3= mice analyzed 1,2,3 weeks after castration, sham= sham-operated). Results are averages ± s.d. (n=10). C. Frozen human prostate sections (normal tissue [n=3], prostatic hyperplasia [n=3], and malignant CaP with Gleason scores 6-8 [n=10]) were stained with CD4, CD8 and CD20 antibodies and DAPI and analyzed by immunofluorescent microscopy. The histogram denotes average frequencies of indicated cell types (n=3 per sample). P values were determined and are depicted as insignificant (ns), significant (*), very significant (**) or highly significant (***).
STAT3 was proposed to promote activation of unliganded AR17. Indeed, STAT3 was activated during CR-CaP emergence, faster than AR was (Fig. S9). Mx1-Cre-mediated IKKβ deletion, which was nearly complete in mature B and T lymphocytes (Fig. S10A), prevented STAT3 activation in regressing tumors, but did not affect ERK and AKT activation (Fig. S10B). Immunohistochemical analysis confirmed STAT3 activation in CaP cells, inhibitable by A490 (Fig. S10C), an inhibitor of STAT3 phosphorylation18 that delayed appearance of CR-CaP (Fig. S10D) but did not inhibit IKK activation (Fig. S11A). Conversely, ML120B did not inhibit STAT3 activation (Fig. S11B).
Ablation of BMDC IKKβ did not prevent leukocyte recruitment into regressing tumors (Fig. S12) but did inhibit cytokine induction (Fig. S13). To further investigate the role of lymphocytes, we used chimeric mice generated by transplantation of BM from lymphocyte-deficient Rag1−/− mice. Although primary tumor growth was identical in mice receiving WT or Rag1−/− BM (Fig. S14A), CR-CaP growth was significantly delayed in mice receiving Rag1−/− BM (Fig. 2A), but not in mice reconstituted with BM from Tcrβ−/−/δ−/− mice (Fig. S14B), which lack only mature T lymphocytes. CR-CaP growth was delayed in mice reconstituted with BM from JH−/− mice (Fig. S14C), which lack mature B cells, or upon B cell depletion with CD20 antibody19 (Fig. S14D). Reconstitution of Rag1−/− FVB mice with splenic B cells, but not T cells of FVB mice, restored rapid CR-CaP re-growth (Fig. 2A). Primary tumors, isolated from Rag1−/− chimeric mice one week after castration, did not show STAT3 activation (Fig. 2B), but reconstitution with B cells, rather than T cells, restored castration-induced STAT3 phosphorylation (Fig. 2C).
Figure 2. Role of B cells and IKKβin STAT3 activation and CR-CaP emergence.
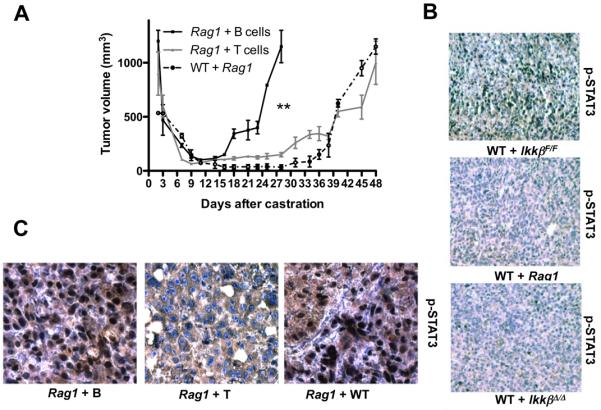
A. Myc-CaP tumors were established in WT mice reconstituted with BM from Rag1−/− males (n=10) or in Rag1−/− males. When tumors reached 1000 mm3, mice were castrated. Three days before castration, Rag1−/− mice (n=10 per group) received via the tail vein purified splenic B or T cells. Tumor volume was measured. Results are averages ± s.e.m.. P values were determined and are indicated as above. B. Tumors were removed from radiation chimeras reconstituted with IkkβF/F, IkkβΔ/Δ, or Rag1−/− BM, one week after castration. STAT3 phosphorylation was analyzed by immunohistochemistry. C. Tumor-bearing Rag1−/− males were injected with WT splenocytes (Rag1 + WT), or purified splenic B (Rag1 + B) or T (Rag1 + T) lymphocytes. One day later, mice were castrated and after one week, tumors were removed and analyzed for STAT3 phosphorylation.
CaP allografts from castrated, but not sham-operated, mice exhibited IKKα nuclear translocation (Fig. 3A,B). Silencing of IKKα in myc-CaP cells using siRNA (Fig. S15A) had little effect on primary tumor growth, but delayed CR-CaP emergence (Fig. 3C). Nuclear translocation of IKKα was dependent on IKKβ in BMDC and on B cells, but not on T cells (Fig. 3D). IKKα nuclear translocation parallels progression of human and murine CaP and coincides with primary tumor infiltration with cells expressing IKKα-activating cytokines, RANK ligand (RANKL) and LTα11. Castration induced LTα and LTβ in regressing myc-CaP allografts, but did not alter RANKL expression (Fig. S7). LT expression in regressing tumors was absent in Rag1−/− mice (Fig. S16A) and flow cytometry localized it to tumor infiltrating B cells (TIBC; Fig. S16B). We characterized TIBC by 7-color flow cytometry with several markers and a LTβR-Ig fusion protein to detect LT. The typical TIBC was a conventional, mature B2 cell that expressed LT on its surface and was negative for B1 markers (Fig. S17). IKKβ deletion abolished LT expression by B cells (Fig. 4A), supporting the previously suggested20 role of NF-κB in LTα/β induction. To examine whether LT production by tumor-infiltrating lymphocytes stimulates CR-CaP growth, we transplanted BM from B-Ltβ−/− or T-Ltβ−/− mice, which lack LTβ in either B or T cells21, into lethally irradiated mice. LTβ ablation in B cells, but not in T cells, delayed growth of CR-CaP (Fig. 4B) and abolished LTβ expression within tumors but did not prevent B cell or macrophage infiltration (Fig. S18). Treatment of mice with the LTβR-Ig decoy22 was as effective as B cell-specific LTβ ablation in delaying CR-CaP growth (Fig. 4C) and prevented IKKα and STAT3 activation (Fig. S19). Silencing of LTβR in Myc-CaP cells (Fig. S15B) also delayed CR-CaP growth (Fig. 4D). Exogenous LT maintained myc-CaP growth in the presence of flutamide, a clinically used AR antagonist3, in a manner dependent on IKKα (Fig. 4E), whose nuclear translocation was LT inducible (Fig. 4F).
Figure 3. Role of IKKα in emergence of CR-CaP.

Tumor-bearing mice were castrated or sham operated as above. A. Tumors were analyzed one week later for nuclear IKKα by immunohistochemistry. B. Tumors removed at indicated times were divided into cytosolic (C) and nuclear (N) fractions and IKKα and histone H3 distribution was determined. C. Tumors were established using myc-CaP cells transduced with lentiviruses expressing scrambled siRNA (sc) or IKKα-specific siRNA. Mice were castrated as above and tumor volume was measured. Results are averages ± s.e.m. (n=10). P values were determined and are indicated as above. D. Tumors were established in lethally irradiated FVB males reconstituted with IkkβF/F or IkkβΔ/Δ BM or in Rag1−/− males reconstituted with either B or T cells. IkkβF/F and IkkβΔ/Δ chimeras were i.p. injected three times with poly(IC) (250 μg) prior to castration to delete IKKβ. One week after castration, tumor samples were analyzed for IKKα distribution by immunohistochemistry. Nuclear IKKα results in punctuate staining, while cytoplasmic IKKα results in diffuse staining.
Figure 4. IKKβ-dependent lymphotoxin production by tumor-infiltrating B cells stimulates IKKα-dependent androgen-free survival.

A. RNA from splenic B cells of IkkβF/F and IkkβΔ/Δ mice was analyzed for LTα and LTβ expression as above. Results are averages ± s.d. (n=3). B. Lethally irradiated FVB males were reconstituted with BM from B-Ltβ−/− or T-Ltβ−/− mice (n=6 per group). After 8 weeks, myc-CaP tumors were established, mice were castrated and tumor volume was measured as above. Results are averages ± s.e.m.. C. FVB mice (n=6 each group) bearing myc-CaP tumors were castrated and given hIgG or LTβR-Ig (100 μg) every 5 days, starting 4 days before castration. Tumor volume was measured as above. Results are averages ± s.e.m.. D. Tumors were established using myc-CaP cells transduced with lentiviruses expressing scrambled (sc) siRNA or LTβ-specific siRNA. Mice were castrated and tumor volume was measured. Results are averages ± s.e.m. (n=10). E. Myc-CaP cells (previously infected with lentiviruses expressing scrambled or IKKα siRNAs) were plated at 40% confluency. After 6 hrs, the cells were cultured with or without flutamide (10 μM) in the absence or presence of LTα2β1, and cell number was determined. F. Myc-CaP cells were plated at 60% confluency. After 12 hrs, cells were stimulated for 1 hr with LTα2β1, collected, divided into cytosolic (C) and nuclear (N) fractions and IKKα and histone H3 distribution was determined. In A, B, C, D and E, P values were determined and are indicated as above.
CR-CaP is a major complication that limits the success of androgen ablation therapy and is responsible for most prostate cancer mortality2. CR-CaP was studied mainly at the level of AR function, the central player in this process4. Our results suggest that an inflammatory response triggered by death of androgen-deprived primary cancer is another important contributor to emergence of CR-CaP. In addition to dying CaP cells, critical participants in this response are tumor infiltrating B cells, which produce LTα:β heterotrimers that stimulate LTβR on CaP cells to induce IKKα nuclear translocation and STAT3 activation, thereby enhancing androgen-independent growth (Fig. S20). Interference with any component of this response results in a significant and reproducible 3-4 week delay in appearance of CR-CaP. Although these inhibitory effects are not absolute, extrapolation from “mouse time” to “human time” suggests that interventions that prevent LT production or signaling may delay appearance of CR-CaP in patients undergoing androgen ablation therapy by 2.3 to 3.1 years. Importantly, our results suggest that, at least for CaP, the inflammatory response elicited by the dying primary tumor, contributes to the failure rather than the previously proposed success of anti-cancer therapy23. Although we have not determined how death of androgen-deprived CaP triggers the inflammatory response described above, necrotic cell death releases mediators, such as HMGB124 and IL-1α25, that activate IKKβ and NF-κB and stimulate production of chemokines, one of which, CXCL13, recruits B cells into the regressing tumor. Notably, TIBC were detected not only in androgen-deprived mouse CaP, but also in human CaP. Although B cells were reported to promote progression of skin carcinomas9 and exert immunosuppressive effects through activation of inhibitory Fc receptors on myeloid cells26, the critical tumor promoting B cell function in our experimental model is production of LT, an IKKα-activating cytokine27, which promotes survival of androgen-deprived CaP. Another important function of TIBC is activation of STAT3, an anti-apoptotic and pro-tumorigenic transcription factor28. Although the critical STAT3-activating cytokine in this system remains to be identified, castration induces expression of STAT3-activating IL-6 and IL-12 family members. Furthermore, CaP cells use autocrine IL-6 to stimulate their progression29 and activated STAT3 promotes ligand-independent AR activation29. LT is also involved in the etiology of human CaP. An epidemiological study revealed that reduced CaP risk due to consumption of non-steroidal anti-inflammatory drugs, such as aspirin, is limited to men who express a common polymorphic LTα allele that specifies high LT production30. Further work should examine the effect of LTα polymorphism on the response to androgen ablation. Our results predict that individuals who are high LT producers are more likely to develop CR-CaP and should therefore be the main beneficiaries of anti-LT therapy.
Methods Summary
A detailed Methods section is available in Supplementary Information. Mice were handled according to institutional and NIH guidelines. Tumors were grown in FVB mice. Where indicated, lethally irradiated FVB mice were reconstituted with BM from different strains that were backcrossed into the FVB background for at least two generations. Ltβ knockout strains were, however, in the BL6 background which does not elicit a graft vs. host response in FVB mice. Conditions for antibody used were posted to http://biorating.com. Human material was obtained from the Cooperative Human Tissue Network (CHTN) along with pathology reports. Histology, gene expression and cell signaling were analyzed as described11, 25.
Supplementary Material
Acknowledgements
We thank C. Sawyers for myc-CaP cells, L. Coussens for JH−/− (FVB) mice, Y.X. Fu for LTβR-Ig fusion protein, R. Rickert for B cell phenotyping help, H. Cheroutre for flow cytometer use and C. Ware for bone marrow. M.A. was supported by Fondazione Italiana per la Ricerca sul Cancro (F.I.R.C.) and American-Italian Cancer Foundation (A.I.C.F.) fellowships. J.-L.L. was supported by Life Science Research Fellowship. Work in M.K.’s laboratory was supported by grants from the NIH, the US Army Medical Research and Materiel Command and Prostate Cancer Foundation. M.K. is an American Cancer Society Research Professor.
Appendix
METHODS
Mice and cell culture
IkkβF/F (BL6) mice were crossed to TRAMP (BL6x129) mice31 and PB-Cre4 (BL6)32 or Mx1-Cre (BL6) mice33 to generate TRAMP+/−/Ikkβ+/F/PB-Cre4+/− and TRAMP+/−/Ikkβ+/F/PB-Cre4+/+ or TRAMP+/−/Ikkβ+/F/Mx1-Cre+/− and TRAMP+/−/Ikkβ+/F/Mx1-Cre+/+ progeny that were intercrossed with TRAMP mice for six generations. After that, TRAMP+/−/Ikkβ+/F/PB-Cre4+/− and TRAMP+/−/Ikkβ+/F/PB-Cre4+/+ or TRAMP+/−/Ikkβ+/F/Mx1-Cre+/− and TRAMP+/−/Ikkβ+/F/Mx1-Cre+/+ mice were intercrossed to generate TRAMP+/−/IkkβF/F/PB-Cre4+/−, TRAMP+/−/IkkβF/F/PB-Cre4+/+, TRAMP+/−/IkkβF/F/Mx1-Cre+/− and TRAMP+/−/IkkβF/F/Mx1Cre+/+ mice. Only male littermates were used. FVB, Tcrβ−/−δ−/− (BL6), Mx1-Cre and Rag1−/− (BL6x129) mice were from the Jackson Laboratory. JH−/− mice (FVB) were kindly provided by L. Coussens (Cancer Research Institute and Anatomic Pathology, UCSF, San Francisco, CA). Bone marrow from B-Ltβ−/− or T-Ltβ−/− mice21 was kindly provided by C.F. Ware (La Jolla Institute for Allergy and Immunology, La Jolla, CA). PB-Cre4 and TRAMP mice were from MMHCC (Mouse Models of Human Cancer Consortium). Mice were maintained under specific pathogen-free conditions, and experimental protocols were approved by the UCSD Animal Care Program, following NIH guidelines. Radiation chimeras were generated as described34. In general, irradiated FVB mice were reconstituted with bone marrow from different strains that have been backcrossed to the FVB background for at least 2 generations. However, in the case of B-Ltβ−/− and T-Ltβ−/− mice, bone marrow donors were of the BL6 background, whose bone marrow did not lead to a graft vs. host response in irradiated FVB mice. Myc-CaP cells derived from the FVB background were provided by C. Sawyers (UCLA and Memorial Sloan Kettering Cancer Center)35 and were cultured under standard conditions and confirmed to be mycoplasma free. Myc-CaP cells were injected subcutaneously into the flank of male FVB mice as described35. Tumor growth was measured with a caliper. Surgical procedures were as described35.
Human specimens
Anonymous human prostate, benign prostatic hyperplasia and prostate cancer frozen sections were provided by the Cooperative Human Tissue Network (CHTN). Pathology reports were provided by CHTN for each sample.
CXCL13 and B cell depletion and LT inhibition
CXCL13 neutralizing antibody was purchased from R&D and administered i.p. at 200 mg/mouse as described36. Anti-CD20 was kindly provided by Genentech (Oceanside, CA) and was administered i.p. at 250 μg/mouse. LTβR-Ig fusion protein was a kind gift from Yang-Xin Fu (University of Chicago, Chicago, IL) and was administered as described22. hIgG and mouse IgG2a were purchased from Sigma-Aldrich and were used as controls.
IKKβ inhibitors
ML120 was kindly provided by Millenium Inc. and administered orally as described37. IKKβ Inhibitor IV was purchased from Calbiochem and was tail vein injected as described14.
Histological procedures
Mouse prostate and CaP tissues and dissected metastatic tumors were immersed in 10% neutral buffered formalin before sectioning and paraffin embedding. Sections were stained and processed as described11, using H&E stain, TUNEL assay kit or antibodies for IKKα (Imgenex), phospho-STAT3 (Cell Signaling) and CD19 (eBioscience) as described38. Frozen sections of human and mouse origins were fixed in acetone and processed as described 11, using antibodies for AR (Santa Cruz), B220 (BD),CD20 (BD), CD4 (BD) and CD8 (BD).
Analysis of RNA and protein expression
Total tissue RNA was prepared using RNAeasy (Qiagen). Quantitative PCR was performed as described38. Cells and tumors were lysed and analyzed by SDS-polyacrylamide gel electrophoresis and immunoblotting38 with antibodies to histone H3, α-tubulin, STAT3 (Santa Cruz Biotechnology), ERK, phospho-ERK, AKT, phospho-AKT, phospho-STAT3 (Cell Signaling). Nuclear extracts were prepared and analyzed for NF-κB DNA-binding as described39.
Lentiviral and retroviral transduction
siRNAs to mouse IKKα, IKKβand LTβR mRNAs were cloned into pLSLPw, provided by I. Verma (The Salk Institute), and lentivirus stocks were prepared as described11. Virus-containing supernatants were added to myc-CaP cells for 2 days with polybrene, and transduced cells were selected in 5 μg ml−1 puromycin (Invitrogen).
Leukocytes purification and flow cytometry
Peripheral blood mononuclear cells (PBMCs) were isolated by centrifugation on a double-layered Histopaque-Ficoll (GE Lifescience) gradient. Splenic B and T lymphocytes were isolated by magnetic cell sorting (MACS) with CD4, CD8 or CD19 antibodies conjugated to magnetic beads. Tumor infiltrating leukocytes were stained with CD45, B220, LTβR-Ig, TCRβ, Gr1, CD4 and CD8 fluorescent antibodies, as well as Aqua LIVE/DEAD dye (Molecular Probes) and analyzed on a flow cytometer (Accuri C6 or Becton Dickinson LSR II).
Statistical analyses
Results are expressed as means ± s.e.m. or s.d. Data were analyzed by Student’s t-test and Kaplan-Meier survival analysis using GraphPad Prism statistical program. Error bars depict s.e.m. or s.d. P values >0.05 were considered insignificant (ns), 0.01 to 0.05 were considered significant (*), 0.001 to 0.01 were considered very significant (**) and < 0.001 were considered as highly significant (***).
References
- 31.Greenberg NM, et al. Prostate cancer in a transgenic mouse. Proc Natl Acad Sci USA. 1995;92:3439–3443. doi: 10.1073/pnas.92.8.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu X, et al. Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech Dev. 2001;101:61–69. doi: 10.1016/s0925-4773(00)00551-7. [DOI] [PubMed] [Google Scholar]
- 33.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 34.Kim S, et al. Carcinoma-produced factors activate myeloid cells through TLR2 to stimulate metastasis. Nature. 2009;457:102–106. doi: 10.1038/nature07623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watson PA, et al. Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res. 2005;65:11565–11571. doi: 10.1158/0008-5472.CAN-05-3441. [DOI] [PubMed] [Google Scholar]
- 36.Zheng B, et al. CXCL13 neutralization reduces the severity of collagen-induced arthritis. Arthritis Rheum. 2005;52:620–626. doi: 10.1002/art.20768. [DOI] [PubMed] [Google Scholar]
- 37.Izmailova ES, et al. Use of molecular imaging to quantify response to IKK-2 inhibitor treatment in murine arthritis. Arthritis Rheum. 2007;56:117–128. doi: 10.1002/art.22303. [DOI] [PubMed] [Google Scholar]
- 38.Luo JL, Maeda S, Hsu LC, Yagita H, Karin M. Inhibition of NF-κB in cancer cells converts inflammation- induced tumor growth mediated by TNFα to TRAIL-mediated tumor regression. Cancer Cell. 2004;6:297–305. doi: 10.1016/j.ccr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 39.Senftleben U, et al. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
References
- 1.Isaacs JT. The biology of hormone refractory prostate cancer. Why does it develop? Urol Clin North Am. 1999;26:263–273. doi: 10.1016/s0094-0143(05)70066-5. [DOI] [PubMed] [Google Scholar]
- 2.Gulley J, Figg WD, Dahut WL. Treatment options for androgen-independent prostate cancer. Clin Adv Hematol Oncol. 2003;1:49–57. [PubMed] [Google Scholar]
- 3.Tran C, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jin RJ, et al. The nuclear factor-kappaB pathway controls the progression of prostate cancer to androgen-independent growth. Cancer Res. 2008;68:6762–6769. doi: 10.1158/0008-5472.CAN-08-0107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karin M, Lawrence T, Nizet V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell. 2006;124:823–835. doi: 10.1016/j.cell.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 6.Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 7.Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7:211–217. doi: 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–867. doi: 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Visser KE, Korets LV, Coussens LM. De novo carcinogenesis promoted by chronic inflammation is B lymphocyte dependent. Cancer Cell. 2005;7:411–423. doi: 10.1016/j.ccr.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 10.Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. 2008;454:436–444. doi: 10.1038/nature07205. [DOI] [PubMed] [Google Scholar]
- 11.Luo JL, et al. Nuclear cytokine activated IKKα controls prostate cancer metastasis by repressing maspin. Nature. 2007;446:690–694. doi: 10.1038/nature05656. [DOI] [PubMed] [Google Scholar]
- 12.Bai A, Higham E, Eisen HN, Wittrup KD, Chen J. Rapid tolerization of virus-activated tumor-specific CD8+ T cells in prostate tumors of TRAMP mice. Proc Natl Acad Sci USA. 2008;105:13003–13008. doi: 10.1073/pnas.0805599105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ellwood-Yen K, et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell. 2003;4:223–238. doi: 10.1016/s1535-6108(03)00197-1. [DOI] [PubMed] [Google Scholar]
- 14.Park BK, et al. NF-kappaB in breast cancer cells promotes osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSF. Nat Med. 2007;13:62–69. doi: 10.1038/nm1519. [DOI] [PubMed] [Google Scholar]
- 15.Wen D, et al. A selective small molecule IkappaB Kinase beta inhibitor blocks nuclear factor kappaB-mediated inflammatory responses in human fibroblast-like synoviocytes, chondrocytes, and mast cells. J Pharmacol Exp Ther. 2006;317:989–1001. doi: 10.1124/jpet.105.097584. [DOI] [PubMed] [Google Scholar]
- 16.Legler DF, et al. B cell-attracting chemokine 1, a human CXC chemokine expressed in lymphoid tissues, selectively attracts B lymphocytes via BLR1/CXCR5. J Exp Med. 1998;187:655–660. doi: 10.1084/jem.187.4.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen T, Wang LH, Farrar WL. Interleukin 6 activates androgen receptor-mediated gene expression through a signal transducer and activator of transcription 3-dependent pathway in LNCaP prostate cancer cells. Cancer Res. 2000;60:2132–2135. [PubMed] [Google Scholar]
- 18.Eriksen KW, et al. Constitutive STAT3-activation in Sezary syndrome: tyrphostin AG490 inhibits STAT3-activation, interleukin-2 receptor expression and growth of leukemic Sezary cells. Leukemia. 2001;15:787–793. doi: 10.1038/sj.leu.2402093. [DOI] [PubMed] [Google Scholar]
- 19.Hamel K, et al. Suppression of proteoglycan-induced arthritis by anti-CD20 B Cell depletion therapy is mediated by reduction in autoantibodies and CD4+ T cell reactivity. J Immunol. 2008;180:4994–5003. doi: 10.4049/jimmunol.180.7.4994. [DOI] [PubMed] [Google Scholar]
- 20.Worm MM, Tsytsykova A, Geha RS. CD40 ligation and IL-4 use different mechanisms of transcriptional activation of the human lymphotoxin alpha promoter in B cells. Eur J Immunol. 1998;28:901–906. doi: 10.1002/(SICI)1521-4141(199803)28:03<901::AID-IMMU901>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 21.Tumanov AV, et al. Dissecting the role of lymphotoxin in lymphoid organs by conditional targeting. Immunol Rev. 2003;195:106–116. doi: 10.1034/j.1600-065x.2003.00071.x. [DOI] [PubMed] [Google Scholar]
- 22.Lee Y, et al. Recruitment and activation of naive T cells in the islets by lymphotoxin beta receptor-dependent tertiary lymphoid structure. Immunity. 2006;25:499–509. doi: 10.1016/j.immuni.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 23.Ghiringhelli F, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 24.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 25.Sakurai T, et al. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell. 2008;14:156–165. doi: 10.1016/j.ccr.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nimmerjahn F, Ravetch JV. Fc-receptors as regulators of immunity. Adv Immunol. 2007;96:179–204. doi: 10.1016/S0065-2776(07)96005-8. [DOI] [PubMed] [Google Scholar]
- 27.Bonizzi G, et al. Activation of IKKα target genes depends on recognition of specific κB binding sites by RelB:p52 dimers. Embo J. 2004;23:4202–4210. doi: 10.1038/sj.emboj.7600391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kortylewski M, Yu H. Stat3 as a potential target for cancer immunotherapy. J Immunother. 2007;30:131–139. doi: 10.1097/01.cji.0000211327.76266.65. [DOI] [PubMed] [Google Scholar]
- 29.Smith PC, Hobisch A, Lin DL, Culig Z, Keller ET. Interleukin-6 and prostate cancer progression. Cytokine Growth Factor Rev. 2001;12:33–40. doi: 10.1016/s1359-6101(00)00021-6. [DOI] [PubMed] [Google Scholar]
- 30.Liu X, Plummer SJ, Nock NL, Casey G, Witte JS. Nonsteroidal antiinflammatory drugs and decreased risk of advanced prostate cancer: modification by lymphotoxin alpha. Am J Epidemiol. 2006;164:984–989. doi: 10.1093/aje/kwj294. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.