

Abstract
Transplant vasculopathy (TV) is an accelerated form of atherosclerosis resulting in chronic rejection of vascularized allografts. The causes of TV are multifactorial and integrate at the level of the vascular wall, leading to a phenotypic switch of endothelial (EC) and smooth muscle cells (SMC). A20 is a NF-κB dependent stress response gene in EC and SMC with potent anti-inflammatory effect in both cell types through blockade of NF-κB. A20 expression in EC and SMC correlates with the absence of TV in rat kidney allografts and long term functioning human kidney allografts.
We demonstrate that A20 protects EC from TNF, Fas and NK cell-mediated apoptosis by inhibiting proteolytic cleavage of caspase 8. A20 also safeguards EC from complement-mediated necrosis. Hence, effectively shutting down cell death pathways initiated by inflammatory and immune offenders associated with TV.
In contrast, A20 sensitizes SMC to cytokine and Fas mediated apoptosis through a novel nitric oxide (NO)-dependent mechanism. The unexpected pro-apoptotic effect of A20 in SMC translates in vivo by the regression of established neointimal carotid lesions following balloon angioplasty in rats. Antedating apoptosis of SMC, expression of the inducible nitric oxide synthase increases in A20 expressing neointimal SMC, corroborating the involvement of NO in causing the pro-apoptotic effect of A20 in SMC.
Combined anti-inflammatory and anti or pro-apoptotic functions of A20 in EC and SMC respectively qualify the positive effect of A20 upon vascular remodeling and healing. We propose that A20 based therapies may be effective in prevention and treatment of TV.
INTRODUCTION
The availability of effective immunosuppressive regimens in transplantation has drastically reduced the failure of vascularized grafts due to acute rejection (<10%). Despite this progress, the United States National Organ Sharing reports indicate that kidney and heart allografts continue to fail at a rate of approximately 5% each year post-transplantation (1, 2). Late graft failure is secondary to chronic rejection, characterized by the development of a pathognomonic occlusive vasculopathy, termed transplant-vasculopathy (TV). TV is particularly dramatic in cardiac transplant recipients and has become the principle cause of late death and allograft dysfunction (3). Pathologically, with the exception of foam cell development, occlusive lesions of TV closely resemble that of atherosclerotic plaques. TV is viewed as a form of accelerated atherosclerosis, resulting from chronic inflammatory processes initiated by the immune response to the allograft (4). Two cardinal manifestations underscore the pathogenesis of TV: Acquisition of an inflammatory phenotype by EC (5). EC apoptosis may ensue, resulting in exposure of the subendothelial matrix and enhancement of the inflammatory reaction; Increased SMC proliferation with the acquisition by the SMC of a synthetic instead of a contractile phenotype with increased matrix production lead to hyperplasia of the neointima (6). Defective SMC apoptosis amplifies the proliferative process (7). Numerous approaches to treating TV have been largely unsuccessful (8). Recent data suggest that the fate of the graft is not only controlled by the intensity of the host immune and non-immune responses but also by the ability of the graft to protect itself from injury (9). The active participation of the graft to its own protection from TV has long been suggested, however it is only recently that a partial insight to the molecular basis of this protective phenotype has been unveiled (10). We have evidence that he 7Zn finger protein A20 is part of the protective phenotype of the vessel wall against TV. A20 is a NF-κB dependent stress response gene in EC and SMC with potent anti-inflammatory effect in both cell types through blockade of NF-κB(11, 12). However, its effect on apoptosis is rather cell type and stimulus specific. A20 expression in EC and SMC correlates with the absence of TV in rat kidney allografts and long term functioning human kidney allografts (13).
METHODS AND RESULTS
In EC, A20 serves a broad anti-apoptotic function. A20 protects EC from TNF, Fas and NK cell-mediated apoptosis by inhibiting proteolytic cleavage of the initiator caspase 8. A20 also safeguards EC from complement-mediated necrosis. As such, A20 affords broad EC protective functions by effectively shutting down cell death pathways initiated by inflammatory and immune offenders associated with TV(14).
In contrast, A20 sensitizes SMC to cytokine and Fas mediated apoptosis. By FACS analysis of DNA contact, we demonstrate that the percentage of apoptotic human aortic smooth muscle cells (SMC) 48h following cytokine treatment was negligible in non-transduced (NT) and recombinant adenovirus (rAd,.β-galactoside-transduced SMC (<6%) while it increased significantly in A20 expressing SMC from 4.0±0.5% to 36.6±4.2% 48h following cytokine treatment (P<0.0001). Despite equivalent NF-κB inhibition, S3MC expressing IκBα demonstrated less apoptosis than A20 expressing cells. The percentage of apoptosis in SMC expressing IκBα increased from 3.0±1.0% to 13.0±3.0% (P=0.001) (Table I). This unexpected pro-apoptotic effect of A20 in SMC extended to the cross-linking of the death receptor Fas. The percentage of apoptotic cells following treatment with 1μg/ml of αFas was negligible in NT and rAd.β-gal-transduced SMC (<2.2%) while a high rate of apoptosis was detected in A20 expressing SMC, increasing from 2.5±0.5% to 26.0±8.0% after addition of αFas (n=3; P<0.0001; Table II). This novel pro-apoptotic function of A20 in SMC is nitric oxide (NO)-dependent. Pretreatment of rAd.A20 transduced SMC with the NO synthase inhibitor L-NAME for 6h prior to adding cytokines led to a dose-dependent reversal of the pro-apoptotic effect of A20. The rate of apoptosis increased from 4.5±0.8% to 16.0±1.3% (n=4; P<0.0001) following addition of cytokines. Pre-incubation of SMC with 2.5mM and 5.0mM of L-NAME prior to adding cytokines decreased SMC apoptosis to 9.4±0.9% (n=4; P<0.0003) and 5.2±0.2% (n=3; P<0.0001), respectively (Table III). The unexpected pro-apoptotic effect of A20 in SMC translates in vivo by the regression of established neointimal carotid lesions following balloon angioplasty injury in rats. Gene transfer of A20 to neointimal SMC within occlusive carotid lesions leads to increased apoptosis of neointimal SMC as evaluated by TUNEL within 3 to 5 days of gene transfer, followed by a remarkable regression of neointimal lesions as evaluated by the drastic decrease in intima over media (I/M) ratio (Figure 1 and 2). The I/M ratio 14 days following gene transfer in saline and rAd.β-gal transduced carotids reached 1.06±0.10 (n=8) and 1.04±0.16 (n=6), respectively, and was not different from the I/M ratio measured prior to gene transfer. In sharp contrast, injured carotids expressing A20 showed a reduction of their I/M ratio to 0.49±0.06 (n=7; P=0.0003 vs. NT and P=0.008 vs. rAd.β-gal). Antedating apoptosis of SMC, expression of the inducible NOS increases in A20 expressing neointimal SMC as shown by immunohistochemistry, corroborating the involvement of NO in causing the pro-apoptotic effect of A20 in SMC (Figure 2).
Table I.
Percentage of apoptosis in non-transduced (NT), rAd,A20, rAd.βgal and rAd.IκBα transduced SMC before and 48 hours following addition of cytokines (100U/ml of IL-1β, 400U/ml of TNF and 400U/ml IFNγ)
| Cytokines | NT | rAd.A20 | rAd.βgal | rAd.IκBα |
|---|---|---|---|---|
| 0 hours | <1% | 4±0.5% | <2% | 3±1% |
| 48 hours | <6% | 36.6±4.2% | <6% | 13±3% |
Table II.
Percentage of apoptosis in non-transduced (NT), rAd,A20 and rAd.βgal transduced SMC before and 48 hours following addition of anti-Fas antibody (1μg/ml).
| α- FAS | NT | rAd.A20 | rAd.βgal |
|---|---|---|---|
| 0 hours | <1% | 2.5±0.5% | <1% |
| 48 hours | <2.2% | 26±8% | <2.2% |
Table III.
Percentage of apoptosis in rAd.A20 transduced SMC 48 hours following treatment with cytokines in the presence or absence of the nitric synthase inhibitor L-NAME.
| L-NAME | 0 | 2.5 mM | 5mM |
|---|---|---|---|
| 48 hours Cytokines | 16±1.3% | 9.4±0.9% | 5.2±0.2% |
Figure 1.
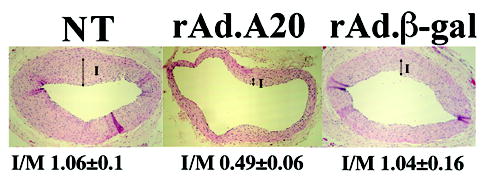
Evaluation of I/M ratio on H&E stained rat carotid arteries sections at 28+14 days following balloon angioplasty demonstrates regression of neointima in rAd.A20 transduced vessels as compared to NT and rAd.β-gal transduced vessels. Arrows define the Intima (I) (100X).
Figure 2.
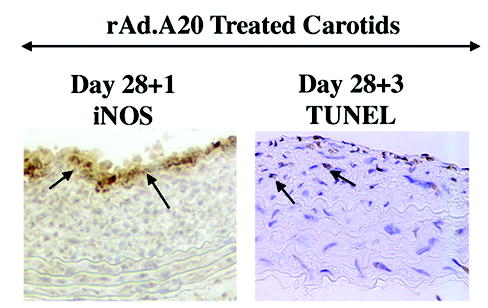
Significant iNOS immunostaining is detected in rAd.A20 transduced vessels at day 28+1, antedating increased TUNEL positive cells at day 28+3 within the neointima of these vessels. Representative sections are shown (400X).
DISCUSSION
EC are the first target of the inflammatory and immune attack encountered by vascularized organs. Damage to EC is at the center of the pathophysiology of TV (15). A therapy aimed at protecting from TV should display anti-inflammatory and anti-death functions. Our data indicate that A20 fulfills these criteria. We have demonstrated that A20 has broad and potent anti-inflammatory functions in EC via blockade of NF-κB in response to stimuli relevant to TV including TNF, CD40/CD40L cognate interactions, oxidative stress and thrombin (11, 16, 17). We have also dissected the cytoprotective function of A20 in EC and demonstrated that it protects from death stimuli encountered by vascularized allografts. All these data justify the pursuit A20-based therapies to halt TV at the level of the grafted endothelium.
In addition, we demonstrate that expression of A20 in SMC equally inhibits NF-κB activation. In SMC, NF-κB is essential for the acquisition of a pro-inflammatory SMC phenotype, promoting migration into the neointima, proliferation and leukocytes migration into TV lesions. Concommitant to NF-κB activation, A20 inhibits the subsequent up-regulation of pro-atherogenic NF-κB dependent genes such as ICAM-1 and MCP-1 and also A20 SMC proliferation via increased expression of the cyclin dependent kinase inhibitors p21waf1 and p27kip1(12). Further, we demonstrate that A20 expression in SMC results in sensitization to cytokine and Fas mediated apoptosis through a novel NO-dependent mechanism.
Combined anti-inflammatory and anti or pro-apoptotic functions of A20 in EC and SMC respectively qualify the positive effect of A20 upon vascular remodeling and healing. We propose that A20 based therapies may be highly effective in prevention and treatment of TV.
Acknowledgments
This work was supported by grant RO1 HL080130-A1 from the National Institutes of Health (NIH/NHLBI) and grant 124720319 from the Roche Organ Transplant Research Foundation (ROTRF) to CF. VIP; DTS, GVS and MDF are recipients of NRSA fellowship awards from the T32 NIH/NHLBI T32 HL07734
References
- 1.Cecka JM. The UNOS scientific renal transplant registry. Clin Transpl. 1999;13:1. [PubMed] [Google Scholar]
- 2.Keck BM, Bennett LE, Rosendale J, Daily OP, Novick RJ, Hosenpud JD. Worldwide thoracic organ transplantation: a report from the UNOS/ISHLT international registry for thoracic organ transplantation. Clin Transplant. 1999;13:35. [PubMed] [Google Scholar]
- 3.Behrendt D, Ganz P, Fang JC. Cardiac allograft vasculopathy. Curr Opin Cardiol. 2000;15 (6):422. doi: 10.1097/00001573-200011000-00009. [DOI] [PubMed] [Google Scholar]
- 4.Tullius SG, Tilney NL. Both alloantigen-dependent and -independent factors influence chronic allograft rejection. Transplantation. 1995;59:2126. [PubMed] [Google Scholar]
- 5.Gimbrone MAJ. Vascular endothelium: an integrator of pathophysiologic stimuli in atherosclerosis. Am J Card. 1995;75 (6):67B. doi: 10.1016/0002-9149(95)80016-l. [DOI] [PubMed] [Google Scholar]
- 6.Rekhter MD, Gordon D. Active proliferation of different cell types, including lymphocytes, in human atherosclerotic plaques. Am J Pathol. 1995;147:668. [PMC free article] [PubMed] [Google Scholar]
- 7.Pollman MJ, Hall JL, Mann MJ, Zhang L, Gibbons GH. Inhibition of neointimal cell bcl-x expression induces apoptosis and regression of vascular disease. Nature Med. 1998;4:222. doi: 10.1038/nm0298-222. [DOI] [PubMed] [Google Scholar]
- 8.Hayry P. New targets for the prevention of transplant vascular disease. Transplant Proc. 2001;33 (3):2332. doi: 10.1016/s0041-1345(01)02012-7. [DOI] [PubMed] [Google Scholar]
- 9.Bach FH, Hancock WW, Ferran C. Protective genes expressed in endothelial cells: a regulatory response to injury. Immunol Today. 1997;18:483. doi: 10.1016/s0167-5699(97)01129-8. [DOI] [PubMed] [Google Scholar]
- 10.Ferran C. Protective genes in the vessel wall: modulators of graft survival and function. Transplantation. 2006;82 (Suppl 1):S36. doi: 10.1097/01.tp.0000231445.62162.d5. [DOI] [PubMed] [Google Scholar]
- 11.Cooper JT, Stroka DM, Brostjan C, Palmetshofer A, Bach FH, Ferran C. A20 blocks endothelial cell activation through a NF-κB-dependent mechanism. J Biol Chem. 1996;271:18068. doi: 10.1074/jbc.271.30.18068. [DOI] [PubMed] [Google Scholar]
- 12.Patel VI, Daniel S, Longo CR, et al. A20, a modulator of smooth muscle cell proliferation and apoptosis, prevents and induces regression of neointimal hyperplasia. Faseb J. 2006;20 (9):1418. doi: 10.1096/fj.05-4981com. [DOI] [PubMed] [Google Scholar]
- 13.Kunter U, Floege J, von Jurgensonn AS, et al. Expression of A20 in the vessel wall of rat-kidney allografts correlates with protection from transplant arteriosclerosis. Transplantation. 2003;75 (1):3. doi: 10.1097/00007890-200301150-00002. [DOI] [PubMed] [Google Scholar]
- 14.Daniel S, Arvelo MB, Patel VI, et al. A20 protects endothelial cells from TNF-, Fas-, and NK-mediated cell death by inhibiting caspase 8 activation. Blood. 2004;104 (8):2376. doi: 10.1182/blood-2003-02-0635. [DOI] [PubMed] [Google Scholar]
- 15.Libby P, Pober JS. Chronic rejection. Immunity. 2001;14 (4):387. doi: 10.1016/s1074-7613(01)00119-4. [DOI] [PubMed] [Google Scholar]
- 16.Ferran C, Stroka DM, Badrichani AZ, et al. A20 inhibits NF-κB activation in endothelial cells without sensitizing to TNF-mediated apoptosis. Blood. 1998;91:2249. [PubMed] [Google Scholar]
- 17.Longo CR, Arvelo MB, Patel VI, et al. A20 protects from CD40-CD40 ligand-mediated endothelial cell activation and apoptosis. Circulation. 2003;108 (9):1113. doi: 10.1161/01.CIR.0000083718.76889.D0. [DOI] [PubMed] [Google Scholar]