

Abstract
Aberrant activation of the Wnt/β-catenin signaling pathway plays a crucial role in oncogenesis of various human malignancies. It has been demonstrated that there is a direct interaction between β-catenin and PPARγ. Here we examined the effects of fifteen reported PPAR ligands in a reporter gene assay that is dependent on β-catenin activation of TCF/LEF transcription factors; only the thiazolidinedione PPARγ agonists troglitazone, rosiglitazone and pioglitazone, and a non-thiazolidinedione PPARγ activator GW1929 inhibited β-catenin-induced transcription in a PPARγ dependent fashion. The results from mammalian one-hybrid experiments showed that functional PPARγ was necessary for ligand-dependent inhibition of β-catenin transactivation. However, a PPARγ activator Fmoc-Leu could not repress β-catenin-mediated signaling and its transactivation activity. These results indicate that activation of PPARγ is necessary, but not sufficient, for the β-catenin antagonistic activity of a PPARγ agonist, and that the inhibitory compounds interfere directly with β-catenin transactivation activity.
Keywords: PPARγ, β-catenin, troglitazone, rosiglitazone, GW1929
1. Introduction
The Wnt/β-catenin signaling pathway has a central role in development and tissue homeostasis, and is abnormally regulated in many human diseases, including cancer. The β-catenin molecule plays a crucial function in Wnt signal transduction (Clevers, 2006; Moon et al., 2004; Nusse, 2005; Polakis, 2000). The stability of free cytoplasmic β-catenin is tightly regulated by a destruction complex, which is composed of the adenomatosis polyposis coli protein (APC), the glycogen synthetase kinase-3β enzyme (GSK-3β), and the scaffolding protein axin. In the canonical Wnt pathway, extracellular Wnt proteins bind to a receptor complex, consisting of a member of the Frizzled (Fzd) family, and the low-density lipoprotein-receptor-related proteins (LRP) 5 or LRP6. Subsequently the cytoplasmic adapter protein disheveled (Dvl) binds to axin in the destruction complex, and prevents GSK-3β phosphorylation of β-catenin. The unphosphorylated β-catenin accumulates in the cytoplasm and translocates to the nucleus, where it interacts with T cell (TCF) and lymphoid-enhancing (LEF) factors to activate transcription of Wnt target genes including c-myc, cyclin D1 and many others (Crawford et al., 1999; He et al., 1998; Shtutman et al., 1999).
The peroxisome proliferator activated receptors (PPARs) belong to the superfamily of nuclear receptor transcription factors, and consist of three members, PPARα, PPARβ/δ and PPARγ (Kliewer et al., 2001). Upon activation by specific ligands, PPAR molecules from heterodimers with the retinoid-X receptor (RXR), to form a complex that regulates the transcriptional activity of target genes by binding to specific PPAR response element sequences (PPRE) within promoter regions. PPARγ influences diverse biological processes, including glucose homeostasis, lipid metabolism, cellular proliferation, and differentiation (Debril et al., 2001; Lehrke and Lazar, 2005). PPARγ ligands encompass a wide range of structurally diverse compounds, including naturally occurring and synthetic long chain polyunsaturated fatty acids, thiazolidinediones and some nonsteroidal anti-inflammatory drugs (NSAIDs) (Forman et al., 1995; Kliewer et al., 1995; Kliewer et al., 1997; Lehmann et al., 1995). In addition to its ligand-dependent activator function, PPARγ can repress other signaling pathways, such as AP-1, NFAT, NFκB, and STAT1, by direct interaction with other transcription factors, competition for coregulators, and blockade of corepressor clearance (Pascual et al., 2005; Welch et al., 2003).
Previous studies have demonstrated crosstalk between the β-catenin and PPARγ signaling pathways. Girnun et al. reported that PPARγ could suppress β-catenin levels and colon carcinogenesis, but only before mutational damage to the APC/β-catenin pathway was evident (Girnun et al., 2002). Other studies showed that β-catenin could directly interact with PPARγ and RXRα (Jansson et al., 2005; Liu et al., 2006; Lu et al., 2005; Xiao et al., 2003). The functional interaction between β-catenin and PPARγ involved the TCF/LEF binding domain of β-catenin and a catenin binding domain (CBD) within PPARγ itself (Liu et al., 2006). Earlier experiments from our laboratory indicated that repression of β-catenin signaling by NSAIDs required high-level expression of PPARγ and RXRα (Lu et al., 2005). These studies indicated that anti-cancer activity of PPARγ agonist may relate to its ability to inhibit β-catenin signaling. The PPARγ ligands with the potential to inhibit β-catenin signaling should be candidate agents for chemoprevention and chemotherapy. Thus, it is important to determine the effects of PPARγ ligands with diverse chemical structures on the β-catenin signaling pathway.
In this study, we examined the effects of a series of PPAR ligands on β-catenin signaling. The thiazolidinedione PPARγ agonists, including troglitazone, rosiglitazone and pioglitazone, and a non-thiazolidinedione PPARγ activator, GW1929, all inhibited β-catenin-mediated signaling. These PPARγ ligands also inhibited the transactivation activity of β-catenin. Both effects required expression of a functional PPARγ protein. However, other compounds that activated a PPARγ dependent reporter gene, as well as a PPARα agonist, had no β-catenin antagonistic activity. The results indicate that activation of PPARγ is necessary but not sufficient for PPARγ ligand-mediated repression of β-catenin signaling.
2. Materials and Methods
2.1. Chemical reagents
GW1929 (N-(2-Benzoylphenyl)-O-[2-(methyl-2-pyridinylamino)ethyl ]-L-tyrosine hydrochloride), GW9662 (2-Chloro-5-nitro-N-phenylbenzamide), tetradecylthioacetic acid, Fmoc-Leu, L-165.041 (4-[3-(4-Acetyl-3-hydroxy-2-propylphenoxy)propoxy]phenoxyacetic acid) and Azelaoyl PAF were purchased from Sigma-Aldrich (St. Louis, MO). T0070907 (2-chloro-5-nitro-N-4-pyridinyl-benzamide) and LY171883 (1-[2-hydroxy-3-propyl-4-[4-(1H-tetrazol-5-yl)butoxy]phenyl]-ethanone) were purchased from Biomol Research Laboratories (Plymouth Meeting, PA). Rosiglitazone, pioglitazone, MCC-555 (5-[[6-[(2-fluorophenyl)methoxy]-2-naphthalenyl]methyl]-2,4-thiazolidinedione), and GW501516 ([2-methyl-4-[[[4-methyl-2-[4-(trifluoromethyl)phenyl]-5-thiazolyl]methyl]thio]phenoxy]-acetic acid) were obtained from Axxora (San Diego, CA). Pseudolaric acid B and PPARγ antagonist III (G3335, dipeptide H-Trp-Glu-OH) were obtained from Calbiochem (La Jolla, CA). Troglitazone was a gift from Dr. David W. Rose (University of California San Diego).
2.2. Plasmids
The reporter plasmids TOPflash, (AOX)3-TK-Luc and UAS-TK-Luc, and the expression vectors for disheveled (Dvl), β-catenin, PPARγ, PPARγEA469, RXRα, VP16-PPARγ and Gal4-PBP were previously described (Lu et al., 2005; Lu et al., 2004). The Gal4-β-catenin expression vector was prepared by ligating β-catenin cDNA into the SalI and BamHI sites of a Gal4 expression vector. The construct was sequenced to confirm its identity.
2.3. Cell culture and Transfection
The human embryonic kidney cell line HEK293 (American Type Culture Collection, Rockville, MD) was transfected using the FuGene transfection reagent (Roche Diagnostics GmbH, Mannheim, Germany) according to the manufacturer's instructions.
HEK293 cells were grown for at least 24 h in 12-well plates prior to transfection. At about 50% confluence, cells were transfected with 0.5 μg of reporter plasmid, 0.1 μg of control plasmid pCMXβgal, 0.1–0.2μg expression plasmids, and carrier DNA pcDNA3 plasmid for a total of 1 μg/well. After overnight incubation, the cells were washed and given fresh medium that contained 0.5% FBS supplemented with the different PPARγ ligands or solvent (DMSO). After 24 h, the cells were lysed in isotonic potassium phosphate buffer, pH 7.8, containing 1% Triton X-100, and luciferase activities were assayed in the presence of substrate using a microtiter plate luminometer (MicroBeta TriLux, Gaithersburg, MD). The luciferase values were normalized for variations in transfection efficiency using the β-galactosidase internal control. In the Results section, data are expressed as percentage of control luciferase or fold stimulation of luciferase activity compared to the basal level. All the transfection results represent means of a minimum of three independent transfections assayed in duplicate, ± the standard error of the mean (S.E.M.).
3. Results
3.1. Repression of β–catenin signaling by PPARγ ligands
Our previous study demonstrated that NSAIDs could inhibit β-catenin signaling in a PPARγ-dependent manner. The presence of RXRα strongly enhanced the β-catenin antagonistic activity of the NSAIDs (Lu et al., 2005). In order to determine if other PPAR ligands could modulate β-catenin signaling, 15 reported PPAR ligands were tested in a cell-based TOPflash reporter assay as described previously (Lu et al., 2005). The TOPflash reporter construct was transfected into HEK293 cells with expression plasmids for Dvl, PPARγ and RXRα. As noted earlier, Dvl is an upstream activator of the β-catenin pathway and activates the TCF/LEF response elements by elevating β-catenin levels. Cells were treated with increasing concentrations of PPAR ligands (0.625-10 μM) for 24 h. As shown in Table 1, the thiazolidinedione PPARγ agonists, including troglitazone, rosiglitazone and pioglitazone, and a non-thiazolidinedione PPARγ activator, GW1929, significantly inhibited Dvl-stimulated transcription. In contrast, PPARγ antagonists and selective agonists for PPARα and PPARδ had little or no effect on Dvl-mediated TOPflash reporter activity (Table 1); nor did the PPARγ activators azelaoyl PAF, MCC-555, pseudolaric acid B or Fmoc-Leu repress β-catenin-mediated signaling. Since troglitazone, rosiglitazone and GW1929 displayed the most potent inhibitory effects on β-catenin signaling, they were chosen for further experimentation. Fmoc-Leu was also chosen for further studies because of its opposite effect on β-catenin-mediated signaling.
Table 1.
Inhibition of β–catenin signaling by PPAR ligands
| Drugs | Functions | Percent control (%) | ||||
|---|---|---|---|---|---|---|
| 0.625 μM | 1.250 μM | 2.500 μM | 5.000 μM | 10.00 μM | ||
| Troglitazone | PPARγ agonist | 47.5±3.5 | 35.9±0.1 | 32.7±0.1 | 29.3±0.6 | 28.1±0.3 |
| Rosiglitazone | PPARγ agonist | 29.9±1.6 | 27.8±1.4 | 27.6±0.4 | 27.1±1.1 | 27.8±0.9 |
| Pioglitazone | PPARγ agonist | 40.7±1.6 | 35.0±1.3 | 34.2±0.7 | 35.0±1.8 | 40.7±1.0 |
| GW1929 | PPARγ agonist | 30.2±1.6 | 29.1±1.1 | 28.7±1.6 | 27.4±2.1 | 27.4±1.5 |
| Azelaoyl PAF | PPARγ agonist | 107.6±6.6 | 102.0±6.3 | 95.6±6.4 | 97.5±9.8 | 93.2±4.6 |
| Fmoc-Leu | PPARγ agonist | 126.0±1.4 | 139.9±1.1 | 166.9±2.6 | 183.7±11.8 | 192.6±6.3 |
| MCC-555 | PPARγ agonist | 164.6±3.4 | 132.5±4.0 | 108.6±0.8 | 109.0±3.0 | 123.7±3.3 |
| Pseudolaric acid B | PPARγ & PPAR δ agonist | 114.1±5.0 | 82.2±5.5 | 63.3±3.2 | 61.2±7.0 | 60.7±5.2 |
| Tetradecylthioacetic acid | PPAR agonist | 90.6±5.9 | 92.4±2.4 | 95.2±3.2 | 97.8±4.8 | 94.9±3.3 |
| L165.041 | PPARδ agonist | 95.7±4.5 | 83.6±2.2 | 74.8±4.3 | 64.4±1.9 | 53.7±0.6 |
| GW501516 | PPARδ agonist | 90.6±6.2 | 87.2±3.6 | 84.9±4.9 | 80.0±2.9 | 75.5±4.5 |
| LY171883 | PPARα agonist | 82.6±14.7 | 88.7±3.2 | 88.7±2.4 | 78.3±4.0 | 70.9±3.5 |
| GW9662 | PPARγ antagonist | 97.7±0.1 | 110.8±3.4 | 136.5±7.7 | 167.5±2.2 | 212.8±1.5 |
| T0070907 | PPARγ antagonist | 108.8±6.3 | 106.6±10.5 | 115.0±4.3 | 118.1±3.8 | 120.1±7.5 |
| PPAR antagonist III (G3335) | PPARγ antagonist | 108.5±3.0 | 95.4±3.3 | 94.5±5.3 | 91.2±4.7 | 93.7±3.1 |
HEK293 cells were transfected with TOPflash reporter and expression plasmids encoding Dvl, PPARγ, and RXRα. After overnight incubation, the cells were treated for 24 h with increasing concentrations of PPAR ligands. The reported data are the mean percentages of TOPflash activity relative to cells treated with the vehicle alone (vehicle treatment = 100%) ±S.E.M.
The activation function 2 (AF-2) domain of nuclear receptors is necessary for ligand-dependent transactivation. To determine the role of AF-2 in PPARγ ligand-mediated inhibition of β-catenin signaling, a mutant PPARγ, which contains a point mutation in the AF2 domain that destroys its transactivation, transrepression, and coactivator activity, was analyzed in cotransfection assays (Li et al., 2000). The PPARγ ligands were tested over a wide range of concentrations (5 nM–5 μM). As shown in Figure 1, in the presence of wild-type PPARγ and RXRα, three PPARγ agonists (Troglitazone, Rosiglitazone and GW1929) inhibited β-catenin signaling in a concentration dependent manner. In contrast, their inhibitory effects were completely blocked by the mutant PPARγ (Fig. 1B, 1C and 1D). This result reveals that PPARγ agonist-induced repression of β–catenin signaling requires a functional activated PPARγ. Interestingly, the PPARγ activator Fmoc-Leu had the opposite effect on β-catenin-mediated transcription in the presence of wild-type PPARγ or mutant PPARγ (Fig. 1E).
Fig. 1.
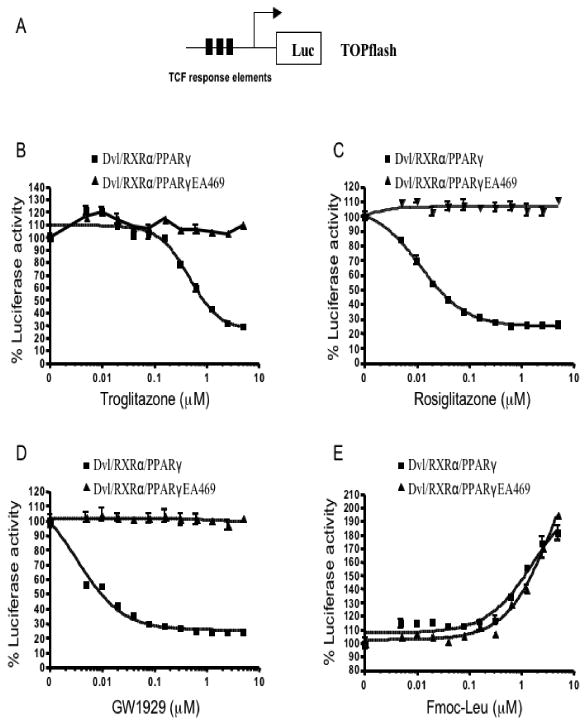
PPARγ agonists repress β-catenin-mediated signaling in a PPARγ dependent manner. (A) A schematic representation of the TOPflash reporter used in this study. TOPflash reporter was transfected into HEK293 cells with expression plasmids for Dvl, RXRα and PPARγ or PPARγEA469 as indicated. After overnight incubation, transfected cells were treated for 24 h with increasing concentrations of troglitazone (B), rosiglitazone (C), GW1929 (D) and Fmoc-Leu (E), after which the reporter gene activity was measured. The results are expressed as % control TOPflash reporter activity ±S.E.M.
3.2. Transcriptional activity of PPARγ and β-catenin inhibition
To determine whether PPARγ agonist-induced repression of β-catenin signaling correlated with the capacity to activate PPARγ dependent transcription, we compared the effects of troglitazone, rosiglitazone, GW1929 and Fmoc-Leu on PPARγ activation using a peroxisome proliferator response element (PPRE)-driven promoter reporter, (AOX)3-TK-Luc. All four PPARγ activators increased the transcriptional activity of the PPARγ reporter (Fig. 2). Fmoc-Leu was the most potent inducer in the PPRE-luciferase reporter system, but had no repressive effect on β-catenin-mediated signaling (Table 1 & Fig. 1E). Thus, activation of PPARγ dependent transcription is necessary but not sufficient for the β-catenin antagonistic activity of PPARγ agonists.
Fig. 2.
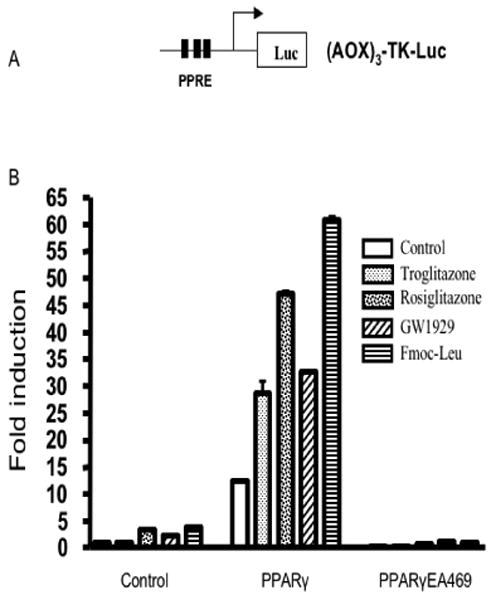
PPARγ agonists stimulate PPRE reporter gene transcription. (A) A schematic representation of the PPRE reporter (AOX)3-TK-Luc. (B) HEK293 cells were transfected with (AOX)3-TK-Luc reporter with or without PPARγ or PPARγEA469, and then were incubated with 5 μM of different PPARγ ligands, as indicated. The fold increase in luciferase activities ±S.E.M. was determined.
3.3. PPARγ agonist-induced interaction of PPARγ with PBP
The PPAR-binding protein (PBP) act as an anchor for the thyroid hormone receptor-associated protein/vitamin D3 receptor-interacting protein (DRIP)/activator-recruited cofactor complex. To examine the effects of PPARγ agonists on the interaction of PPARγ with PBP, a mammalian two-hybrid assay comprised of Gal4-PBP and VP16-PPARγ was used. In this assay, the reporter plasmid UAS-TK-Luc contains five copies of the Gal4 binding element. As shown in Figure 3, treatment with troglitazone, rosiglitazone and GW1929 induced an interaction between PPARγ and PBP that was 3.8-7.6 fold above the activity observed in the absence of ligands. No signal was detected in the presence of the VP16-PPARγ fusion factor alone. The PPARγ activator Fmoc-Leu, however, had no effect in this reporter assay (Fig. 3), suggesting that its binding to PPARγ could not induce its interaction with PBP.
Fig. 3.
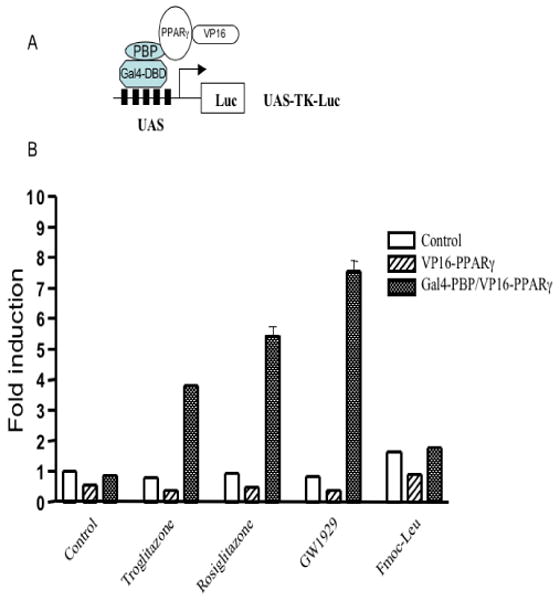
PPARγ agonists induce the interaction of PPARγ with its binding protein PBP. (A) Schematic representation of the mammalian two-hybrid system used in this study. (B) The reporter plasmid UAS-TK-Luc was transfected into HEK293 cells with a VP16-PPARγ expression plasmid, either alone or with VP16-PPARγ and Gal4-PBP. Transfected cells were treated with different PPARγ activators (5 μM each) as indicated. Cells were harvested 24 h after treatment, and then luciferase activities were determined.
3.4. PPARγ agonist-induced repression of the transactivation activity of β-catenin
To determine directly the effects of PPARγ agonists on β–catenin transactivation activity, a plasmid encoding a Gal4 DNA-binding domain (DBD) fused to β–catenin (Gal4-β-catenin) was constructed. In this system, the Gal4-β-catenin fusion protein-induced activation of a UAS-TK-Luc reporter was dependent on the activation domain of β-catenin. Transfection of an expression plasmid for Gal4-β-catenin led to more than 10-fold increase in expression of reporter gene (Fig. 4). Treatment with PPARγ agonists had little or no direct effect on reporter gene activity (Fig. 4). However, in the presence of PPARγ and RXRα, troglitazone, rosiglitazone and GW1929, but not Fmoc-Leu, suppressed the transactivation activity of β-catenin. Mutant PPARγ completed abolished PPARγ agonist-mediated repression of β-catenin transactivation (Fig. 4). These results confirm that PPARγ ligand-mediated repression of β-catenin transactivation depends on a functional PPARγ.
Fig. 4.
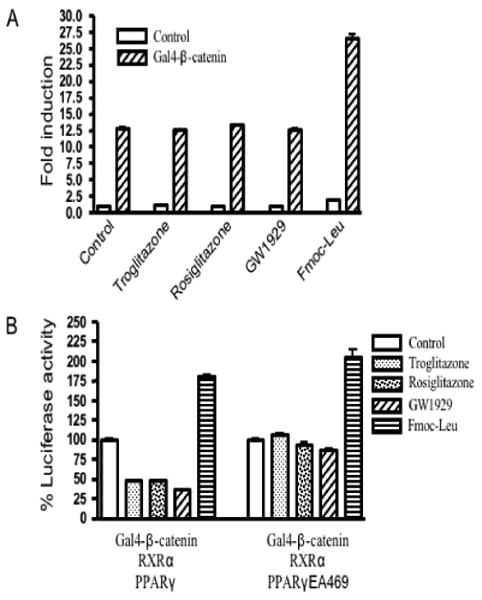
PPARγ agonists repress the transactivation activity of β–catenin in the presence of functional PPARγ. (A) The reporter gene UAS-TK-Luc was transfected into HEK293 cells, together with a Gal4-β-catenin expression plasmid. Then cells were exposed to 5 μM troglitazone, 5 μM rosiglitazone, 5 μM GW1929, 5 μM Fmoc-Leu or vehicle alone for 24 h, harvested, and extracted for determination of luciferase activity. The results are expressed as fold induction of luciferase activity compared to the basal level ±S.E.M. (B) HEK293 cells were transfected with UAS-TK-Luc, the reporter construct, along with expression plasmids for Gal4-β-catenin, RXRα, PPARγ or PPARγEA469, as indicated. After transfection, cells were grown with the PPARγ agonists troglitazone, rosiglitazone, GW1929 and Fmoc-Leu (5 μM each), and assayed for luciferase activity 24 h later. The results are expressed as % control reporter activity ±S.E.M.
4. Discussion
Our previous studies demonstrated that NSAIDs inhibited Wnt/β-catenin signaling in a PPARγ and RXRα dependent manner (Lu et al., 2005). However, the mechanisms involved in PPARγ-mediated repression of β-catenin signaling are still largely unknown. Although PPARγ activation was reported to induce β-catenin degradation, we did not observe significant PPARγ-induced changes in β-catenin expression at the tested concentrations (data not shown). Instead, results from mammalian one-hybrid experiments showed that a functional PPARγ confers ligand-specific inhibition of β-catenin transactivation.
The PPARγ ligands have chemopreventative and/or chemotherapeutic activity against a wide variety of cancer cells (Grommes et al., 2004; Koeffler, 2003). There may be multiple mechanisms by which the PPARγ ligands exert their anti-cancer effect, such as by increasing expression of the cyclin dependent kinase inhibitors p21waf1 and p27kip1, and by decreasing levels of protein phosphatase 2A, cyclins D1 and E, inflammatory cytokines, and nuclear factor κB (Altiok et al., 1997; Koeffler, 2003; Koga et al., 2001; Wang et al., 2001). Here we showed that three thiazolidinedione PPARγ activators, troglitazone, rosiglitazone and pioglitazone, as well as a non-thiazolidinedione PPARγ activator, GW1929, all repressed β-catenin dependent transcription. The inhibition of the Wnt/β-catenin signaling mediated by PPARγ ligands, may contribute to the anti-cancer activity of these drugs. Importantly, the PPARγ ligand-induced repression of β–catenin signaling may be cell type-specific and depends on cellular levels of β–catenin, PPARγ and RXR. Further studies are needed to test the effects of PPARγ ligands on the Wnt/β-catenin signaling pathway in cancer cells with high levels of β-catenin, PPARγ and RXR.
Our studies reveal that PPARγ activation and β-catenin inhibition are not equivalent. Some PPARγ agonists such as Fmoc-Leu, azelaoyl PAF, MCC-555 and pseudolaric acid B had little or no repressive effects on β-catenin signaling. These results suggest that PPARγ ligand-induced inhibition of β-catenin signaling is not simply a consequence of the nuclear hormone receptor's transcriptional activity. Therefore, it would be useful to examine the repressive effect of all known PPARγ ligands on β-catenin signaling, in order to determine the structure activity relationship required for Wnt/β-catenin antagonism.
The PPARγ activator Fmoc-Leu is a N-protected leucine analogue, structurally different from the glitazones, which binds to and induces a change of PPARγ confirmation, resulting in the differential recruitment of co-regulators (Rocchi et al., 2001). Consistent with these previous findings, Fmoc-leu was incapable of inducing the interaction of PPARγ with PBP, unlike troglitazone, rosiglitazone and GW1929. Fmoc-Leu strongly activated the transcriptional activity of a PPRE reporter, but had no inhibitory effects on both β-catenin-mediated signaling and its transactivation activity. It is noteworthy that Fmoc-Leu actually enhanced β-catenin-mediated activation of TCF/LEF and its transactivation activity. This enhancing effect did not seem to depend on PPARγ binding because a similar effect was observed in the absence of PPARγ or in the presence of mutant PPARγ. Like Fmoc-Leu, a PPARγ antagonist GW9662 also exhibited an enhancing effect on β–catenin signaling (Table 1). Our preliminary result suggests that the enhancing effect of GW9662 is not dependent on PPARγ (data not shown).
Collectively, these results confirm that activation of PPARγ is not sufficient for ligand-induced repression of β-catenin signaling. One potential explanation for the data is that ligand-induced conformational changes of PPARγ lead to altered cofactor recruitment. The cofactors required for repression of β-catenin signaling may be different from those that regulate PPARγ transcriptional activity.
In summary, our studies provide insight but raise new questions regarding the mechanisms involved in PPARγ ligand-induced repression of β-catenin signaling. Some PPARγ agonists may bind to a β-catenin/PPARγ/RXRα complex and induce conformational changes that alter the pattern of cofactor recruitment. Further studies are required to characterize the roles of specific cofactors in PPARγ ligand-induced inhibition of the Wnt/β-catenin signal transduction pathway.
Acknowledgments
*We thank Howard B. Cottam, Guanyi Jin, and Michael Rosenbach for advice and technical assistance, and Nancy Noon for secretarial support. This work was supported in part by a Leukemia and Lymphoma Society SCOR grant (CA81534-06) and a NCI-DDG grant (CA113318-01).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Altiok S, Xu M, Spiegelman BM. PPARgamma induces cell cycle withdrawal: inhibition of E2F/DP DNA-binding activity via down-regulation of PP2A. Genes Dev. 1997;11:1987–1998. doi: 10.1101/gad.11.15.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Crawford HC, Fingleton BM, Rudolph-Owen LA, Goss KJ, Rubinfeld B, Polakis P, Matrisian LM. The metalloproteinase matrilysin is a target of beta-catenin transactivation in intestinal tumors. Oncogene. 1999;18:2883–2891. doi: 10.1038/sj.onc.1202627. [DOI] [PubMed] [Google Scholar]
- Debril MB, Renaud JP, Fajas L, Auwerx J. The pleiotropic functions of peroxisome proliferator-activated receptor gamma. Journal of molecular medicine (Berlin, Germany) 2001;79:30–47. doi: 10.1007/s001090000145. [DOI] [PubMed] [Google Scholar]
- Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell. 1995;83:803–812. doi: 10.1016/0092-8674(95)90193-0. [DOI] [PubMed] [Google Scholar]
- Girnun GD, Smith WM, Drori S, Sarraf P, Mueller E, Eng C, Nambiar P, Rosenberg DW, Bronson RT, Edelmann W, Kucherlapati R, Gonzalez FJ, Spiegelman BM. APC-dependent suppression of colon carcinogenesis by PPARgamma. Proc Natl Acad Sci U S A. 2002;99:13771–13776. doi: 10.1073/pnas.162480299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grommes C, Landreth GE, Heneka MT. Antineoplastic effects of peroxisome proliferator-activated receptor gamma agonists. The lancet oncology. 2004;5:419–429. doi: 10.1016/S1470-2045(04)01509-8. [DOI] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science New York, NY. 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- Jansson EA, Are A, Greicius G, Kuo IC, Kelly D, Arulampalam V, Pettersson S. The Wnt/beta-catenin signaling pathway targets PPARgamma activity in colon cancer cells. Proc Natl Acad Sci U S A. 2005;102:1460–1465. doi: 10.1073/pnas.0405928102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Sundseth SS, Jones SA, Brown PJ, Wisely GB, Koble CS, Devchand P, Wahli W, Willson TM, Lenhard JM, Lehmann JM. Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc Natl Acad Sci U S A. 1997;94:4318–4323. doi: 10.1073/pnas.94.9.4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Xu HE, Lambert MH, Willson TM. Peroxisome proliferator-activated receptors: from genes to physiology. Recent progress in hormone research. 2001;56:239–263. doi: 10.1210/rp.56.1.239. [DOI] [PubMed] [Google Scholar]
- Koeffler HP. Peroxisome proliferator-activated receptor gamma and cancers. Clin Cancer Res. 2003;9:1–9. [PubMed] [Google Scholar]
- Koga H, Sakisaka S, Harada M, Takagi T, Hanada S, Taniguchi E, Kawaguchi T, Sasatomi K, Kimura R, Hashimoto O, Ueno T, Yano H, Kojiro M, Sata M. Involvement of p21(WAF1/Cip1), p27(Kip1), and p18(INK4c) in troglitazone-induced cell-cycle arrest in human hepatoma cell lines. Hepatology Baltimore, Md. 2001;33:1087–1097. doi: 10.1053/jhep.2001.24024. [DOI] [PubMed] [Google Scholar]
- Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma) J Biol Chem. 1995;270:12953–12956. doi: 10.1074/jbc.270.22.12953. [DOI] [PubMed] [Google Scholar]
- Lehrke M, Lazar MA. The many faces of PPARgamma. Cell. 2005;123:993–999. doi: 10.1016/j.cell.2005.11.026. [DOI] [PubMed] [Google Scholar]
- Li M, Pascual G, Glass CK. Peroxisome proliferator-activated receptor gamma-dependent repression of the inducible nitric oxide synthase gene. Molecular and cellular biology. 2000;20:4699–4707. doi: 10.1128/mcb.20.13.4699-4707.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wang H, Zuo Y, Farmer SR. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Molecular and cellular biology. 2006;26:5827–5837. doi: 10.1128/MCB.00441-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Cottam HB, Corr M, Carson DA. Repression of beta-catenin function in malignant cells by nonsteroidal antiinflammatory drugs. Proc Natl Acad Sci U S A. 2005;102:18567–18571. doi: 10.1073/pnas.0509316102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu D, Zhao Y, Tawatao R, Cottam HB, Sen M, Leoni LM, Kipps TJ, Corr M, Carson DA. Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc Natl Acad Sci U S A. 2004;101:3118–3123. doi: 10.1073/pnas.0308648100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5:691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- Nusse R. Wnt signaling in disease and in development. Cell Res. 2005;15:28–32. doi: 10.1038/sj.cr.7290260. [DOI] [PubMed] [Google Scholar]
- Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, Rose DW, Willson TM, Rosenfeld MG, Glass CK. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–763. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837–1851. [PubMed] [Google Scholar]
- Rocchi S, Picard F, Vamecq J, Gelman L, Potier N, Zeyer D, Dubuquoy L, Bac P, Champy MF, Plunket KD, Leesnitzer LM, Blanchard SG, Desreumaux P, Moras D, Renaud JP, Auwerx J. A unique PPARgamma ligand with potent insulin-sensitizing yet weak adipogenic activity. Molecular cell. 2001;8:737–747. doi: 10.1016/s1097-2765(01)00353-7. [DOI] [PubMed] [Google Scholar]
- Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96:5522–5527. doi: 10.1073/pnas.96.10.5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Fu M, D'Amico M, Albanese C, Zhou JN, Brownlee M, Lisanti MP, Chatterjee VK, Lazar MA, Pestell RG. Inhibition of cellular proliferation through IkappaB kinase-independent and peroxisome proliferator-activated receptor gamma-dependent repression of cyclin D1. Molecular and cellular biology. 2001;21:3057–3070. doi: 10.1128/MCB.21.9.3057-3070.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch JS, Ricote M, Akiyama TE, Gonzalez FJ, Glass CK. PPARgamma and PPARdelta negatively regulate specific subsets of lipopolysaccharide and IFN-gamma target genes in macrophages. Proc Natl Acad Sci U S A. 2003;100:6712–6717. doi: 10.1073/pnas.1031789100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao JH, Ghosn C, Hinchman C, Forbes C, Wang J, Snider N, Cordrey A, Zhao Y, Chandraratna RA. Adenomatous polyposis coli (APC)-independent regulation of beta-catenin degradation via a retinoid X receptor-mediated pathway. J Biol Chem. 2003;278:29954–29962. doi: 10.1074/jbc.M304761200. [DOI] [PubMed] [Google Scholar]