

Abstract
The use of sub-nanometer resolution electron density as spatial constraints for denovo and ab-initio structure prediction requires knowledge of protein boundaries to accurately segment the electron density for the prediction algorithms. Here we present a procedure where even poorly segmented density can be used to determine the fold of the protein. The method is automated, fast, capable of searching for multiple copies of a protein fold, and accessible to densities encompassing more than a thousand residues. The automation is particularly powerful as it allows the procedure to take full advantage of the expanding repository in the Protein Data Bank. We have tested the method on nine segmented sub-nanometer image reconstruction electron densities. The method successfully identifies the correct fold for the six densities for which an atomic structure is known, identifies one fold that agrees with prior structural data, one fold that agrees with predictions from the Fold & Function Assignment server, and one fold that correlates with secondary structure prediction. The identified folds in the last three examples can be used as templates for comparative modeling of the bacteriophage P22 tail-machine (a 3 MDa complex composed of 39 proteins).
Keywords: Cryo-electron microscopy, fold prediction, comparative modeling
Introduction
Cryo-electron microscopy (cryo-EM) allows for direct visualization of large macromolecular complexes in their near native state. The automation of image acquisition and processing, image reconstruction, has allowed cryo-EM to routinely produce sub-nanometer resolution electron density of the molecule of interest (Bubeck et al., 2005; Frank et al., 1996; Lander et al., 2009b; Ludtke et al., 1999; Suloway et al., 2005; Yan et al., 2007). At sub-nanometer resolution it is possible to identify α-helices and β-sheets in the density (Jiang et al., 2001; Kong and Ma, 2003). Hence if a general mechanism of action involves secondary structure reorganization, it can be described from image reconstructions of the different functional states of a macromolecular complex. Accurate incorporation of atomic models into the image reconstruction can provide a more detailed interpretation of the complex's mechanism of action, its architecture and assembly, and its evolutionary history (Baker and Johnson, 1996; Khayat et al., 2005). There are currently four procedures to incorporate atomic models into the image reconstruction: 1) docking the crystal or NMR structure of individual subunits, 2) docking of comparative models of individual subunits, 3) docking de novo or ab-initio predicted structures, or 4) tracing the polypeptide chain from the electron density (Baker and Johnson, 1996; Chen et al., 2009; Das and Baker, 2008; Jiang et al., 2008). Comparative modeling requires an initial atomic structure with sufficient sequence identity to the protein under study to be used as a template. De novo or ab-initio predicted structures are currently limited to proteins smaller than 180 residues possessing a single domain (Helles, 2008). Tracing the polypeptide chain into the cryo-EM derived electron density is currently restricted to 4.5 Å or better resolution electron density (Chen et al., 2009; Jiang et al., 2008). Here we describe an additional procedure where a database of structures is systematically mined for matches to sub-nanometer resolution electron density. Conceptually this is the molecular replacement method first described by Rossman and Blow in 1962 (Rossmann and Blow, 1962). We call our procedure FREDS (fold recognition electron density search). FREDS is independent of sequence information, and is therefore advantageous for systems where limited sequence homology exists such that comparative modeling becomes difficult. FREDS can be used for densities of any size, as it searches through a non-redundant database of protein structures for an atomic counterpart to the electron density rather than attempt to predict the structure. Lastly, FREDS is particularly powerful as it does not discriminate between α-helical, β-sheet, or an α/β class of proteins.
The procedure is remarkably simple, automated, accessible to sub-nanometer resolution electron density, applicable to symmetrical or multi-subunit densities, parallelized for multiple processors, and, with constant updates from the Protein Data Bank (PDB) (http://www.pdb.org/), has an ever-increasing searchable database. FREDS is modular and uses a series of available software. This provides flexibility for the user to use alternative programs, and allows for FREDS to grow as more powerful software packages become available. FREDS only requires as input a sub-nanometer resolution electron density, preferably segmented to temporally facilitate the search.
The overall goal of FREDS is similar to that of SPI-EM–the prediction of a domain's fold by parsing through a database of domain structures looking for a match to the user provided electron density. However, the algorithms used by FREDS and SPI-EM are different. FREDS attempts to identify the structures that best describes the user provided electron density, whereas SPI-EM attempts to identify the CATH superfamily that best describes the user provided electron density (Velazquez-Muriel et al., 2005). This single distinction allows FREDS to freely search any protein structure database, while it restricts SPI-EM to a pre-categorized protein structure database. We will discuss this further below.
Fig. 1 is a flow chart outlining the strategy used in FREDS. A non-redundant database of protein chains, containing a single representative from clusters of chains with more than 30% sequence identity, is updated from the PDB on a monthly basis (Altschul et al., 1990). A domain database is automatically generated from the chain database to define the searchable database. All structures from the domain database are fitted to the user provided electron density and a raw cross correlation coefficient (rCC) is calculated. Each rCC is normalized, a Z-score is calculated, and the solutions are then sorted from highest to lowest Z-score.
Figure 1.

A flowchart of the described search procedure. As of Oct. 2009 the searchable database contains 16,087 domains larger than 25-residues.
A benchmark set of nine segmented densities, derived from six sub-nanometer image reconstructions deposited into the Electron Microscopy Data Bank (EMDB) (www.ebi.ac.uk/pdbe/emdb), is used to test the procedure. FREDS identifies all of the correct folds when the atomic counterpart to the density is known, and identifies three convincing folds for densities with unreported atomic structures. FREDS will be available for download ((http://www.scripps.edu/~rkhayat).
Materials and Methods
Generating and maintaining a non-redundant parent database
A list of clustered PDB chains, based on sequence identity, is made available for download by the PDB. We have been using the list with 30% sequence identity threshold to remove homologous folds when generating our database. The first entry for each cluster is subjected to a number of conditions prior to being deleted or inserted into the parent database. These include: 1) removal if non-protein entries, 2) removal of chains with only Cα entries, (3) mutation of UNK to ALA residues, and 4) removal of all but the first (lowest energy) model of an NMR entry. The parent database is updated as a new list becomes available from the PDB each month. The update involves applying the four conditions mentioned above to the first entries of clusters with no previously downloaded entries. Hence, only new clusters have a representative added to the database.
Domain Detection and Searching the Database
The domains in each protein chain are automatically detected from the parent database using PDP: protein domain parser (Alexandrov and Shindyalov, 2003). Each domain is used in a real space molecular replacement with the program MOLREP (Vagin and Teplyakov, 1997). The atomic structures of domains from GroEL and bacteriophage Lambda gpD were used to empirically optimize the parameters for a successful MOLREP search of the segmented electron densities from the corresponding normalized image reconstructions. The success and quality of each search was based on the MOLREP correlation coefficient score of the solution and visual assessment of the solution's fit into the density. A number of MOLREP parameters were combinatorially tested. These include the resolution, the density contour level, the search modes, and the scoring modes. The resolutions tested were 9.0 Å, 8.0 Å, 7.0 Å, and that shown in table 1. The density contour levels tested varied from 0 to 3-sigma –in 1-sigma increments. The parameters producing the best results were then used in the remaining searches. Each domain is independently fit; therefore the process has been parallelized.
Table 1.
Results obtained with FREDS. The top ten solutions are shown for each search. In italics are the structures with similar folds. The percent identity column was calculated by structure based sequence alignment with the program TM-align (Zhang and Skolnick, 2005).
| Densityℑ | Res. | Solutions | Residues | Z-score | IDℵ | Hours (CPU)ℜ |
|---|---|---|---|---|---|---|
| GroEL * | 6.0 Å | 1GRL_A1 | 254 | 21.4 | 100% | 65 (4) |
| (1457) | 1Q2V_A1 | 258 | 15.4 | 24% | ||
| 1R7R_A4 | 154 | 5.3 | ||||
| 1WGW_A1 | 87 | 4.6 | ||||
| 1DLY_A1 | 121 | 4.5 | ||||
| 2CZ2_A2 | 111 | 4.4 | ||||
| 1Z21_A1 | 69 | 4.3 | ||||
| 3F9V_A3 | 79 | 4.3 | ||||
| 1Q2V_A2 | 104 | 4.2 | ||||
| 2EWF_A3 | 73 | 4.1 | ||||
|
| ||||||
| GroEL † | 6.0 Å | 1GRL_A3 | 158 | 9.6 | 100% | 48 (4) |
| (1457) | 1GRL_A2 | 86 | 8.5 | 100% | ||
| 1Q2V_A3 | 156 | 5.6 | 15% | |||
| 2JPS_A1 | 95 | 5.2 | --- | |||
| 1Z96_A1 | 42 | 5.1 | ||||
| 1XB2_B1 | 43 | 5.0 | ||||
| 2O4T_A1 | 82 | 4.9 | ||||
| 2C5U_A4 | 59 | 4.9 | ||||
| 1L9Z_H4 | 58 | 4.8 | ||||
| 1C03_A1 | 163 | 4.5 | ||||
|
| ||||||
| Metarhodopsin | 6.0 Å | 1GZM_A1 | 328 | 16.4 | 100% | 144 (6) |
| (1079) | 3DDL_A1 | 252 | 6.5 | 6.5% | ||
| 2JMH_A1 | 105 | 4.8 | ||||
| 1K0N_A1 | 85 | 4.8 | ||||
| 1JNV_Z3 | 60 | 4.6 | ||||
| 2IAK_A1 | 120 | 4.5 | ||||
| 2B7M_A3 | 79 | 4.5 | ||||
| 1W63_A3 | 94 | 4.5 | ||||
| 3C72_A2 | 91 | 4.5 | ||||
| 2Z73_A1 | 350 | 4.4 | ||||
|
| ||||||
| RDV ‡ | 8.5 Å | 1UF2_A1 | 302 | 22.4 | 100% | 78 (8) |
| (1378) | 1UF2 A4 | 267 | 14.6 | 100% | ||
| 1UF2_A3 | 193 | 11.1 | 100% | |||
| 1UF2_A2 | 100 | 7.7 | 100% | |||
| 3GZU_A2 | 335 | 6.0 | 7.3% | |||
| 3GZU_A1 | 319 | 5.9 | 8.5% | |||
| 1UF2_A5 | 105 | 5.7 | 100% | |||
| 1WIJ_A1 | 66 | 4.4 | --- | |||
| 2FJI_13 | 115 | 3.9 | ||||
| 1NF1_A2 | 183 | 3.8 | ||||
|
| ||||||
| Stressosome | 8.0 Å | 1TID_B1 | 115 | 6.5 | --- | 45 (4) |
| (1552) | 2VY9_A1 | 112 | 5.8 | 16% | ||
| 3D02_A2 | 156 | 4.8 | ||||
| 1H4X_A1 | 110 | 4.7 | ||||
| 2V64_A1 | 210 | 4.6 | ||||
| 2PFF_H2 | 296 | 4.5 | ||||
| 1TQI_A2 | 101 | 4.4 | ||||
| 3BT7_A1 | 131 | 4.1 | ||||
| 2DFW_A2 | 83 | 4.1 | ||||
| 1U8S_A2 | 85 | 4.1 | ||||
|
| ||||||
| Lambda gpD | 7.0 Å | 1TD3_A1 | 103 | 8.1 | >30% | 70 (8) |
| 2Q5T_A1 | 394 | 4.3 | ||||
| (5012) | 2I8B_A1 | 116 | 3.7 | |||
| 1YA0_A2 | 364 | 3.6 | ||||
| 1K92_A1 | 395 | 3.6 | ||||
| 2Q6T_A1 | 112 | 3.3 | ||||
| 2Z8X_A1 | 348 | 3.2 | ||||
| 1JMJ_A1 | 386 | 3.2 | ||||
| 3C0Y_A1 | 386 | 3.1 | ||||
| 2BP1_A1 | 323 | 3.1 | ||||
|
| ||||||
| P22 gp1 | 8.0 Å | 1GNC_A1 | 164 | 5.8 | UNK | 96 (6) |
| (5051) | 1FXK_C1 | 68 | 5.3 | |||
| 1UCU_A1 | 80 | 5.1 | ||||
| 2K0N_A1 | 85 | 5.0 | ||||
| 1P49_A2 | 58 | 4.9 | ||||
| 2GHO_D3 | 40 | 4.9 | ||||
| 3GI7_A1 | 120 | 4.8 | ||||
| 2S35_A1 | 106 | 4.8 | ||||
| 1Z0K_B1 | 61 | 4.6 | ||||
| 2JES_A2 | 274 | 4.5 | ||||
|
| ||||||
| P22 gp4 | 8.0 Å | 1A6Q_A2 | 62 | 5.5 | UNK | 48 (4) |
| (5051) | 1LLQ_A6 | 95 | 5.0 | |||
| 2EFG_B1 | 55 | 4.5 | ||||
| 1E94_E3 | 109 | 4.4 | ||||
| 2Q5T_A1 | 394 | 4.2 | ||||
| 3CXB_A2 | 82 | 4.2 | ||||
| 2G62_A1 | 223 | 4.1 | ||||
| 1L9Z_H4 | 58 | 4.1 | ||||
| 2QGY_A1 | 385 | 4.0 | ||||
| 2NYY_A2 | 325 | 3.9 | ||||
|
| ||||||
| P22 gp10 | 8.0 Å | 1RI6_A1 | 333 | 6.9 | UNK | 120 (4) |
| (5051) | 1L0Q_A1 | 296 | 6.8 | |||
| 1GXR_A1 | 324 | 6.5 | ||||
| 2EYQ_A1 | 259 | 5.3 | ||||
| 3E11_A5 | 313 | 4.9 | ||||
| 3DAD_A2 | 224 | 4.4 | ||||
| 1PGU_A1 | 278 | 4.3 | ||||
| 3BWS_A2 | 232 | 4.1 | ||||
| 2VDU_B1 | 374 | 3.9 | ||||
| 2BED_A1 | 123 | 3.9 | ||||
Numbers in parenthesis are the electron microscopy data bank (EMDB) identification.
First segmented volume of GroEL
Second segmented volume of GroEL
Rice Dwarf Virus P3 subunit. The two italicized and two underlined structures are homologues of one another.
The percent identity between the crystal structure of the segmented electron density and the identified structure. This is calculated from structure based sequence alignment with the SSM server.
The time required for completing the search. In parenthesis is the number of Intel E5430 XEON-EMT processors used.
UNK Unknown sequence identity, as no crystal structure is available for the segmented electron density.
Calculating the Z-score
The cross correlation coefficient (CC) scores, produced by MOLREP, are dependent on domain size (Fig. S1). We will refer to these scores as the raw CC (rCC). This dependence must be eliminated in order to calculate a statistically meaningful description of the search –a Z-score for the fit of each domain. To model the dependence of rCC on domain size, a plot of rCC versus domain size is fitted to two functions. A linear model is used if the searched domains range from 25 to 400 residues, and a powerlaw model is used if the searched domains possess more than 400 residues. Variables from the fitted plots are used to calculate the average score () for any domain size. The quotient of rCC and is a size independent value –we refer to this as nCC (Fig. S2). A Z-score is then calculated for each fitted domain using the nCC values. The highest Z-score is indicative of the best solution.
Benchmark set
A benchmark set of nine segmented densities was generated from experimental sub-nanometer resolution cryo-EM image reconstructions. Six image reconstructions were used: two segmented densities from GroEL at 6.0 Å (EMDB 1457), Bovine Metarhodopsin I at 6.0 Å (EMDB ID: 1079), the RsbR146-274RsbS stressosome core at 8.0 Å (EMDB ID: 1552), the P3 subunit of Rice Dwarf virus at 8.5 Å (EMDB ID: 1378), gp1, gp4 and gp10 of the bacteriophage P22 tail machine at 8.0 Å (EMDB ID: 5051), and the gpD trimer of the mature bacteriophage Lambda at 7.0 Å (EMDB ID: 5012). The electron densities were low-pass filtered to the mentioned resolution using the EMAN package.
The densities were segmented so as to simulate the segmentation that would occur when no knowledge of the subunit boundaries is known. This was done similar to that described to Zhou et al., 2001. Briefly, subunit boundaries were determined by interactively examining the continuity of density at various contour thresholds using the UCSF Chimera package. Segmentation was carried out with the UCSF Chimera package (Pettersen et al., 2004). Density exterior to the subunit defining boundaries was intentionally included with the segmented density to test the reliability of FREDS. Segmented densities were padded into cubic volumes and centered using the EMAN package, and converted to CCP4 format using BSOFT (Heymann, 2001; Ludtke et al., 1999).
Results
Building the search database
Conformational flexibility in proteins can pose difficulty in X-ray crystallography when searching for a molecular replacement solution (Suhre and Sanejouand, 2004). This problem has also been documented for cryo-EM image reconstructions, where the subunit adopts a conformation that differs from the reported crystal structure (Trabuco et al., 2008). Consequently the conformational difference between the atomic structure and the cryo-EM image reconstruction may be large enough to impede finding a match using a high throughput search. To address this possibility we generated a database of protein domains, with the expectation that domains in general, but not always, do not undergo dramatic conformational changes. Moreover, since domains are the building blocks of proteins and therefore interchangeably used, it would be more suitable to search for domains rather than entire chains.
The purpose of generating a new domain database, as opposed to using an existing database such as SCOP or CATH (Murzin et al., 1995; Orengo et al., 1997), is to take advantage of the continually expanding PDB. Both SCOP and CATH are updated either annually or once every several years, and therefore lag behind the PDB. There are also a number of bacteriophage and viral protein structures with no entries in SCOP or CATH. While these examples may be exclusive, they do raise the important point that both SCOP and CATH have limitations that should be considered.
A non-redundant database is important for expediting the search by removing nearly identical structures. We currently use sequence identity to identify chains with homologous folds; however, we are implementing a structural similarity algorithm to further distill the database and facilitate the search.
There are a number of automatic methods for protein domain decomposition: PDP, PUU, Domain-Parser, and DDomain (Alexandrov and Shindyalov, 2003; Guo et al., 2003; Holm and Sander, 1994; Zhou et al., 2007). DDomain is restricted to contiguous domains and therefore not preferred. An evaluation between PDP, PUU, and Domain-Parser suggested that PDP was the most accurate method (Holland et al., 2006). Consequently, we chose PDP as our automated method for protein decomposition. Currently there are 16,087 domains in the searchable database.
Running the search
The success of the search requires the use of a robust program that can quickly search through a database of atomic structures for a fit to the electron density. It is important to use a program capable of searching in real space, and of searching for multiple copies of the same or different proteins within the electron density. To our knowledge, candidates for a real space search include Situs, 3SOM, MOLREP, MODELLER, COAN, and FOLDHUNTER (Ceulemans and Russell, 2004; Eswar et al., 2007; Jiang et al., 2001; Vagin and Teplyakov, 1997; Volkmann, 2002; Wriggers et al., 1999). While both 3SOM and MOLREP complete a database search quickly enough to make them a suitable search engine (data not shown), only MOLREP manages to return the correct structures for our six control searches –two densities for GroEL, Bovine Metarhodopsin, the P3 subunit of Rice Dwarf Virus, the stressosome core, and the bacteriophage Lambda gpD trimer. Moreover, only MOLREP is capable of searching for multiple copies of proteins in the electron density. Hence, we designed our search protocol using MOLREP as the search engine. Using the corresponding atomic domains as search models, the segmented electron densities of GroEL and the bacteriophage Lambda gpD trimer were used to optimize the MOLREP search parameters.
Identifying the correct solution
Throughout our searches it became evident that there was a general decay in the correlation coefficient with increasing domain size. Fig. S1 is a series of plots of the domain size (number of residues) and the raw cross correlation coefficient (rCC) reported by MOLREP. The decay in rCC as a function of domain size is the background distribution and describes the fit of a randomly selected domain, of a given size, to the electron density –with smaller domains fitting the electron density better than larger domains. To model the background distribution, we fitted each plot to a linear function as well as a number of non-linear functions including: one to three parameter exponential decay functions, two to three order polynomials, a one-parameter power function, and a simplified Bleasdale-Nelder model (data not shown). Our analysis demonstrated that a linear function fit best for domains ranging from 25 to 400 residues (Fig. S1).
A Z-score for each domain can be obtained by eliminating the dependency of the rCC to domain size (see Experimental Procedures). The Z-score of the correct solution should thus be greater than the Z-scores of the remaining domains (see below). The greater the difference between the highest Z-score(s) and the remaining top solutions signifies the greater the confidence that an accurate fold has been identified.
Case Examples
To test the procedure we used segmented density from experimentally determined cryo-EM image reconstructions obtained from the Electron Microscopy Data Bank (EMDB). Each search included all the structures present in the domain database.
GroEL
The GroEL complex is a 0.8 MDa tetradecamer with D7 symmetry (Fig. 2a). The cryo-EM image reconstruction was determined to 5.4 Å resolution using single particle analysis, and several crystal structures have been reported (Braig et al., 1994; Stagg et al., 2008). Two visibly distinguishable volumes of densities can be identified in the image reconstruction –the upper apical domain and the lower equatorial domain. These volumes were manually segmented for the search after the density was low-pass filtered to 6.0 Å resolution with EMAN (see Experimental procedures). Searching the first segmented density at 6.0 Å resolution produced the top 10 solutions shown in Table 1. A sharp decay in Z-scores can be seen between the second and third solutions. The top solution was the corresponding domain from GroEL (PDB ID: 1GRL), and the second solutions was a domain from the GroupII Thermococcus strain KS-1 chaperonin (PDB ID: 1Q2V). Fig. 2a shows the fit of the solutions to the segmented density. Alignment of the domains from 1GRL and 1Q2V with the SSM server produces an RMSD of 1.70 Å and a Z-score of 8.8 –indicating similar folds (Krissinel and Henrick, 2004).
Figure 2.

Surface and ribbon representations of cryo-EM image reconstructions with known atomic structures. Shown are the image reconstructions in cyan, the segmented densities in grey, and the top three solutions identified and fitted with FREDS as blue, cyan, and yellow ribbon cartoons. A) GroEL at 6.0 Å resolution. An ellipse identifies the 2-fold symmetry axis of GroEL. B) Bovine Metarhodopsin at 6.0 Å resolution, C) the P3 subunit of Rice Dwarf Virus at 8.5 Å resolution. D) The Stressosome complex at 8.0 Å resolution. E) The bacteriophage Lambda at 7.0 Å resolutions.
The search for the second segmented density produces a similar trend. The decay in Z-scores is sharpest between solutions two and three (Table 1). The top two Z-scores belong to different domains of GroEL (PDB ID: 1GRL) accounted for by different regions of the density (Fig. 2a). The third solution is a domain from a GroupII Thermococcus strain KS-1 chaperonin (PDB ID: 1Q2V). The comparable domains from GroEL and the Thermococcus chaperonin overlay with an RMSD of 1.73 Å and a Z-score of 5.1 –indicating similar folds.
Bovine Metarhodopsin I
The density for Bovine Metarhodopsin I was determined to 5.5 Å resolution using 2D electron crystallography (Ruprecht et al., 2004). Bovine Metarhodopsin I is a 39 kDa monomer that forms a mainly alpha helix up-down bundle. The search identifies a sharp decay in the Z-scores of the second and third solutions (Table 1). The top solution is the Bovine Rhodopsin (PDB ID: 1GZM), and the second solution is the Xanthorhodopsin (PDB ID: 3DDL) (Fig. 2b).
The Rice Dwarf Virus
Rice Dwarf Virus (RDV) is a 70 MDa double-shelled icosahedral virus. The T=1 inner core is composed of 120 copies of the P3 protein, and the T=13 outer shell is composed of 780 copies of the P8 protein (Naitow et al., 1999). The crystal structure of RDV has been determined to 3.5 Å (Nakagawa et al., 2003). A 8.3 Å cryo-EM image reconstruction of the P3 subunit was obtained using single particle analysis (Liu et al., 2007). The entire P3 density, corresponding to a 114 kDa polypeptide, was used for the search. The top ten solutions identified by FREDS show a sharp decay in the Z-scores of the seventh and eighth solutions (Table 1). The top four and seventh solutions are different domains of the P3 crystal structure (Fig 2c; PDB ID: 1UF2). The fifth and sixth solutions are two domains from the VP7 subunit of Rhesus rotavirus (PDB ID: 3GZU). Structural comparison between the two-rotavirus subunit domains was carried out with the SSM server. Domains 3GZU_A1 and 1UF2_A4 overlap with an RMSD of 3.3 Å, a Z-score of 2.4, and 50% of matched secondary structure elements. Domains 3GZU_A2 and 1UF2_A1 overlap with an RMSD of 3.8 Å, a Z-score of 0.6, and 50% of matched secondary structure elements. The low Z-scores returned by the SSM server suggest that the structures are different, however visual inspection of the aligned structure reveals that the structure are indeed similar (Fig. S3).
RsbR146-274RsbS stressosome core
The stressosome core cryo-EM image reconstruction was determined to 8.0 Å resolution using single particle analysis (Marles-Wright et al., 2008). The 1.8 MDa complex forms an icosahedral shell composed of RsbR and RsbS (Fig. 2d). Both RsbR and RsbS have similar atomic structures (Marles-Wright et al., 2008). The top two solutions include the Geobacillus Stearothermophilus Anti-sigma F factor antagonist (PDB ID: 1TID), and the Sigma B protein from Moorella thermoacetica MtRsbS (PDB ID: 2VY9). MtRsbS is the atomic structure of the segmented density. Structural comparison between the Anti-sigma F factor antagonist and MtRsbS returns a Z-score of 4.3 –indicating that the structures are similar.
The bacteriophage Lamdba gpD
The bacteriophage Lambda gpD is a 31 kDa trimeric assembly that decorates the T=7l icosahedral capsid at the quasi and icosahedral 3-fold axis (Lander et al., 2008). The crystal structure of gpD has been determined to 1.1 Å resolution (Yang et al., 2000). The trimeric density of gpD was used for searching the entire database. Table 1 shows a sharp decay in the Z-scores for the first and second solutions. The top solution is the capsid-stabilizing protein of lamboid phage 21 (PDB ID: 1TD3). Comparison of this structure with the lambda gpD crystal structure returns a Z-score of 9.8 –indicating the structure to be similar (Fig. 2d). PDB entry 1TD3 is the representative of the cluster containing the Lambda gpD.
The bacteriophage P22 tail-machine
The tail machine of P22 is a 3 MDa complex composed of five gene products. The gene products are assembled in a combination of 12-, 6-, and 3-fold symmetry (Fig. 3a) (Lander et al., 2009a). The portal complex is composed of 12 gp1 subunits (80 kDa each) and is at the periphery of the tail machine. The atomic structure of the gp1 has not been reported, but structural data suggest that it is homologous to the bacteriophage SPP1 portal protein (Lander et al., 2009a; Lebedev et al., 2007). A search of the database using the segmented density of gp1 produces the top ten-solution list in Table 1. The lack of a sharp drop in the Z-scores suggests that FREDS could not identify a fold for the P22 portal protein with high confidence. However, inspection of the top 10 solutions reveals a 274-residue domain of the SPP1 portal as the tenth solution of the search (Fig. 3c) (PDB ID: 2JES). The top nine solutions are helical and cluster to a helical section of the gp1 density (Fig. 3b).
Figure 3.
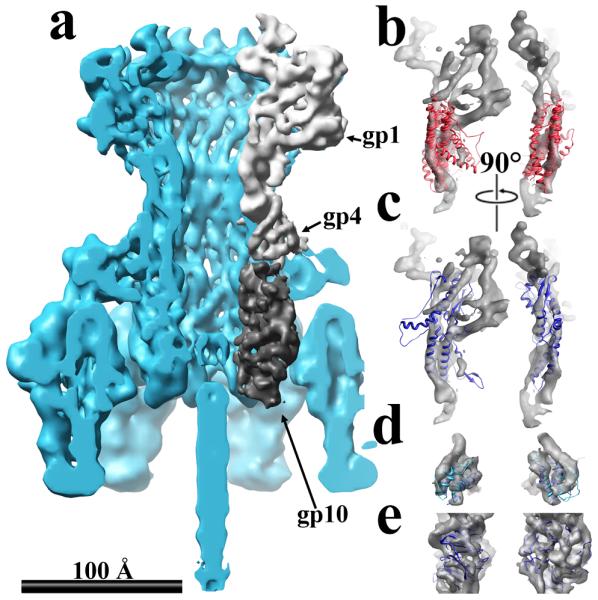
Surface and ribbon representations of cryo-EM image reconstructions with unknown atomic structures. The same coloring scheme is used as in Fig. 2. A) The P22 tail-machine density at 9.4 Å resolution in cyan, the gp1, gp4 and gp10 segmented densities as different shades of grey. B) The P22 gp1 density in grey with the top nine and tenth solutions shown as light red and blue ribbon cartoons –respectively. C) The P22 gp4 density in grey with the second top solution as a blue ribbon cartoon. D) The P22 gp10 density in grey and the top three solutions in blue, cyan, and yellow ribbon cartoons.
Attached to the base of the portal are 12 copies of the 18 kDa scaffold protein gp4 (Strauss and King, 1984). The crystal structure of gp4 has not been reported, and no homologous structures are known. Secondary structure prediction using the Jpred3 server indicates that gp4 is composed of four helices (Cole et al., 2008). The search with the segmented density produced a set of solutions with fairly close Z-scores (Table 1). The top two solutions are helical domains and show good agreement with the density; however, the second solution is visually more coincident with the density (PDB ID: 1LLQ) (Fig. 3d). While the two identified domains are composed of four helices, they do not share a similar structure.
Attached to the gp4 complex are 6 copies of the 52.5 kDa gp10 protein (Strauss and King, 1984). There is a sharp drop in the decay in the Z-scores for the third and fourth solutions (Table 1). The top three solutions share the WD40 fold (PDB IDs: 1RI6, 1L0Q, and 1GXR). Structure prediction of gp10 using the FFAS03 server also produces the WD40 fold with high confidence (data not shown).
Resolution and the Z-score
We tested the effect of the electron density resolution on the Z-score by running FREDS at various resolutions for the two GroEL and the Rice Dwarf Virus P3 subunit electron densities (Table 2). FREDS identified identical solutions for the first segmented density of GroEL at 6, 7, 8, and 9 Å resolutions. Moreover, a sharp drop in the decay of Z-scores could be seen between the second and third solutions at all of these resolutions. For the second segmented density of GroEL, FREDS identified the same top three solutions at 6 and 7 Å resolution -the two GroEL domains corresponding to the segmented density (1GRL_A3 and 1GRL_A2), and the GroupII Thermococcus strain KS-1 chaperonin domain (1QV2_A3) that is similar to a GroEL domain (1GRL_A3) (Tables 1 and 2). The GroupII Thermococcus strain KS-1 chaperonin domain was the seventh solution at the 8 Å resolution search, and solution fifty-five at the 9 Å resolution search.
Table 2.
The effects of resolution on FREDS. Structures in italics have the folds similar to the known crystal structure of the segmented density. The segmented densities were low-pass filtered to the indicated resolutions prior to submission to FREDS.
| Res. | GroEL volume 1 | GroEL volume 2 | Rice Dwarf Virus P3 | |||
|---|---|---|---|---|---|---|
| Solutions | Z-score | Solutions | Z-score | Solutions | Z-score | |
| 7 Å | 1GRL_A1 | 12.4 | 1GRL_A3 | 5.1 | ||
| 1Q2V_A1 | 9.0 | 1GRL_A2 | 4.9 | |||
| 1R7R_A4 | 3.3 | 1Q2V_A3 | 3.0 | |||
| 3GS3_A2 | 3.3 | 3F2E_A1 | 3.0 | |||
| 1WGW_A1 | 3.0 | 1C03_A1 | 2.9 | |||
| 1U2C_A1 | 3.0 | 2PGS_A1 | 2.9 | |||
|
| ||||||
| 8 Å | 1GRL_A1 | 11.1 | 1GRL_A3 | 4.3 | 1UF2_A1 | 13.9 |
| 1Q2V_A1 | 8.1 | 1GLR_A2 | 2.8 | 1UF2_A4 | 9.1 | |
| 3EC6_A1 | 3.1 | 2WLC_A1 | 2.8 | 1UF2_A3 | 7.6 | |
| 1R7R_A4 | 2.9 | 2PGS_A1 | 2.7 | 1UF2_A2 | 5.1 | |
| 1NLX_A1 | 2.8 | 1ZJC_A1 | 2.7 | 3GZU_A2 | 4.3 | |
| 1WGW_A1 | 2.7 | 2O8B_B5 | 2.7 | 1UF2_A5 | 4.2 | |
| 3GS3_A2 | 2.6 | 1Q2V_A3 | 2.6 | 3GZU_A1 | 4.0 | |
| 2J0F_A2 | 2.6 | 1N26_A1 | 2.6 | 1Z2L_A2 | 2.7 | |
|
| ||||||
| 9 Å | 1GRL_A1 | 9.7 | 1GRL_A3 | 3.5 | 1UF2_A1 | 12.3 |
| 1Q2_VA1 | 6.3 | 2PGS_A1 | 2.7 | 1UF2_A4 | 7.8 | |
| 3EC6_A1 | 3.3 | 1O90_A1 | 2.7 | 1UF2_A3 | 5.7 | |
| 2ZOE_A1 | 2.9 | 2WLC_A1 | 2.6 | 3GZU_A2 | 4.4 | |
| 2O18_A1 | 2.7 | 3C9F_A1 | 2.5 | 1UF2_A2 | 3.8 | |
| 1GM5_A6 | 2.7 | 1ZJC_A1 | 2.4 | 1UF2_A5 | 3.6 | |
| 1KEA_A2 | 2.4 | 1AY0_A2 | 2.4 | 3GZU_A1 | 3.6 | |
| 2J0F_A2 | 2.4 | 2VRD_A1 | 2.4 | 2FJI_13 | 2.5 | |
The effect of electron density resolution on FREDS. Solutions in italics are the correc fold for the indicated density.
The results from the RDV search at 8 Å are nearly identical to those at 8.5 Å, with the exception that the sixth solution at 8 Å is the seventh solution at 8.5 Å –and vice versa. A similar trend can be seen for the solutions at the 9 Å resolution search. Once again, a sharp drop in the decay of Z-scores between the seventh and eighth solutions at all the resolutions signifies that FREDS has confidently identified the folds for the experimental electron density. Table 2 shows that the Z-scores and the magnitude of sharp decay in Z-scores, associated with confidently identifying the correct fold, are resolution dependent.
Discussion
The incorporation of experimentally and theoretically derived atomic models into cryo-EM image reconstructions can provide a wealth of information that may be inaccessible to either method alone. Here we present a method where the electron density is used to search for an atomic structure counterpart. The identified structure can then be used as a template for comparative modeling. As mentioned earlier, FREDS has similarities to SPI-EM. Both FREDS and SPI-EM rank the folds/superfamily folds that best describe the user provided electron density by comparing the fit of each fold/superfamily fold to a background distribution. FREDS calculates a background distribution for each search using the scores obtained for the fitted structures during that particular search. The FREDS background distribution is therefore dependent on the electron density. SPI-EM however calculates the background distribution by comparing all the members of a CATH superfamily to all the superfamily representatives in the CATH database. The SPI-EM background distribution is therefore independent of the user provided electron density, but heavily influenced by the superfamily classification of the CATH database. Any inaccuracies in the CATH superfamily classification may result in SPI-EM inappropriately ranking superfamily folds that describe the user provided electron density. This is problematic as the CATH superfamily classification is under constant reorganization. Domain structures are regularly reorganized into different CATH superfamilies, and CATH superfamilies are merged or removed with each CATH update.
The densities used in our searches are experimental densities and therefore reflect real case scenarios. Time constraints require the macromolecular densities to be segmented into searchable portions; however, our method is not stringent on accurate segmentation, as demonstrated by the GroEL and Rice Dwarf Virus examples. FREDS successfully identified two distinct domains in the second example of GroEL. The smaller intermediate domain is an 86-residue structure that corresponds to less than 35% of the mass of the density. FREDS also accurately identified the five domains pertaining to the 114 kDa RDV P3 protein. The smallest domain accounts for less than 10% of the entire P3 mass. FREDS can also identify the correct fold from symmetric densities, as shown with the gpD trimer of Lambda.
Table 1 also shows that FREDS can successfully, and with confidence, identify structural folds that are similar to the known solutions. The identity column in table 1 is calculated from a structure based sequence alignment between the known atomic structure of the indicated electron density and the remaining solutions confidently identified by FREDS. The identities between the similar folds vary from 24% for the case of the first segmented density of GroEL to 6.5% for the case of the rhodopsins. The effect of resolution on the success of FREDS has been tabulated in Table 2. There is deterioration in the magnitude of Z-scores with decreasing resolution. FREDS identifies the same solutions at various resolutions for the first segmented density of GroEL and the RDV P3 density. FREDS performs similarly at 6 and 7 Å resolution for the second segmented GroEL density. FREDS identifies the corresponding domain(s) of GroEL to the density at 8 and 9 Å resolution, but has difficulty in identifying the fold similar to GroEL –indicating a resolution limitation to FREDS in some cases.
For densities where atomic structures of the proteins are not available (gp1, gp4 and gp10 of the bacteriophage P22 tail machine) the top solutions identified by FREDS are structurally and biologically convincing. The search for a fit to the gp1 segmented density does not produce a distinct solution; however, visualization of the top ten solutions reveals the top nine solutions to be helical domains that cluster to a helical region of the gp1 density. The tenth solution is the 274-residue domain of the evolutionary related bacteriophage SPP1 portal protein. The structural homology between the P22 and SPP1 portal subunits was demonstrated by Lander et al. 2009, where the crystal structure of the SPP1 subunit was readily docked and refined into the P22 gp1 segmented density. The top two solutions for the P22 gp4 density belong to domains from serine/threonine phosphatase 2C and malic enzyme from Ascaris suum. The role of the domain from serine/threonine phosphatase 2C is unknown, but the domain from malic enzyme acts as a scaffold by engaging two additional malic enzyme domains (Coleman et al., 2002). This function appears to be similar to that of gp4, which is necessary for attachment of gp10 to the growing tail machine complex (Strauss and King, 1984). The domain from A. suum malic enzyme accounts for 66% of the gp4 mass. Docking twelve of the malic enzyme domains into the P22 tail-machine image reconstruction identifies two regions of density where additions to the domain could account for a portion of the missing mass. These include a β-sheet region on the exterior of the tail-machine, and the gp4-gp10 interface (Fig. S4). The WD40 domain identified for gp10 is one of the most abundant structures in eukaryotic proteins and is incorporated into proteins with diverse functions. Regardless of the protein function, the WD40 domain acts as a scaffold for protein assembly and disassembly. Similarly, the WD40 domain of gp10 appears to act as a scaffold for the attachment of the needle-like gp26 of the P22 tail-machine (Fig. 3a and 3e).
Conclusion
As with all methods, there are limitations to FREDS. For the case of P22 gp4, FREDS was unable to distinguish the difference between two proteins with different topologies. This indicates that there may be situations where FREDS is unable to identify the accurate fold by discerning the difference between distinct topologies belonging to the same architecture. For example, there are a number of topologically different β-sandwich structures in the PDB and it is foreseeable that it would be difficult for FREDS to identify the proper topology at resolutions where the connectivity of loops cannot be resolved. In such instances, the secondary structure connectivity obtained from secondary structure prediction could eliminate the ambiguity. An additional foreseeable failure of FREDS would involve a scenario where the user provided electron density pertains to a protein with a fold that is not represented in the database –a novel fold. A solution to this problem could be to incorporate FREDS into the ab-initio structure prediction process of Rosetta. The initial low-resolution decoys produced by Rosetta can be processed with FREDS to identify the candidate(s) correlating best with the electron density. These decoys can then be processed through the high-resolution decoy procedure in Rosetta to identify the most likely structure(s) (Rohl et al., 2004).
Supplementary Material
Determining the Z-score. Plots of the domain size (number of residues) and the MOLREP correlation coefficient (rCC) for the GroEL segment 1, GroEL segment 2, Bovine Metarhodopsin, Rice dwarf virus, Stressosome, P22 gp1, P22 gp10, and Lambda gpD searches. The rCC score for each domain is shown as a tick mark, and the linear fit to the data is shown as a dark bold line. The rCC Z-scores suggesting a solution have been circled for clarity.
Plots of the nCC scores and domain sizes. Plots belong to the GroEL segment 1, GroEL segment 2, Bovine Metarhodopsin, Rice dwarf virus, Stressosome, P22 gp1, P22 gp10, and Lambda gpD searches.
Ribbon representation of folds identified by FREDS for the RDV P3 subunit electron density. Structural overlap showing the similar folds shared between domains from the RDV P3 subunit (shown in green) and domains from the Rhesus rotavirus VP7 (shown in yellow).
Surface representation of the P22 tail machine at 9.4 Å resolution. A) Colored in dark grey are protein densities for which a crystal structure is available. Also colored in dark grey is the portal (gp1) complex. Colored in light grey are densities for gp10 and gp4. The folds identified for gp10 and gp4 have been docked into their corresponding densities. Colored in red are regions of the density for which a fold could not be determined. B) Segmented density of the gp4.
Acknowledgements
We would like to thank Dr. Jeff Lee and Dr. Edward Brignole for careful reading of the manuscript and providing important suggestions. This work was supported by the National Institutes of Health Grant R01 GM54076 (to J.E.F and G.C.L.). R.K. was supported by National Institutes of Health Postdoctoral Fellowship F32 AI065071. The 3D reconstructions of GroEL, the P22 tail machine, and bacteriophage Lambda were conducted at the National Resource for Automated Molecular Microscopy (NRAMM), which is supported by the National Institutes of Health through the National Center for Research Resources P41 program (RR17573).
Abbreviations
- FREDS
fold recognition electron density search
- EM
electron microscopy
- NMR
nuclear magnetic resonance
- PDB
protein data bank
- EMDB
electron microscopy data bank
- CC
correlation coefficient
- SCOP
structural classification of proteins
- CATH
class, architecture, topology and homologous superfamily
- PDP
protein domain parser
- gp
gene product
- RMSD
root mean standard deviation
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexandrov N, Shindyalov I. PDP: protein domain parser. Bioinformatics. 2003;19:429–30. doi: 10.1093/bioinformatics/btg006. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Baker TS, Johnson JE. Low resolution meets high: towards a resolution continuum from cells to atoms. Curr Opin Struct Biol. 1996;6:585–94. doi: 10.1016/s0959-440x(96)80023-6. [DOI] [PubMed] [Google Scholar]
- Braig K, Otwinowski Z, Hegde R, Boisvert DC, Joachimiak A, Horwich AL, Sigler PB. The crystal structure of the bacterial chaperonin GroEL at 2.8 A. Nature. 1994;371:578–86. doi: 10.1038/371578a0. [DOI] [PubMed] [Google Scholar]
- Bubeck D, Filman DJ, Cheng N, Steven AC, Hogle JM, Belnap DM. The structure of the poliovirus 135S cell entry intermediate at 10-angstrom resolution reveals the location of an externalized polypeptide that binds to membranes. J Virol. 2005;79:7745–55. doi: 10.1128/JVI.79.12.7745-7755.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceulemans H, Russell RB. Fast fitting of atomic structures to low-resolution electron density maps by surface overlap maximization. J Mol Biol. 2004;338:783–93. doi: 10.1016/j.jmb.2004.02.066. [DOI] [PubMed] [Google Scholar]
- Chen JZ, Settembre EC, Aoki ST, Zhang X, Bellamy AR, Dormitzer PR, Harrison SC, Grigorieff N. Molecular interactions in rotavirus assembly and uncoating seen by high-resolution cryo-EM. Proc Natl Acad Sci U S A. 2009;106:10644–8. doi: 10.1073/pnas.0904024106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole C, Barber JD, Barton GJ. The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 2008;36:W197–201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman DE, Rao GS, Goldsmith EJ, Cook PF, Harris BG. Crystal structure of the malic enzyme from Ascaris suum complexed with nicotinamide adenine dinucleotide at 2.3 A resolution. Biochemistry. 2002;41:6928–38. doi: 10.1021/bi0255120. [DOI] [PubMed] [Google Scholar]
- Das R, Baker D. Macromolecular modeling with rosetta. Annu Rev Biochem. 2008;77:363–82. doi: 10.1146/annurev.biochem.77.062906.171838. [DOI] [PubMed] [Google Scholar]
- Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using MODELLER. Curr Protoc Protein Sci. 2007 doi: 10.1002/0471140864.ps0209s50. Chapter 2, Unit 2 9. [DOI] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–9. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Guo JT, Xu D, Kim D, Xu Y. Improving the performance of DomainParser for structural domain partition using neural network. Nucleic Acids Res. 2003;31:944–52. doi: 10.1093/nar/gkg189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helles G. A comparative study of the reported performance of ab initio protein structure prediction algorithms. J R Soc Interface. 2008;5:387–96. doi: 10.1098/rsif.2007.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heymann JB. Bsoft: image and molecular processing in electron microscopy. J Struct Biol. 2001;133:156–69. doi: 10.1006/jsbi.2001.4339. [DOI] [PubMed] [Google Scholar]
- Holland TA, Veretnik S, Shindyalov IN, Bourne PE. Partitioning protein structures into domains: why is it so difficult? J Mol Biol. 2006;361:562–90. doi: 10.1016/j.jmb.2006.05.060. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Parser for protein folding units. Proteins. 1994;19:256–68. doi: 10.1002/prot.340190309. [DOI] [PubMed] [Google Scholar]
- Jiang W, Baker ML, Ludtke SJ, Chiu W. Bridging the information gap: computational tools for intermediate resolution structure interpretation. J Mol Biol. 2001;308:1033–44. doi: 10.1006/jmbi.2001.4633. [DOI] [PubMed] [Google Scholar]
- Jiang W, Baker ML, Jakana J, Weigele PR, King J, Chiu W. Backbone structure of the infectious epsilon15 virus capsid revealed by electron cryomicroscopy. Nature. 2008;451:1130–4. doi: 10.1038/nature06665. [DOI] [PubMed] [Google Scholar]
- Khayat R, Tang L, Larson ET, Lawrence CM, Young M, Johnson JE. Structure of an archaeal virus capsid protein reveals a common ancestry to eukaryotic and bacterial viruses. Proc Natl Acad Sci U S A. 2005;102:18944–9. doi: 10.1073/pnas.0506383102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong Y, Ma J. A structural-informatics approach for mining beta-sheets: locating sheets in intermediate-resolution density maps. J Mol Biol. 2003;332:399–413. doi: 10.1016/s0022-2836(03)00859-3. [DOI] [PubMed] [Google Scholar]
- Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr. 2004;60:2256–68. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- Lander GC, Evilevitch A, Jeembaeva M, Potter CS, Carragher B, Johnson JE. Bacteriophage lambda stabilization by auxiliary protein gpD: timing, location, and mechanism of attachment determined by cryo-EM. Structure. 2008;16:1399–406. doi: 10.1016/j.str.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Khayat R, Li R, Prevelige PE, Potter CS, Carragher B, Johnson JE. The P22 tail machine at subnanometer resolution reveals the architecture of an infection conduit. Structure. 2009a;17:789–99. doi: 10.1016/j.str.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, Lyumkis D, Potter CS, Carragher B. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol. 2009b;166:95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebedev AA, Krause MH, Isidro AL, Vagin AA, Orlova EV, Turner J, Dodson EJ, Tavares P, Antson AA. Structural framework for DNA translocation via the viral portal protein. Embo J. 2007;26:1984–94. doi: 10.1038/sj.emboj.7601643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Marles-Wright J, Grant T, Delumeau O, van Duinen G, Firbank SJ, Lewis PJ, Murray JW, Newman JA, Quin MB, Race PR, Rohou A, Tichelaar W, van Heel M, Lewis RJ. Molecular architecture of the “stressosome,” a signal integration and transduction hub. Science. 2008;322:92–6. doi: 10.1126/science.1159572. [DOI] [PubMed] [Google Scholar]
- Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–40. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- Naitow H, Morimoto Y, Mizuno H, Kano H, Omura T, Koizumi M, Tsukihara T. A low-resolution structure of rice dwarf virus determined by ab initio phasing. Acta Crystallogr D Biol Crystallogr. 1999;55:77–84. doi: 10.1107/S0907444998007677. [DOI] [PubMed] [Google Scholar]
- Nakagawa A, Miyazaki N, Taka J, Naitow H, Ogawa A, Fujimoto Z, Mizuno H, Higashi T, Watanabe Y, Omura T, Cheng RH, Tsukihara T. The atomic structure of rice dwarf virus reveals the self-assembly mechanism of component proteins. Structure. 2003;11:1227–38. doi: 10.1016/j.str.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Orengo CA, Michie AD, Jones S, Jones DT, Swindells MB, Thornton JM. CATH--a hierarchic classification of protein domain structures. Structure. 1997;5:1093–108. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Rohl CA, Strauss CE, Misura KMS, Baker D. Protein structure prediction using Rosetta. Enzymology. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- Rossmann MG, Blow DM. The Detection of Sub-Units within the Crystallographic Asymmetric Unit. Acta Crystallogr. 1962;15:24–32. [Google Scholar]
- Ruprecht JJ, Mielke T, Vogel R, Villa C, Schertler GF. Electron crystallography reveals the structure of metarhodopsin I. Embo J. 2004;23:3609–20. doi: 10.1038/sj.emboj.7600374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg SM, Lander GC, Quispe J, Voss NR, Cheng A, Bradlow H, Bradlow S, Carragher B, Potter CS. A test-bed for optimizing high-resolution single particle reconstructions. J Struct Biol. 2008;163:29–39. doi: 10.1016/j.jsb.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss H, King J. Steps in the stabilization of newly packaged DNA during phage P22 morphogenesis. J Mol Biol. 1984;172:523–43. doi: 10.1016/s0022-2836(84)80021-2. [DOI] [PubMed] [Google Scholar]
- Suhre K, Sanejouand YH. On the potential of normal-mode analysis for solving difficult molecular-replacement problems. Acta Crystallogr D Biol Crystallogr. 2004;60:796–9. doi: 10.1107/S0907444904001982. [DOI] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Trabuco LG, Villa E, Mitra K, Frank J, Schulten K. Flexible fitting of atomic structures into electron microscopy maps using molecular dynamics. Structure. 2008;16:673–83. doi: 10.1016/j.str.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 1997;30:1022–1025. [Google Scholar]
- Velazquez-Muriel JA, Sorzano CO, Scheres SH, Carazo JM. SPI-EM: towards a tool for predicting CATH superfamilies in 3D-EM maps. J Mol Biol. 2005;345:759–71. doi: 10.1016/j.jmb.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Volkmann N. A novel three-dimensional variant of the watershed transform for segmentation of electron density maps. J Struct Biol. 2002;138:123–9. doi: 10.1016/s1047-8477(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Wriggers W, Milligan RA, McCammon JA. Situs: A package for docking crystal structures into low-resolution maps from electron microscopy. J Struct Biol. 1999;125:185–95. doi: 10.1006/jsbi.1998.4080. [DOI] [PubMed] [Google Scholar]
- Yan X, Sinkovits RS, Baker TS. AUTO3DEM--an automated and high throughput program for image reconstruction of icosahedral particles. J Struct Biol. 2007;157:73–82. doi: 10.1016/j.jsb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Forrer P, Dauter Z, Conway JF, Cheng N, Cerritelli ME, Steven AC, Pluckthun A, Wlodawer A. Novel fold and capsid-binding properties of the lambda-phage display platform protein gpD. Nat Struct Biol. 2000;7:230–7. doi: 10.1038/73347. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–9. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Xue B, Zhou Y. DDOMAIN: Dividing structures into domains using a normalized domain-domain interaction profile. Protein Sci. 2007;16:947–55. doi: 10.1110/ps.062597307. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Determining the Z-score. Plots of the domain size (number of residues) and the MOLREP correlation coefficient (rCC) for the GroEL segment 1, GroEL segment 2, Bovine Metarhodopsin, Rice dwarf virus, Stressosome, P22 gp1, P22 gp10, and Lambda gpD searches. The rCC score for each domain is shown as a tick mark, and the linear fit to the data is shown as a dark bold line. The rCC Z-scores suggesting a solution have been circled for clarity.
Plots of the nCC scores and domain sizes. Plots belong to the GroEL segment 1, GroEL segment 2, Bovine Metarhodopsin, Rice dwarf virus, Stressosome, P22 gp1, P22 gp10, and Lambda gpD searches.
Ribbon representation of folds identified by FREDS for the RDV P3 subunit electron density. Structural overlap showing the similar folds shared between domains from the RDV P3 subunit (shown in green) and domains from the Rhesus rotavirus VP7 (shown in yellow).
Surface representation of the P22 tail machine at 9.4 Å resolution. A) Colored in dark grey are protein densities for which a crystal structure is available. Also colored in dark grey is the portal (gp1) complex. Colored in light grey are densities for gp10 and gp4. The folds identified for gp10 and gp4 have been docked into their corresponding densities. Colored in red are regions of the density for which a fold could not be determined. B) Segmented density of the gp4.