

Abstract
In 1997, we identified a novel peptide, catestatin (CST: bovine chromogranin A [CHGA]344-364: RSMRLSFRARGYGFRGPGLQL; human CHGA352-372: SSMKLSFRARGYGFRGPGPQL), which is a potent inhibitor of nicotinic cholinergic-stimulated catecholamine secretion. CST shows characteristic inhibitory effects on nicotinic cationic (Na+, Ca2+) signal transduction, which are specific to the neuronal nicotinic receptor. Utilizing systematic polymorphism discovery at the human CHGA locus we discovered three human variants of CST: G364S, P370L, and R374Q that showed differential potencies towards inhibition of catecholamine secretion. In humans, CHGA is elevated and its processing to CST is diminished in hypertension. Diminished CST is observed not only in hypertensive individuals but also early-normotensive offspring of patients with hypertension, suggesting that an early deficiency of CST might play a pathogenic role in the subsequent development of the disease. Consistent with human findings, prevention of endogenous CST expression by targeted ablation (knockout) of the mouse Chga locus (Chga-KO) resulted in severe hypertension that can be “rescued” specifically by replacement of the CST peptide. CST acts directly on the heart to inhibit the inotropic and lusitropic properties of the rodent heart and also acts as a potent vasodilator in rat and human. While the G364S CST variant caused profound changes in human autonomic activity and seemed to reduce risk of developing hypertension, CST replacement rescued Chga-KO mice from dampened baroreflex sensitivity. In addition, CST has been shown to induce chemotaxis and acts as an antimicrobial as well as an antimalarial peptide. The present review summarizes these multiple actions of CST.
Keywords: Chromaffin, chromogranin, nicotine, blood pressure, chemotaxis, innate immunity, baroreflex sensitivity, and myocardial contractility
Introduction
Chromogranin A (CHGA (MIM 118910)), the index member of the chromogranin/secretogranin protein family, is a 48-kDa acidic polypeptide, which is the major protein found in the core of catecholamine storage vesicles of chromaffin cells and postganglionic sympathetic axons [1-4]. This protein is stored and released from the same secretory vesicles that contain catecholamines in chromaffin cells and noradrenergic neurons [5, 6]. CHGA is required for formation of catecholamine secretory vesicles in chromaffin cells and its expression may be sufficient to induce a regulated secretory system even in non-secretory cells [7]. Although initially detected in chromaffin granules, this protein was later found to be distributed ubiquitously in secretory vesicles of endocrine, neuroendocrine, and neuronal cells [1, 8-10]. Because of the presence of 8-10 dibasic sites [11-15], CHGA also serves as a pro-hormone that gives rise to biologically active peptides such as the dysglycemic peptide pancreastatin (hCHGA250-301) [16-18], the antimicrobial peptide prochromacin (bCHGA79-431) [19], the vasodilator vasostatin (hCHGA1-76) [20], and CST (bCHGA344-364; hCHGA352-372) that acts to inhibit catecholamine release [21]. CST also inhibits desensitization of catecholamine release induced by nicotine [22].
CHGA is overexpressed by chromaffin cells in rodent models of both genetic (spontaneously hypertensive rat) [23, 24] and acquired (renovascular) [25] hypertension, and twin studies demonstrate the heritability of both plasma CHGA and CST concentration (44-60%) in humans [26, 27]. In clinical practice, CHGA is used as a marker of pheochromocytomas [28-30], carcinoid tumors [31-33], neuroblastomas, neuroendocrine tumors, and neurodegerative diseases. Circulating CHGA levels are elevated in patients with chronic heart failure [34] and after acute myocardial infarction [35], which is partially supported by the finding of myocardial production of CHGA in humans with dilated and hypertrophic cardiomyopathy [36]. In addition, CHGA has been shown to increase in proportion to clinical severity and to be associated with prognosis in patients with both chronic and post-infarction heart failure [37]. In fact, CHGA is now claimed to be an independent predictor of long-term mortality and heart failure hospitalizations across the spectrum of acute coronary syndromes [38]. CHGA is also a good marker of sepsis and systemic inflammatory response syndrome [39, 40] and an independent indicator of prognosis in critically ill nonsurgical patients [41]. In human essential (hereditary) hypertension, the plasma concentration of CST is diminished in not only established cases, but also in the early-normotensive offspring of patients with hypertension (FH+) suggesting that an early deficiency of this catecholamine release inhibitory peptide might play a pathogenic role in the subsequent development of the disease [42]. Consistent with human findings, prevention of endogenous CST expression by targeted ablation (knockout) of the mouse Chga locus (Chga-KO) results in severe hypertension that can be “rescued” specifically by replacement of the CST peptide [43].
CST causes vasodilation in rats [44] and humans [45], acting directly on the heart to regulate myocardial contractility and relaxation in rodent heart [46] and to improve baroreflex sensitivity in Chga-KO mice [47]. CST also induces chemotaxis of human monocytes [48] and acts as an antibacterial peptide [49, 50]. The present review focuses on these multiple actions of CST: effects on catecholamine secretion, cardiovascular physiology, chemotaxis and innate immunity.
1. Background and discovery of CST as the potent nicotinic-cholinergic antagonist
Although it was reported in 1988 that CHGA-proteolytic products inhibit catecholamine secretion from primary cultures of bovine chromaffin cells [51], the identity of the peptide remained elusive for about a decade. It was in 1997 when we identified the catecholamine release-inhibitory domain within CHGA by synthesizing 15 peptides (average length, 22 residues; range, 19-25 residues) spanning 78% of the length of the bovine CHGA (431 amino acid) and tested their effects (at 10 μM dose) on nicotine-evoked catecholamine secretion from PC12 cells [21]. Of the 15 peptides tested only one domain (bovine CHGA344-364) was found to inhibit nicotine-evoked catecholamine secretion [21]. We coined the term “catestatin” to describe the catecholamine secretion inhibitory property of this peptide. In PC12 cells, CST showed an IC50 of 200 nM (Fig. 1) [21]. Given that the concentration of CHGA in chromaffin granules was ~2-4 mM and the concentration of chromaffin granule core contents in the extracellular space in the vicinity of the exocytotic pore was 10-fold lower than in the granule, the CHGA concentration was expected to be ~ 0.2-0.4 mM in the local extracellular space just after exocytosis. Therefore, our experimentally determined IC50 of ~ 200 nM in PC12 cells appeared to be physiologically relevant [21]. Similar results were obtained in bovine chromaffin cells, neurite-bearing (post-nerve growth factor treatment) PC12 cells [21] and in primary cultures of hippocampal neurons [52].
Figure 1.
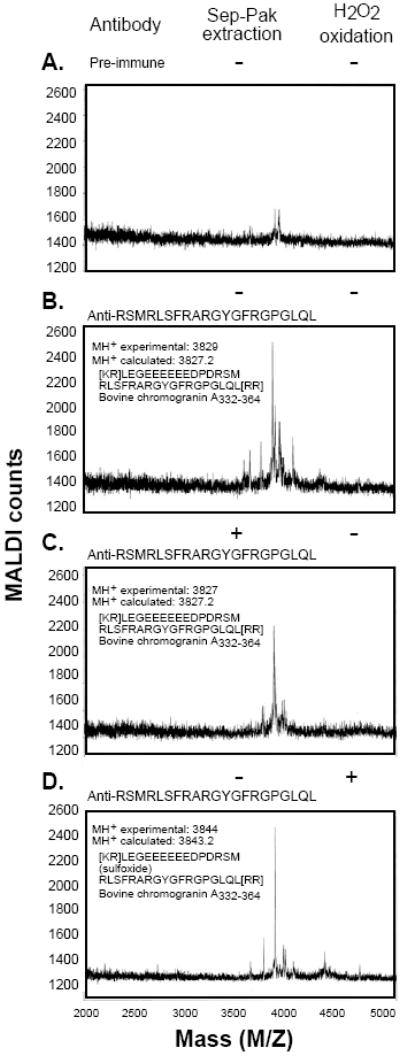
Identification of CST in immunoprecipitated bovine adrenal medullary chromaffin granules. Aliquots (200 ml) of the low molecular weight chromaffin granule peptides (devoid of full length CHGA) separated by gel filtration were immunoprecipitated (20 ml of anti-bovine CST antiserum) and then subjected to MALDI mass spectrometry (1–2 ml). (A) Immunoprecipitation by preimmune serum. (B) Immunoprecipitation by rabbit anti-bovine CST antiserum. (C) Immunoprecipitation by rabbit anti-bovine CST antiserum, followed by adsorption and elution from a C-18 (Sep-Pak) cartridge. (D) Immunoprecipitation by rabbit anti-bovine CST after M346 oxidation by 10 μM H2O2. (Reproduced with permission from The American Society for Biochemistry & Molecular Biology).
2. Proteolytic processing of CHGA to generate CST
Like the other pro-hormones, CHGA contains 8-10 dibasic residue sites, which are considered as potential sites for proteolytic cleavage [1]. Our first indication that CST was generated from CHGA came from the observation that low molecular weight chromaffin vesicle peptides, identified with an antiserum directed against synthetic CST, inhibited secretory activity [21]. Subsequently, we found extensive processing of CST in CHGA by evaluating CST radioimmunoassay of size-fractionated chromaffin granules. The major CST form identified by matrix-assisted laser desorption ionization (MALDI) mass spectrometry and confirmed by diagnostic M346 oxidation was bovine CHGA332–364, which is flanked by dibasic sites (Fig. 2A-D) [53]. Of note, the preferred cleavage site for PC1 and PC2 prohormone convertases is at the COOH-terminal sides of paired basic residues [54-56]. We also detected human CHGA340–372 as the major form in human pheochromocytoma chromaffin granules [53]. Subsequent studies identified secretion of CST (bCHGA344-364: RSMRLSFRARGYGFRGPGLQL; calculated m/z = 2425.8) from primary cultures of bovine chromaffin cells in response to KCl depolarization [57]. Besides the well-established subtilisin-like serine proteases, we have found that in vitro digestion of recombinant CHGA with a serine protease plasmin generated a 14-amino acid from the C-terminal end of CST (CHGA360-373) [58-60], which showed nicotinic inhibition of catecholamine secretion (IC50 of ~2-3 μM). Because of the recent revelation that cysteine protease cathepsin L (CTSL) acts as a novel enzyme for proteolytic processing of neuropeptides [61-63], we first determined its localization within chromaffin granules/vesicles, and then identified active CST-region fragments (CHGA360-373) by proteolytic cleavage of CHGA [64]. Of note, both plasmin and CTSL were unable to cleave the variant CHGA-P370L to generate functional CST peptide (CHGA360-373).
Figure 2.
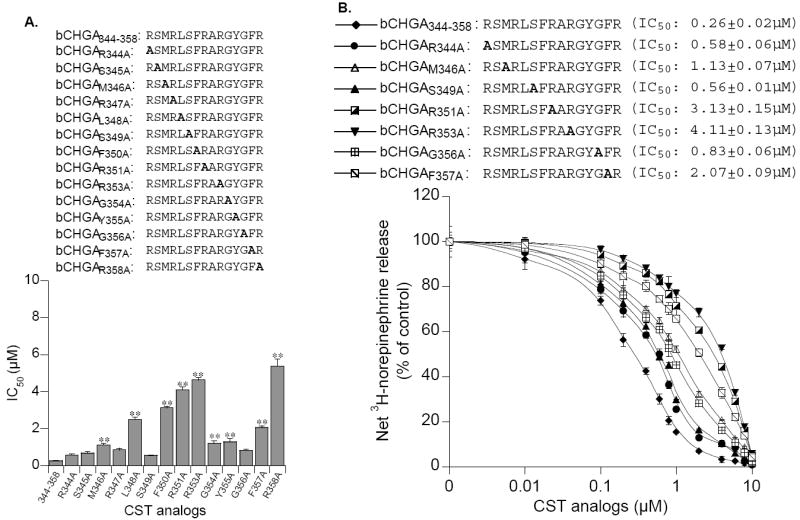
Identification of crucial amino acids in the active core of CST for inhibition of catecholamine release. PC12 cells prelabeled with [3H]L-norepinephrine were incubated with 60 μM nicotine, with or without logarithmically ascending doses (0.01 to 10 μM) of bovine CST for 30 min. (A). Effect of alanine substitution of individual amino acids. Amino acids preceding the numbers 344-358 represent the particular amino acid (and its position) substituted by alanine. For example, in bCHGAR344A, R344 is substituted by A. Alanine substitutions are also shown in bold letters. (B). Graphic comparison of crucial amino acids in the active CST core (bovine CHGA344-358) sequence for blockade of nicotinic cholinergic-stimulated catecholamine release. Control (100%) release is that in the presence of nicotine (60 μM) stimulation alone. Results are shown as the mean ± SEM. IC50 values of each peptide for inhibition of secretion are given in parentheses. bCHGA, Bovine chromogranin A. (Reproduced with permission from The Endocrine Society).
3. CST inhibition of catecholamine release
A. In vitro effects in PC12 and bovine chromaffin cells
Our initial studies with bovine CST (bCHGA344-364) showed dose-dependent inhibition of nicotine-evoked catecholamine secretion from PC12 cells with an IC50 of ~0.2 μM) (Fig. 1) [21]. Similar inhibition of nicotine-induced catecholamine secretion was seen in neurite-(NGF treatment for 5 days) bearing PC12 cells [65], primary cultures of bovine adrenal chromaffin cells [66] and rat hippocampal neurons [67]. In addition, the IC50 values for CST blockade in voltage-clamped oocytes expressing several combinations of neuronal nAChR subunits including α3β4, α3β2, α4β2, and α7 were found to be 0.3 μM for α7, 0.4 μM for both α3β2 and α3β4, and 1.7 μM for α4β2 receptors [68]. Testing N- and C-terminal, as well as bidirectional deletions, of CST in PC12 cells revealed that a completely active core sequence of CST is constituted by the 15 N-terminal amino acids of CST (bCHGA344-358) [69]. Selective substitution of R by A implicated arginine residues at positions 351, 353 and 358 as crucial for inhibitory activity of CST [70], which were supported by our modeling [70] and NMR studies [71]. Further studies utilizing a series of single amino acid truncations or single residue substitutions by alanine, uncovered important roles of the following amino acids for their effects on profound suppression of nicotine-evoked catecholamine secretion compared to wild-type CST: L348A (by ~9.6-fold), and F350A (by ~12-fold), R351A (by ~15.7-fold, R353A (by ~17.8-fold) and R358A (by ~20.7-fold) [69] (Fig. 3).
Figure 3.
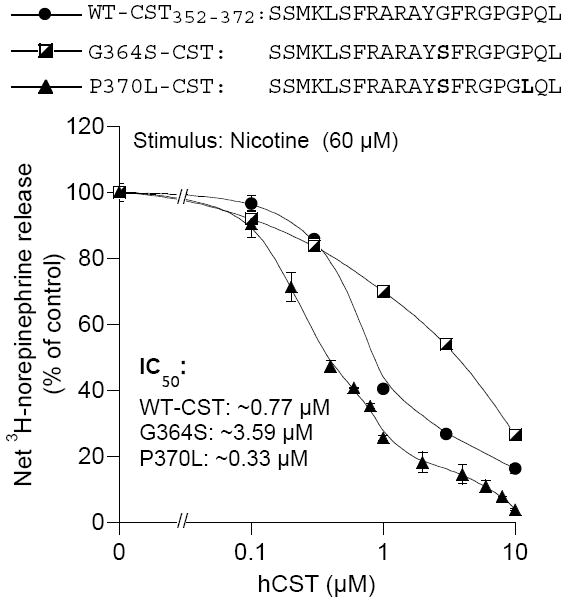
Altered efficacy of nicotinic inhibition by naturally occurring human CST variant peptides. PC12 cells prelabeled with [3H]L-norepinephrine were incubated with 60 μM nicotine, with or without bovine CST (0.01 to 10 μM) for 30 min. Control (100%) release is that in the presence of nicotine (60 μM) stimulation alone. Results are shown as the mean ± SEM. (Reproduced with permission from The University of Chicago Press).
We discovered three human variants of CST: G364S, P370L, and R374Q from resequencing CHGA in 180 individuals (2n = 360 chromosomes) (Table 1). Up to ~4% of the human chromosomes encoded one of these CST amino acid variants. Testing these variants on inhibition of nicotine-induced catecholamine secretion revealed the following rank order of potency: P370L (IC50 0.37±0.03 μM) > wild-type (IC50 0.82±0.02 μM) > G364S (IC50 3.65±0.11 μM) (Fig. 4) [72, 73]. A decrease in potency was paralleled by a decline in the Hill slope, suggesting negative cooperativity at higher doses might underlie the observed loss of potency [73].
Table 1.
Inter-species homologies in the CST sequence in humans and other mammals including sequence variants in human CST (hCHGA352-372). Amino acids at positions variant in human CST are shown in bold type. CHGA: chromogranin A. The typical dibasic proteolytic cleavage site at the carboxy-terminal side of CST is given in brackets, [RR]. For human CHGA, this [RR] site is R373R374.
| Species | Amino acid sequence Variant | CST (frequency) | region in CHGA |
|---|---|---|---|
| Mouse | RSMRLSFRTRGYGFRDPGLQL[RR] | - | CHGA364-384 |
| Rat | RSMRLSFRARGYGFRDPGLQL[RR] | - | CHGA367-387 |
| Cow | RSMRLSFRARGYGFRGPGLQL[RR] | - | CHGA344-364 |
| Pig | RSMRLSFRAPAYGFRGPGLQL[RR] | - | CHGA343-363 |
| Horse | RSMKLSFRARAYGFRGPGLQL[RR] | - | CHGA343-363 |
| Chimp | SSMKLSFRARAYGFRGPGPQL[RR] | - | CHGA354-374 |
| Human | |||
| Wild-type | SSMKLSFRARAYGFRGPGPQL[RR] | - | CHGA352-372 |
| Variant | SSMKLSFRARAYSFRGPGPQL[RR] | G364S (3.1%) | |
| Variant | SSMKLSFRARAYGFRGPGLQL[RR] | P370L (0.6%) | |
| Variant | SSMKLSFRARAYGFRGPGPQL[RQ] | R374Q (0.3%) | |
Figure 4.
CST effects on catecholamine release ex vivo from the superfused rat adrenal gland and in vivo from mouse adrenal gland. (A). Catecholamine secretion from superfused rat adrenal glands was induced by electrical stimulation of the splanchnic nerve (10 Hz, 30 sec), acetylcholine (10 μM, 2 min), and nicotine (10 mM, 2 min), and compared to basal secretion. Perfusates were collected for 2 min for catecholamine assay. Experiments were conducted on 3 different days, and the results were averaged (mean ± SEM) after subtraction of basal (unstimulated) release. *, P < 0.03; **, P < 0.003; ***, P < 0.0001 (stimulation with CST or without [no CST]). (Reproduced with permission from The Endocrine Society). (B). Catecholamine release by nicotinic-cholinergic stimulation and blockade by nicotinic-cholinergic antagonists, including CST. The sympathoadrenal system was activated by the nicotinic-cholinergic agonist nicotine (2.5 mg/kg intraperitoneally) for comparison with vehicle alone (mock). Animals were pretreated 30 min prior to nicotine or vehicle alone (mock) or with nicotinic cholinergic antagonists (either the classical antagonist chlorisondamine 5 mg/kg intraperitoneally, or the novel peptide antagonist CST, 2.5 mg/kg intraperitoneally) to achieve an extracellular target concentration of ~4 μM. In each experiment, n = 6 males were studied, at age 60–70 days. 30 min after nicotine or vehicle, animals were anesthetized (ketamine, 60 mg/kg of body weight; xylazine, 6.4 mg/kg of body weight; acepromazine, 1.2 mg/kg of body weight), and blood was collected for determination of plasma catecholamines. Results are shown as mean ± S.E. (Reproduced with permission from The American Society for Biochemistry & Molecular Biology).
CST inhibition of catecholamine secretion was found to be specific to stimulation by nicotine. CST was unable to inhibit catecholamine secretion when it was induced by secretagogues that bypass the nicotinic-cholinergic receptor (nAChR), including membrane depolarization (by 55 mM KCl) [21] to open voltage-gated calcium channels, an alkaline earth (2 mM BaCl2) to block cell surface K+ channels and thereby depolarize the cell membrane [21], and a Ca2+ ionophore (1 μM ionomycin) to admit extracellular Ca2+ to the cytosol [21] as well as by secretagogues that target different receptors to the nAChR, such as ATP (100 μM) acting on the P2x purinergic receptor [21], or PACAP (0.2 μM) acting on the PAC1 G-protein-coupled receptor [21, 74]. CST inhibition of catecholamine secretion was found to be at the very 1st step in nicotinic cationic signal transduction i.e., at the level of Na+ uptake followed by inhibition of Ca2+ uptake. Since CST inhibition of catecholamine secretion remained unaltered by log10-ascending doses of nicotine (10-1000 μM) we concluded that CST antagonism of nicotine action was non-competitive in nature [21].
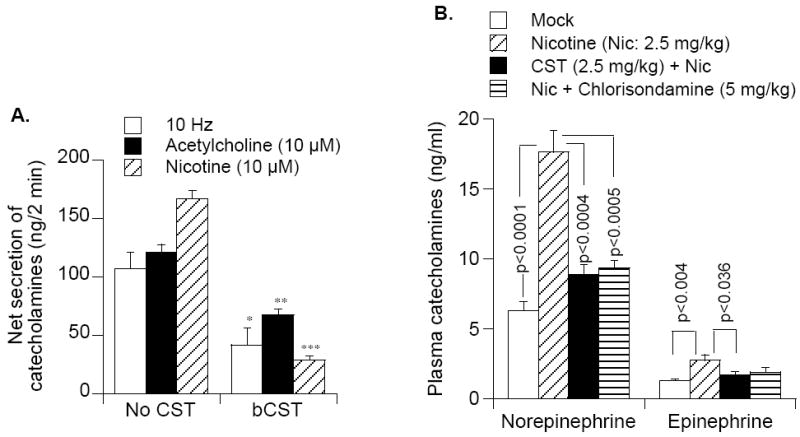
B. Ex vivo effect in superfused rat adrenal gland
The superfused rat adrenal gland model has been widely used by Wakade’s group to distinguish contribution of cholinergic (e.g., acetylcholine) and peptidergic (vasoactive intestinal peptide and pituitary adenylyl cyclase activating polypeptide) in evoking catecholamine secretion [75-77]. In superfused rat adrenal gland, we found that CST caused inhibition of catecholamine secretion induced by both the stimulation of the splanchnic nerve (10 Hz, 30 sec) and by nicotinic cholinergic agonists (acetylcholine or nicotine), although the inhibition was most efficient (80%) for secretion caused by nicotine itself (Fig. 5) [69]. Since nerve stimulation causes release of acetylcholine that may trigger catecholamine secretion by activating both muscarinic and nicotinic receptors in the rodent [78], the finding is consistent with the idea that CST is selective for the nicotinic cholinergic mechanism [21, 69].
Figure 5.
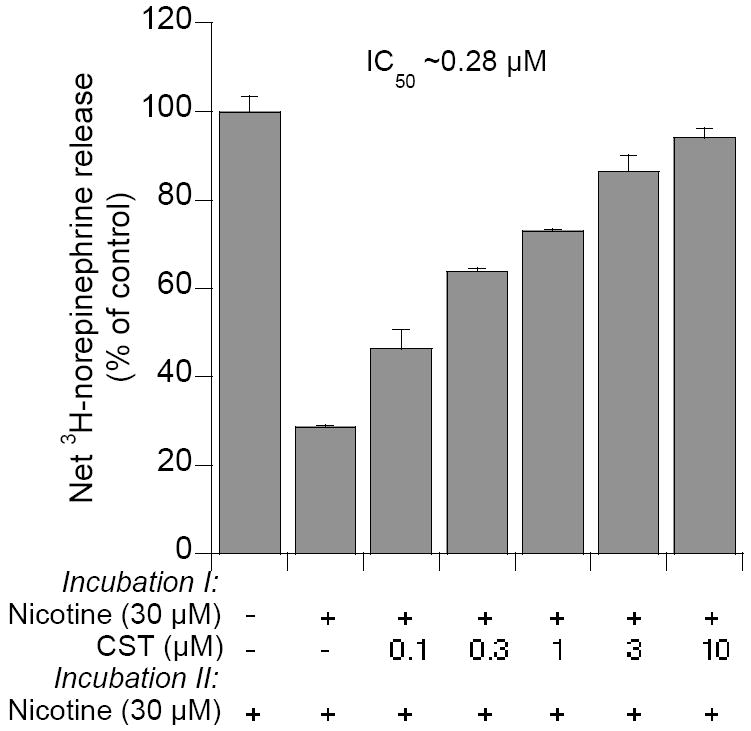
CST inhibition of desensitization of catecholamine release. L-[3H]-norepinephrine preloaded cells were treated with the nicotinic cholinergic agonist nicotine (30 μM) either alone or in combination with logarithmically ascending doses (0.01 to 10 μM) of bovine CST analogs or substance P (0.1 to 10 μM) for 10 min (incubation I), washed twice (6 min each), and rechallenged with nicotine (10 μM) for 10 min (incubation II) before measurement of norepinephrine secretion. Control cells received nicotine only in incubation II. (Reproduced with permission from The American Society for Biochemistry & Molecular Biology).
C. In vivo effect in mice
Direct activation of nAChR by nicotine (2.5 mg/Kg, IP) in transgenic mice (Chga promoter driving expression of the luciferase gene) caused an acute (30 min) 2.7-fold release of catecholamines (norepinephrine and epinephrine) into the bloodstream [79]. Pretreatment with CST (20 nmol/25 g, IP) resulted in 80% inhibition of nicotine-induced catecholamine secretion (Fig. (6)), indicating that this peptide can act in vivo as a nicotinic antagonist. This in vivo finding extends the significance of our in vitro studies of nicotinic signaling to catecholamine secretion [21, 22, 69] and establishes a fundamental role for CHGA and its CST fragment at the nexus of nicotinic cholinergic signaling.
Figure 6.
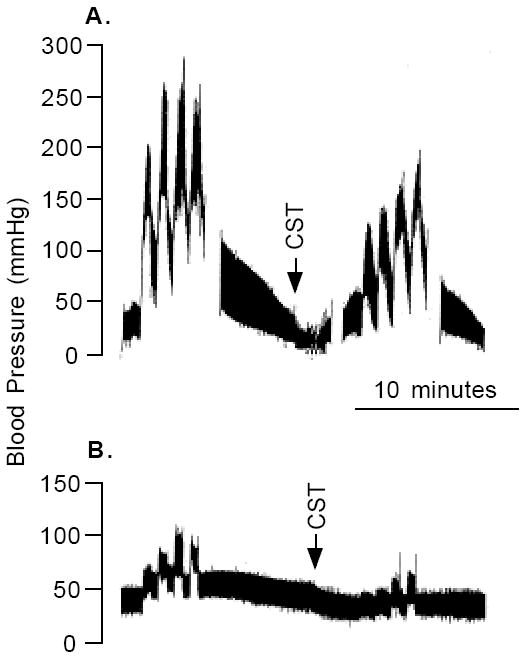
CST effect on BP in vivo in rat. Typical BP response during 7.5 V 20 Hz stimulation of the sympathetic nervous system of a pithed rat, before and after treatment with intravenous CST (0.3 μmol), without (A), or with (B) prior adrenergic blockade by propranolol (2 mg/kg) and phenoxybenzamine (20 mg/kg).
4. CST inhibition of desensitization of catecholamine release
Nicotinic receptors undergo desensitization upon prolonged or repeated exposure to agonist [22]. CST was found to inhibit nicotine-induced desensitization of catecholamine release with an IC50 of ~0.28 μM (Fig. 7) [22]. As with inhibition of nicotine-evoked secretion of catecholamines by CST, inhibition of desensitization by CST was found to be specific to nAChR activation and non-competitive in nature with respect to agonist [22]. Prior nicotinic desensitization caused an 82% diminution of 22Na+ uptake that was markedly inhibited by CST, with an IC50 ~0.31 μM. We believe that CST blockade of nicotinic desensitization of catecholamine release may advantageous to an organism, particularly during stress when CST might sustain catecholamine release to counteract with the stress situation.
Figure 7.
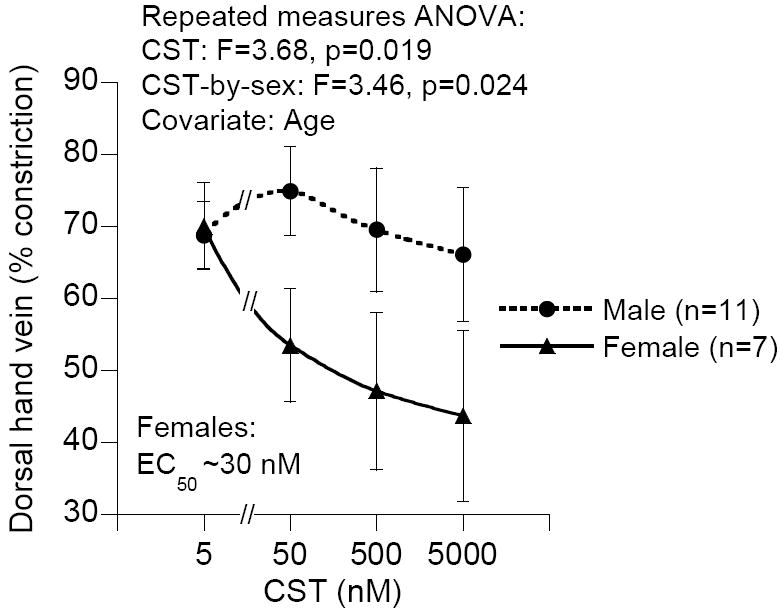
Vasodilation by exogenous CST infusion into the human dorsal hand vein: Stratification by sex. CST exhibited dose-dependent vasodilation (p=0.019) in phenylephrine-induced venoconstriction (~70%), with the effect most prominent in female subjects (p=0.024; covariate: age). The F value (>1) indicates that the means are significantly different from one another. Assuming maximal venodilation with the highest concentration of CST (~5000 nM), the EC50 (semi-maximal effective concentration) for females was ~30 nM.
5. CST modulation of transcription of Chga gene
In cultured chromaffin cells in vitro, exocytotic stimuli promoted the resynthesis of newly released catecholamine storage vesicle contents, a process known as “stimulus-secretion-synthesis coupling” or “stimulus-transcription coupling” [80, 81]. We sought to determine whether this phenomenon occur in vivo. Treatment with nicotine caused ~2-fold increase in expression of the Chga/luciferase transgene, confirming the phenomenon in vivo, and this increase was inhibited >90% by intraperitoneal injection of CST [79]. This establishes an entirely new role for CST on gene expression in vivo.
6. CST stimulation of histamine release from mast cells
Because of the potent vasodilator action of CST in rats [44] we tested whether CST can induce release of the vasodilator histamine from mast cells. The most active N-terminal domain of CST (bCHGA344-358: RSMRLSFRARGYGFR) caused a concentration-dependent (0.01 – 5 μM) release of histamine from peritoneal and pleural mast cells [82]. CST was found to be the most potent activator of histamine release than the wasp venom mastoparan and the neuropeptide substance P. Since CST-evoked histamine release was suppressed by pertussis toxin we suggested involvement of a Gi subunit in CST signaling to histamine release that is distinct from the mechanism of inhibition of catecholamine release from chromaffin cells as described above.
7. CST induction of chemotaxis and the underlying signaling cascade
CHGA, an important constituent of the plaques in Alzheimer’s disease [83, 84], activates monocyte-derived microglia that invade and surround the plaques [85-87]. Based on these findings, we reasoned that CST would regulate monocyte migration. Consistent with our hypothesis we found that CST caused dose-dependent induction of chemotaxis in human monocytes, exerting its maximal effect at 1 nM, which is comparable to the established formylated chemoattractant fMLP [48]. At the receptor level, CST acts through a tyrosine kinase and a G-protein-coupled receptor involving sphingosine 1-phosphate. At the post-receptor signaling pathway level, CST signals through phosphoinositide 3-kinase, nitric oxide and mitogen activated protein kinase dependent pathways. Evaluation of CST effects in animal models of inflammation or Alzheimer’s disease is crucial to determine the biological relevance of the chemotactic effect of CST.
8. CST action on innate immunity
The innate immunity refers to the inborn system of first defense against microorganisms. It is triggered by a range of natural cationic peptides isolated from insect lymph, frog skin, mammalian neutrophil granules and plants, and several CHGA peptides, including vasostatin-I [88] and prochromacin [89]. Because of the highly cationic nature of CST, a characteristic feature of the antibacterial compound, we reasoned that CST would act as an antibacterial peptide. The N-terminal domain of CST (CHGA344-358), containing arginine-rich amphiphilic domain, inhibited growth of Gram-positive and Gram-negative bacteria, a variety of filamentous fungi and several forms of yeasts [25], without showing any hemolytic activity. This peptide rapidly passes through the cell membrane, accumulates in the inner part of the cells and possibly acts on intracellular targets. By using Western blot with specific antibody we have also identified several CST-containing fragment (hCHGA340-394) in the leukotoxin class S Panton-Valentine (2.3 nM) and leukotoxin class F Panton-Valentine (0.6 nM)-stimulated secretion medium of human polymorphonuclear neutrophils (PMN) [49]. Recently, it has been shown by Metz-Boutigue’s group that CST can penetrate into PMN, which basically qualifies CST to be a new member of the cell penetrating peptide family. In addition, they have shown that CST can penetrate into PMNs by a calmodulin-regulated calcium independent phospholipase A2 pathway [39]. Subsequently, we have detected CHGA in keratinocytes and demonstrated it’s processing to CST in human skin, penetrating through human epidermis and exhibiting inhibitory potencies against skin pathogens [102]. CST expression in murine skin was found to have increased in response to injury and infection, which showed potential for increased protection against infection [50]. These findings demonstrate a direct link between the neuroendocrine and immune systems.
Because of the inhibitory effects of antimicrobial peptides such as scorpine, magainin 2, cecropin B, defensin, and dermaseptin S3 and S4 on inhibition of growth of malarial parasite [90-93], Metz-Boutique and Candolfi’s groups recently tested in vitro the effects of CST on growth of Plasmodium falciparum and found that CST acts as a potent inhibitor of the chroloquine-sensitive strain of P. falciparum 3D7, the chloroquine-resistant strain 7G8 as well as the multidrug-resistant strain W2 [94]. It is believed that CST exerts antimalarial activity possibly by inactivating plasmepsins, the aspartic proteases that are involved in the degradation of the host cell hemoglobin, providing nutrients for the growth of the malarial parasite [95]. They identified that the N-terminal serine residue in hCST is essential for maximal inhibition of growth of the malarial parasite. Measurement of plasma CST concentration in patients with malarial infection would establish a very important function of CST and may establish CST as a new player in protecting humans against malarial disease.
9. CST effect on cardiovascular system
A. Vasodilator effect of CST in rat and human
Because of the potent catecholamine release inhibitory effect of CST, we tested whether CST exerts effects on cardiovascular system. Intravenous administration of CST to rats reduced pressor responses to activation of sympathetic outflow by electrical (7.5 V 20 Hz) stimulation (Fig. 8A-B) [44]. The CST effect persisted even after adrenergic (α [phenoxybenzamine, 20 mg/kg IP] plus β [propranolol, 2mg/kg IV]) blockade [44]. Since the vasodepressor effect of CST was blocked by a histamine H1 receptor antagonist (hydroxyzine, 5mg/kg IV) and CST elevated endogenous circulating histamine 21-fold coupled with exogenous histamine mimicking the vasodepressor actions of CST, we concluded that CST is a potent vasodilator in vivo whose actions appear to be mediated, at least in part, by histamine release and action at H1 receptors.
Figure 8.
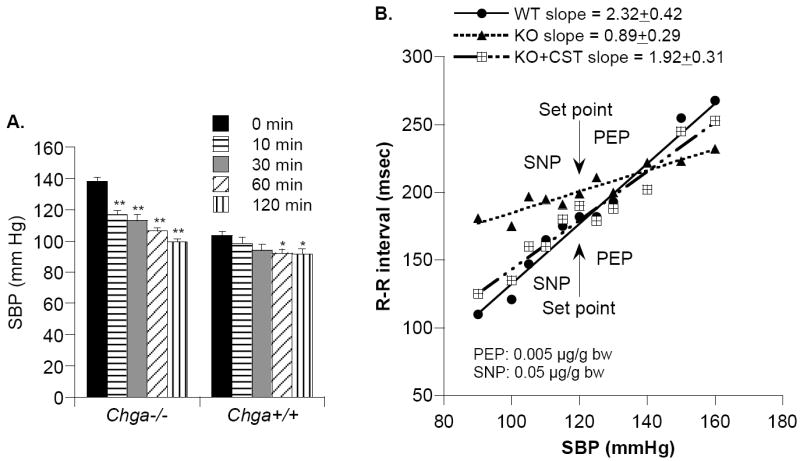
CST reversal of heightened SBP and dampened baroreflex sensitivity in Chga-KO mice. (A). Rescue from elevated SBP by exogenous CST: exaggerated SBP fall in Chga-KO mice. SBP was monitored by telemetry before and after administration of CST (2.5mg/kg body weight, IP) at time 0 in wild type (WT; n=4) and Chga-KO (n=4) mice. Results were analyzed by 2-way, repeated-measures ANOVA, evaluating the effects of time (p<0.001), mouse strain (p=0.009), or strain/peptide interaction (p<0.001). (B). Baroreceptor slope after treatment with phenylephrine (PE: 0.005 μg/g bw iv) or sodium nitroprusside (SNP: 0.05 μg/g bw iv) in unconscious WT and Chga-KO mice, or after supplementation of CST (4 μg/g bw iv) in KO mice. Slopes in line drawings are presented from one representative animal per group. The slope values presented at the top of the Figure are the mean values ± one SEM (msec/mmHg; n=8 animals/group). “Set point” refers to the initial/resting/starting point for each animal (for SBP, in mmHg, and R-R interval, in msec), prior to administration of drugs.
We have also evaluated the potential vasodilator effect of CST in 18 healthy human subjects (male and female) by infusing CST to achieve target concentrations of ~50, ~500, ~5000 nM into dorsal hand veins without systemic counter-regulation, after pharmacological venoconstriction with phenylephrine. CST caused dose-dependent vasodilation predominantly in female subjects after phenylephrine-induced preconstriction to 69% (Fig. 9). Of note, the EC50 (~30 nM) for vasodilation induced by CST was the same order of magnitude to circulating endogenous CST (4.4 nM) [45]. We also found that despite low CHGA precursor concentrations, female subjects had higher plasma CST levels than males, which reflects increased processing of CHGA-to-CST. These findings indicate that CST may contribute to regulation of endogenous vascular tone and influence the complex predisposition to hypertension.
Figure 9.
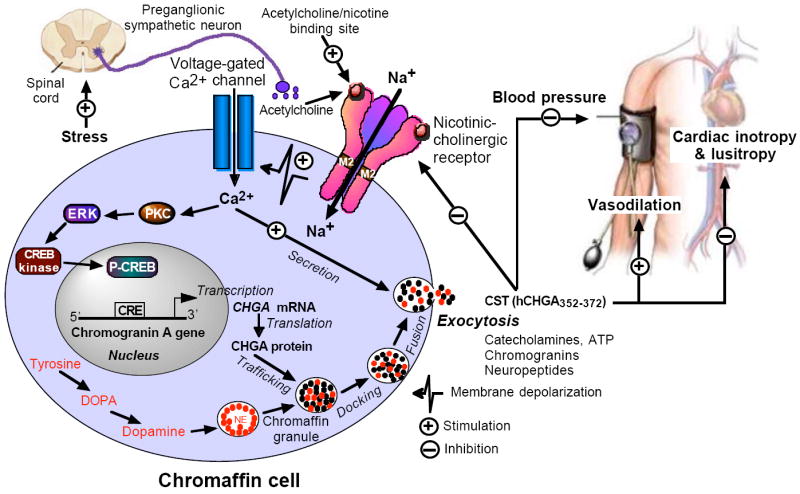
Model showing the autocrine-paracrine homeostatic regulation (negative-feedback) of catecholamine secretion by CST and its regulation of cardiovascular parameters including BP, cardiac contractility and vasodilation. Binding of nicotine (acetylcholine surrogate) to the nicotinic-cholinergic receptor induces extracellular Na+ influx resulting in depolarization of the cell membrane that causes influx of calcium through voltage-gated calcium channels. Influx of calcium induces both catecholamine release by exocytosis (all-or-none secretion) and Chga gene transcription through a pathway involving activation of protein kinase C (PKC) and mitogen activated protein kinase (MAPK). CST formed in and secreted from chromaffin granules inhibits subsequent catecholamine secretion, decreases BP and cardiac contractility, and induces vasodilation. NE, norepinephrine; ERK, extracellular signal-regulated kinase; CRE, cAMP-response element; CREB, cAMP-response element-binding protein; p-CREB: phosphorylated CREB.
B. CST actions on lowering of blood pressure in rodents and humans
Hypertension is a complex trait with an ill-defined genetic predisposition, in which adrenergic mechanisms seem to be involved even at the early stages. Since excess sympathetic activity is implicated in causing hypertension, and alterations in sympathetic responses may occur in normotensive relatives of patients even prior to the onset of hypertension, it is expected that the CST mechanism might be altered in hypertension or in individuals at risk for development of hypertension. Consistent with this hypothesis, we found that the plasma concentration of CST is diminished not only in established cases of essential (hereditary) hypertension, but also in the still-normotensive offspring of patients with hypertension (FH+) [42]. This indicates that an early deficiency in CST might play a pathogenic role in the subsequent development of hypertension, suggesting a pathophysiologic mechanism linking CST to hypertension.
Consistent with the human findings, we detected high blood pressure (BP) and higher plasma catecholamines in mice after targeted ablation of the Chga gene (Chga-KO). Of note, the elevated BP was rescued by insertion of the human CHGA gene in the Chga null background [43], consistent with a hypotensive effect of CHGA. Furthermore, CST replacement rescued Chga-KO mice from the high resting BP (Fig. 8A) and plasma catecholamines. We have not yet determined whether this effect of CST was secondary to a histamine release from mast cells, as we have seen in our studies in the rat. Immobilization stress in telemetered mice caused increments in SBP and HR in both WT and Chga-KO mice, with higher maxima but blunted increments in the KO state [47]. CST replacement selectively diminished stress-induced increments in BP and HR in KO mice, implicating CST as an antihypertensive peptide even in stressful conditions. CST administration (30 min) rescued KO mice from higher plasma catecholamine, indicating that the CST restoration of elevated BP in KO mice [43] likely resulted from CST inhibition of catecholamine secretion from chromaffin cells.
To establish a functional role for CST in the central nervous system, Gaede et al. tested the effects of CST in the spinal cord by intrathecal injection of CST, in conjunction with nicotine and isoproterenol in the anesthetized rat. CST attenuated the hypotensive effect of isoproterenol and the hypertensive effect of nicotine on mean arterial pressure, splanchnic sympathetic nerve activity, and heart rate [96]. Based on the above findings, the authors concluded that CST antagonizes both central nicotinic acetylcholine receptors and β-adrenoceptors that are involved in cardiovascular regulation in vivo.
Based on the above findings we can state that CST regulates BP by acting as an inhibitor of peripheral [43, 45] as well as central [96] nicotinic-cholinergic receptor and β-adrenoceptors [46, 96]. We are yet to ascertain how CST modulates activities both nicotinic-cholinergic and adrenoceptor. However, our unpublished observations seem to suggest that CST can regulate BP by elevating reactive oxygen species as well as lipid peroxidation and depletion of nitric oxide (Gayen JR et al. unpublished observation).
C. Direct cardiovascular effects of CST
We have found that circulating levels of CST decrease in patients with essential hypertension [42] and targeted ablation of the Chga gene in mice increases BP, which can be “rescued” by replacement with CST [43], indicating a direct role of CST in preventing hypertension. This profound vasoreactivity prompted us to test the direct cardiovascular effects of CST on myocardial and coronary functions. In the Langendorff-perfused rat heart, CST dose-dependently increased heart rate and coronary pressure and decreased left ventricular pressure, and both positive and negative LVdP/dt. CST also abolished isoproterenol-induced positive inotropism and lusitropism [46]. In addition, CST inhibited endothelin-1-induced positive inotropism and coronary constriction. Signaling studies indicate that CST acts through β2-ARs-Gi/o protein-NO-cGMP signaling pathways to exert its cardiosuppressive effect. Thus, in addition to its important role in the control of BP, CST is now emerging as a peptide that has direct cardiovascular actions under both basal and stimulated conditions, suggesting that the negative inotropism and lusitropism of CST may be important components of its hypotensive action.
In the hearts of homeotherms (e.g., mammals), the coronary vascular endothelium and endocardial endothelium act in concert to modulate humorally myocardial activity, making contribution of endocardial endothelium in the paracrine regulation of myocardial function difficult to define [97]. In contrast, in the avascular frog heart endocardial endothelium is the only barrier between the superfusing blood and the subjacent myocardial microenvironment and is therefore a unique model to analyze its autocrine/paracrine role in the transduction of blood-borne endoluminal chemical stimuli, which can target the myocardium [98]. Therefore, to delineate CST’s direct myocardiotropic action, we used the avascular frog heart as a bioassay where CST dose-dependently decreased stroke volume and stroke work, with a threshold concentration of 11 nM, which is comparable to the in vivo level of the peptide (~2-4 nM) [99]. In addition, CST inhibited the positive inotropic effect induced by isoproterenol or endothelin-1.
D. CST regulation of autonomic function
Cardiovascular performance is controlled by the autonomic nervous system (ANS). Beat-to-beat fluctuation in the heart rate (HR) is a balanced consequence of ANS tone to the heart, both sympathetic (increasing HR) and parasympathetic (decreasing HR). We found that humans with a genetic variation in the CST region, particularly G364S, displayed alterations in baroreceptor function, both parasympathetic and sympathetic. The G/S heterozygotes displayed increased baroreceptor slope during upward and downward deflections (by ~47 and ~44%, respectively), increased cardiac parasympathetic index (by ~2.4-fold), and decreased cardiac sympathetic index (by ~26%) when compared with the G/G homozygotes. This CST variant seems to reduce risk of developing hypertension, especially in men [52].
Abnormalities in baroreflex sensitivity (BRS) in experimental [100, 101] and human hypertension have been demonstrated, with hypertensive subjects exhibiting diminished BRS compared with their normotensive counterparts [102-104]. Additionally, the family history of hypertension is associated with lower BRS in both normotensive and hypertensive offspring [105]. Since genetic variation at the human CHGA locus results in alterations in BP in the population, and that targeted ablation of the mouse Chga locus results in profound hypertension [43], we studied potential mechanisms of such changes in the ANS, using physiological, biochemical, and pharmacological probes. Consistent with hypertensive subjects, the BRS slope in Chga-KO mice was decreased by ~3-fold in response to reflex bradycardia caused phenylephrine-induced hypertension and reflex tachycardia caused by sodium nitroprusside-induced hypotension (Fig. 11) [47]. In addition, the set point was found to have increased in KO mice. Of note, CST replacement restored dampened BRS slope in KO mice (Fig. 11), indicating that CST resets the entire ANS reflex arc to restore normal cardiovascular function. To probe the relative roles of endogenous/basal sympathetic versus parasympathetic tone in control of BP and HR, we employed the muscarinic-cholinergic antagonist atropine or the b-adrenergic antagonist propranolol where HR and BP responses to each antagonist were found to have exaggerated in Chga-KO animals. These findings may be attributable to either diminished baroreceptor function, or heightened outflow from both the parasympathetic and sympathetic branches, or both. Since the CHGA and CST mechanisms are altered in human hypertension, our results in this experimental animal model may provide insight into the pathogenesis of this common human disorder.
Conclusions and Perspectives
CST (bCHGA344-364; hCHGA352-372) is now established to act as a potent inhibitor of nicotine-evoked catecholamine secretion (IC50 ~0.2-0.4 μM) in three preparations: in vitro in cultured chromaffin cells, ex vivo in the superfused rat adrenal gland and in vivo in mice. Thus, CST represents a novel, autocrine homeostatic (negative-feedback) mechanism controlling catecholamine release from chromaffin cells and noradrenergic neurons (Fig. 12). CST inhibition of desensitization of catecholamine release might be advantageous to an organism during stress, when the peptide might act to sustain catecholamine release. These basic studies on the mechanisms of action of CST in vitro and in vivo are given a physiological perspective with the observations in humans of reduced plasma CST levels associated with an augmented risk of developing hypertension and increased pressor responses to environmental stressors. The results with human CST variants indicate the possibilities for inter-individual variations in human nicotinic signaling as the human carriers of 364S display profound alterations in autonomic activity in both the parasympathetic and sympathetic branches, which in turn may protect the 364Ser carrier against future development of hypertension, especially in males. The vasodilatory effects of CST in human hand vein, especially in females indicate that CST may contribute to sex differences in endogenous vascular tone and influence the complex predisposition to hypertension. Because of the potent inhibition of the inotropic and lusitropic properties of the rodent heart, CST is now considered as a novel cardiac modulator, which could protect the heart against against excessive sympathetic drive such as that seen in hypertensive cardiomyopathy (Fig. 12). Restoration (elevation) of BRS sensitivity after exogenous CST in Chga-KO mice indicates that CST resets the entire autonomic nervous reflex arc to restore normal cardiovascular function. Evaluation of CST effects in animal models of inflammation or Alzheimer’s disease is crucial to determine the biological relevance of the chemotactic effect of CST. The antimicrobial function of CST against a wide assortment of skin pathogens and its upregulation upon skin injury provide a direct link between the antimocrobial defense of the skin and the neuroendocrine peptide CST. CST is thus emerging as a very important peptide regulating multiple functions and bears all the potentials to be a therapeutic agent to treat multiple diseases like hypertension, cardiomyopathy, inflammation, malaria and skin infection.
Acknowledgments
The authors are deeply indebted to all the co-workers for their immense contributions in this field. This work was supported by grants from the Department of Veterans Affairs and the National Institutes of Health.
Grant support: Department of Veterans Affairs and the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Winkler H, Fischer-Colbrie R. The chromogranins A and B: the first 25 years and future perspectives. Neuroscience. 1992;49:497–528. doi: 10.1016/0306-4522(92)90222-N. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taupenot L, Harper KL, O’Connor DT. Mechanisms of disease: The chromogranin-secretogranin family. New Engl J Med. 2003;348:1134–49. doi: 10.1056/NEJMra021405. [DOI] [PubMed] [Google Scholar]
- 3.Helle KB, Corti A, Metz-Boutigue MH, Tota B. The endocrine role for chromogranin A: A prohormone for peptides with regulatory properties. Cell Mol Life Sci. 2007 doi: 10.1007/s00018-007-7254-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montero-Hadjadje M, Vaingankar S, Elias S, Tostivint H, Mahata SK, Anouar Y. Chromogranins A and B and secretogranin II: evolutionary and functional aspects. Acta Physiol (Oxf) 2008;192:309–24. doi: 10.1111/j.1748-1716.2007.01806.x. [DOI] [PubMed] [Google Scholar]
- 5.Takiyyuddin MA, Cervenka JH, Hsiao RJ, Barbosa JA, Parmer RJ, O’Connor DT. Chromogranin A. Storage and release in hypertension. Hypertension. 1990;15:237–46. doi: 10.1161/01.hyp.15.3.237. [DOI] [PubMed] [Google Scholar]
- 6.Takiyyuddin MA, Cervenka JH, Sullivan PA, Pandian MR, Parmer RJ, Barbosa JA, O’Connor DT. Is physiologic sympathoadrenal catecholamine release exocytotic in humans? Circulation. 1990;81:185–95. doi: 10.1161/01.cir.81.1.185. [DOI] [PubMed] [Google Scholar]
- 7.Kim T, Tao-Cheng J, Eiden LE, Loh YP. Chromogranin A, an “On/Off” Switch Controlling Dense-Core Secretory Granule Biogenesis. Cell. 2001;106:499–509. doi: 10.1016/s0092-8674(01)00459-7. [DOI] [PubMed] [Google Scholar]
- 8.Simon JP, Aunis D. Biochemistry of the chromogranin A protein family. Biochem J. 1989;262:1–13. doi: 10.1042/bj2620001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huttner WB, Gerdes HH, Rosa P. The granin (chromogranin/secretogranin) family. Trends Biochem Sci. 1991;16:27–30. doi: 10.1016/0968-0004(91)90012-k. [DOI] [PubMed] [Google Scholar]
- 10.O’Connor DT. Chromogranin: widespread immunoreactivity in polypeptide hormone producing tissues and in serum. Regul Pept. 1983;6:263–80. doi: 10.1016/0167-0115(83)90145-3. [DOI] [PubMed] [Google Scholar]
- 11.Iacangelo A, Affolter HU, Eiden LE, Herbert E, Grimes M. Bovine chromogranin A sequence and distribution of its messenger RNA in endocrine tissues. Nature. 1986;323:82–6. doi: 10.1038/323082a0. [DOI] [PubMed] [Google Scholar]
- 12.Benedum UM, Baeuerle PA, Konecki DS, Frank R, Powell J, Mallet J, Huttner WB. The primary structure of bovine chromogranin A: a representative of a class of acidic secretory proteins common to a variety of peptidergic cells. Embo J. 1986;5:1495–502. doi: 10.1002/j.1460-2075.1986.tb04388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konecki DS, Benedum UM, Gerdes HH, Huttner WB. The primary structure of human chromogranin A and pancreastatin. J Biol Chem. 1987;262:17026–30. [PubMed] [Google Scholar]
- 14.Wu HJ, Rozansky DJ, Parmer RJ, Gill BM, O’Connor DT. Structure and function of the chromogranin A gene. Clues to evolution and tissue-specific expression. J Biol Chem. 1991;266:13130–4. [PubMed] [Google Scholar]
- 15.Rozansky DJ, Wu H, Tang K, Parmer RJ, O’Connor DT. Glucocorticoid activation of chromogranin A gene expression. Identification and characterization of a novel glucocorticoid response element. J Clin Invest. 1994;94:2357–68. doi: 10.1172/JCI117601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tatemoto K, Efendic S, Mutt V, Makk G, Feistner GJ, Barchas JD. Pancreastatin, a novel pancreatic peptide that inhibits insulin secretion. Nature. 1986;324:476–8. doi: 10.1038/324476a0. [DOI] [PubMed] [Google Scholar]
- 17.O’Connor DT, Cadman PE, Smiley C, Salem RM, Rao F, Smith J, Funk SD, Mahata SK, Mahata M, Wen G, Taupenot L, Gonzalez-Yanes C, Harper KL, Henry RR, Sanchez-Margalet V. Pancreastatin: multiple actions on human intermediary metabolism in vivo, variation in disease, and naturally occurring functional genetic polymorphism. J Clin Endocrinol Metab. 2005;90:5414–25. doi: 10.1210/jc.2005-0408. [DOI] [PubMed] [Google Scholar]
- 18.Gayen JR, Saberi M, Schenk S, Biswas N, Vaingankar SM, Cheung WW, Najjar SM, O’Connor DT, Bandyopadhyay G, Mahata SK. A novel pathway of insulin sensitivity in chromogranin a null mice: A crucial role for pancreastatin in glucose homeostasis. J Biol Chem. 2009;284:28498–509. doi: 10.1074/jbc.M109.020636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strub JM, Goumon Y, Lugardon K, Capon C, Lopez M, Moniatte M, Van Dorsselaer A, Aunis D, Metz-Boutigue MH. Antibacterial activity of glycosylated and phosphorylated chromogranin A-derived peptide 173-194 from bovine adrenal medullary chromaffin granules. J Biol Chem. 1996;271:28533–40. doi: 10.1074/jbc.271.45.28533. [DOI] [PubMed] [Google Scholar]
- 20.Aardal S, Helle KB, Elsayed S, Reed RK, Serck-Hanssen G. Vasostatins, comprising the N-terminal domain of chromogranin A, suppress tension in isolated human blood vessel segments. J Neuroendocrinol. 1993;5:405–12. doi: 10.1111/j.1365-2826.1993.tb00501.x. [DOI] [PubMed] [Google Scholar]
- 21.Mahata SK, O’Connor DT, Mahata M, Yoo SH, Taupenot L, Wu H, Gill BM, Parmer RJ. Novel autocrine feedback control of catecholamine release. A discrete chromogranin A fragment is a noncompetitive nicotinic cholinergic antagonist. J Clin Invest. 1997;100:1623–33. doi: 10.1172/JCI119686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahata SK, Mahata M, Parmer RJ, O’Connor DT. Desensitization of catecholamine release: The novel catecholamine release-inhibitory peptide catestatin (chromogranin A344-364) acts at the receptor to prevent nicotinic cholinergic tolerance. J Biol Chem. 1999;274:2920–8. doi: 10.1074/jbc.274.5.2920. [DOI] [PubMed] [Google Scholar]
- 23.Schober M, Howe PR, Sperk G, Fischer-Colbrie R, Winkler H. An increased pool of secretory hormones and peptides in adrenal medulla of stroke-prone spontaneously hypertensive rats. Hypertension. 1989;13:469–74. doi: 10.1161/01.hyp.13.5.469. [DOI] [PubMed] [Google Scholar]
- 24.O’Connor DT, Takiyyuddin MA, Printz MP, Dinh TQ, Barbosa JA, Rozansky DJ, Mahata SK, Wu H, Kennedy BP, Ziegler MG, Wright FA, Schlager G, Parmer RJ. Catecholamine storage vesicle protein expression in genetic hypertension. Blood Press. 1999;8:285–95. doi: 10.1080/080370599439508. [DOI] [PubMed] [Google Scholar]
- 25.Takiyyuddin MA, De Nicola L, Gabbai FB, Dinh TQ, Kennedy B, Ziegler MG, Sabban EL, Parmer RJ, O’Connor DT. Catecholamine secretory vesicles. Augmented chromogranins and amines in secondary hypertension. Hypertension. 1993;21:674–9. doi: 10.1161/01.hyp.21.5.674. [DOI] [PubMed] [Google Scholar]
- 26.Takiyyuddin MA, Parmer RJ, Kailasam MT, Cervenka JH, Kennedy B, Ziegler MG, Lin MC, Li J, Grim CE, Wright FA, et al. Chromogranin A in human hypertension. Influence of heredity. Hypertension. 1995;26:213–20. doi: 10.1161/01.hyp.26.1.213. [DOI] [PubMed] [Google Scholar]
- 27.O’Connor DT, Zhu G, Rao F, Taupenot L, Fung MM, Das M, Mahata SK, Mahata M, Wang L, Zhang K, Greenwood TA, Shih PA, Cockburn MG, Ziegler MG, Stridsberg M, Martin NG, Whitfield JB. Heritability and genome-wide linkage in US and australian twins identify novel genomic regions controlling chromogranin a: implications for secretion and blood pressure. Circulation. 2008;118:247–57. doi: 10.1161/CIRCULATIONAHA.107.709105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Connor DT, Frigon RP, Sokoloff RL. Human chromogranin A. Purification and characterization from catecholamine storage vesicles of human pheochromocytoma. Hypertension. 1984;6:2–12. doi: 10.1161/01.hyp.6.1.2. [DOI] [PubMed] [Google Scholar]
- 29.Kimura N, Miura W, Noshiro T, Mizunashi K, Hanew K, Shimizu K, Watanabe T, Shibukawa S, Sohn HE, Abe K, Miura Y, Nagura H. Plasma chromogranin A in pheochromocytoma, primary hyperparathyroidism and pituitary adenoma in comparison with catecholamine, parathyroid hormone and pituitary hormones. Endocr J. 1997;44:319–27. doi: 10.1507/endocrj.44.319. [DOI] [PubMed] [Google Scholar]
- 30.Stridsberg M, Husebye ES. Chromogranin A and chromogranin B are sensitive circulating markers for phaeochromocytoma. Eur J Endocrinol. 1997;136:67–73. doi: 10.1530/eje.0.1360067. [DOI] [PubMed] [Google Scholar]
- 31.O’Connor DT, Deftos LJ. Secretion of chromogranin A by peptide-producing endocrine neoplasms. N Engl J Med. 1986;314:1145–51. doi: 10.1056/NEJM198605013141803. [DOI] [PubMed] [Google Scholar]
- 32.Syversen U, Mignon M, Bonfils S, Kristensen A, Waldum HL. Chromogranin A and pancreastatin-like immunoreactivity in serum of gastrinoma patients. Acta Oncol. 1993;32:161–5. doi: 10.3109/02841869309083906. [DOI] [PubMed] [Google Scholar]
- 33.Nikou GC, Lygidakis NJ, Toubanakis C, Pavlatos S, Tseleni-Balafouta S, Giannatou E, Mallas E, Safioleas M. Current diagnosis and treatment of gastrointestinal carcinoids in a series of 101 patients: the significance of serum chromogranin-A, somatostatin receptor scintigraphy and somatostatin analogues. Hepatogastroenterology. 2005;52:731–41. [PubMed] [Google Scholar]
- 34.Ceconi C, Ferrari R, Bachetti T, Opasich C, Volterrani M, Colombo B, Parrinello G, Corti A. Chromogranin A in heart failure; a novel neurohumoral factor and a predictor for mortality. Eur Heart J. 2002;23:967–74. doi: 10.1053/euhj.2001.2977. [DOI] [PubMed] [Google Scholar]
- 35.Omland T, Dickstein K, Syversen U. Association between plasma chromogranin A concentration and long-term mortality after myocardial infarction. Am J Med. 2003;114:25–30. doi: 10.1016/s0002-9343(02)01425-0. [DOI] [PubMed] [Google Scholar]
- 36.Pieroni M, Corti A, Tota B, Curnis F, Angelone T, Colombo B, Cerra MC, Bellocci F, Crea F, Maseri A. Myocardial production of chromogranin A in human heart: a new regulatory peptide of cardiac function. Eur Heart J. 2007;28:1117–27. doi: 10.1093/eurheartj/ehm022. [DOI] [PubMed] [Google Scholar]
- 37.Estensen ME, Hognestad A, Syversen U, Squire I, Ng L, Kjekshus J, Dickstein K, Omland T. Prognostic value of plasma chromogranin A levels in patients with complicated myocardial infarction. Am Heart J. 2006;152:927, e1–6. doi: 10.1016/j.ahj.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 38.Jansson AM, Rosjo H, Omland T, Karlsson T, Hartford M, Flyvbjerg A, Caidahl K. Prognostic value of circulating chromogranin A levels in acute coronary syndromes. Eur Heart J. 2009;30:25–32. doi: 10.1093/eurheartj/ehn513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang D, Shooshtarizadeh P, Laventie BJ, Colin DA, Chich JF, Vidic J, de Barry J, Chasserot-Golaz S, Delalande F, Van Dorsselaer A, Schneider F, Helle K, Aunis D, Prevost G, Metz-Boutigue MH. Two chromogranin a-derived peptides induce calcium entry in human neutrophils by calmodulin-regulated calcium independent phospholipase A2. PLoS One. 2009;4:e4501. doi: 10.1371/journal.pone.0004501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang D, Lavaux T, Sapin R, Lavigne T, Castelain V, Aunis D, Metz-Boutigue MH, Schneider F. Serum concentration of chromogranin A at admission: an early biomarker of severity in critically ill patients. Ann Med. 2009;41:38–44. doi: 10.1080/07853890802199791. [DOI] [PubMed] [Google Scholar]
- 41.Zhang D, Lavaux T, Voegeli AC, Lavigne T, Castelain V, Meyer N, Sapin R, Aunis D, Metz-Boutigue MH, Schneider F. Prognostic value of chromogranin A at admission in critically ill patients: a cohort study in a medical intensive care unit. Clin Chem. 2008;54:1497–503. doi: 10.1373/clinchem.2007.102442. [DOI] [PubMed] [Google Scholar]
- 42.O’Connor DT, Kailasam MT, Kennedy BP, Ziegler MG, Yanaihara N, Parmer RJ. Early decline in the catecholamine release-inhibitory peptide catestatin in humans at genetic risk of hypertension. J Hypertens. 2002;20:1335–45. doi: 10.1097/00004872-200207000-00020. [DOI] [PubMed] [Google Scholar]
- 43.Mahapatra NR, O’Connor DT, Vaingankar SM, Sinha Hikim AP, Mahata M, Ray S, Staite E, Wu H, Gu Y, Dalton N, Kennedy BP, Ziegler MG, Ross J, Jr, Mahata SK. Hypertension from targeted ablation of chromogranin A can be rescued by the human ortholog. J Clin Invest. 2005;115:1942–52. doi: 10.1172/JCI24354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kennedy BP, Mahata SK, O’Connor DT, Ziegler MG. Mechanism of cardiovascular actions of the chromogranin A fragment catestatin in vivo. Peptides. 1998;19:1241–8. doi: 10.1016/s0196-9781(98)00086-2. [DOI] [PubMed] [Google Scholar]
- 45.Fung MM, Salem RM, Mehtani P, Thomas B, Lu CF, Perez B, Rao F, Stridsberg M, Ziegler M, Mahata SK, OC DT. Direct vasoactive effects of the chromogranin A (CHGA) peptide catestatin in humans in vivo. Clin Exp Hypertens. 2009 doi: 10.3109/10641960903265246. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Angelone T, Quintieri AM, Brar BK, Limchaiyawat PT, Tota B, Mahata SK, Cerra MC. The antihypertensive chromogranin a peptide catestatin acts as a novel endocrine/paracrine modulator of cardiac inotropism and lusitropism. Endocrinology. 2008;149:4780–93. doi: 10.1210/en.2008-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gayen JR, Gu Y, O’Connor DT, Mahata SK. Global disturbances in autonomic function yield cardiovascular instability and hypertension in the chromogranin A null mouse. Endocrinology. 2009;150:5027–35. doi: 10.1210/en.2009-0429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Egger M, Beer AG, Theurl M, Schgoer W, Hotter B, Tatarczyk T, Vasiljevic D, Frauscher S, Marksteiner J, Patsch JR, Schratzberger P, Djanani AM, Mahata SK, Kirchmair R. Monocyte migration: a novel effect and signaling pathways of catestatin. Eur J Pharmacol. 2008;598:104–11. doi: 10.1016/j.ejphar.2008.09.016. [DOI] [PubMed] [Google Scholar]
- 49.Briolat J, Wu SD, Mahata SK, Gonthier B, Bagnard D, Chasserot-Golaz S, Helle KB, Aunis D, Metz-Boutigue MH. New antimicrobial activity for the catecholamine release-inhibitory peptide from chromogranin A. Cell Mol Life Sci. 2005;62:377–85. doi: 10.1007/s00018-004-4461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Radek KA, Lopez-Garcia B, Hupe M, Niesman IR, Elias PM, Taupenot L, Mahata SK, O’Connor DT, Gallo RL. The neuroendocrine peptide catestatin is a cutaneous antimicrobial and induced in the skin after injury. J Invest Dermatol. 2008;128:1525–34. doi: 10.1038/sj.jid.5701225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simon JP, Bader MF, Aunis D. Secretion from chromaffin cells is controlled by chromogranin A-derived peptides. Proc Natl Acad Sci U S A. 1988;85:1712–6. doi: 10.1073/pnas.85.5.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rao F, Wen G, Gayen JR, Das M, Vaingankar SM, Rana BK, Mahata M, Kennedy BP, Salem RM, Stridsberg M, Abel K, Smith DW, Eskin E, Schork NJ, Hamilton BA, Ziegler MG, Mahata SK, O’Connor DT. Catecholamine release-inhibitory peptide catestatin (chromogranin A(352-372)): naturally occurring amino acid variant Gly364Ser causes profound changes in human autonomic activity and alters risk for hypertension. Circulation. 2007;115:2271–81. doi: 10.1161/CIRCULATIONAHA.106.628859. [DOI] [PubMed] [Google Scholar]
- 53.Taylor CV, Taupenot L, Mahata SK, Mahata M, Wu H, Yasothornsrikul S, Toneff T, Caporale C, Jiang Q, Parmer RJ, Hook VY, O’Connor DT. Formation of the catecholamine release-inhibitory peptide catestatin from chromogranin A. Determination of proteolytic cleavage sites in hormone storage granules. J Biol Chem. 2000;275:22905–15. doi: 10.1074/jbc.M001232200. [DOI] [PubMed] [Google Scholar]
- 54.Benjannet S, Rondeau N, Day R, Chretien M, Seidah NG. PC1 and PC2 are proprotein convertases capable of cleaving proopiomelanocortin at distinct pairs of basic residues. Proc Natl Acad Sci U S A. 1991;88:3564–8. doi: 10.1073/pnas.88.9.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dhanvantari S, Seidah NG, Brubaker PL. Role of prohormone convertases in the tissue-specific processing of proglucagon. Mol Endocrinol. 1996;10:342–55. doi: 10.1210/mend.10.4.8721980. [DOI] [PubMed] [Google Scholar]
- 56.Azaryan AV, Krieger TJ, Hook VY. Purification and characteristics of the candidate prohormone processing proteases PC2 and PC1/3 from bovine adrenal medulla chromaffin granules. J Biol Chem. 1995;270:8201–8. doi: 10.1074/jbc.270.14.8201. [DOI] [PubMed] [Google Scholar]
- 57.Lee JC, Taylor CV, Gaucher SP, Toneff T, Taupenot L, Yasothornsrikul S, Mahata SK, Sei C, Parmer RJ, Neveu JM, Lane WS, Gibson BW, O’Connor DT, Hook VY. Primary sequence characterization of catestatin intermediates and peptides defines proteolytic cleavage sites utilized for converting chromogranin a into active catestatin secreted from neuroendocrine chromaffin cells. Biochemistry. 2003;42:6938–46. doi: 10.1021/bi0300433. [DOI] [PubMed] [Google Scholar]
- 58.Parmer RJ, Mahata M, Gong Y, Mahata SK, Jiang Q, O’Connor DT, Xi X-P, Miles LA. Processing of chromogranin A by plasmin provides a novel mechanism for regulating catecholamine secretion. J Clin Invest. 2000;106:907–15. doi: 10.1172/JCI7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jiang Q, Taupenot L, Mahata SK, Mahata M, O’Connor DT, Miles LA, Parmer RJ. Proteolytic cleavage of chromogranin A (CgA) by plasmin: selective liberation of a specific bioactive CgA fragment that regulates catecholamine release. J Biol Chem. 2001;276:25022–9. doi: 10.1074/jbc.M101545200. [DOI] [PubMed] [Google Scholar]
- 60.Biswas N, Vaingankar SM, Mahata M, Das M, Gayen JR, Taupenot L, Torpey JW, O’Connor DT, Mahata SK. Proteolytic cleavage of human chromogranin a containing naturally occurring catestatin variants: differential processing at catestatin region by plasmin. Endocrinology. 2008;149:749–57. doi: 10.1210/en.2007-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hook V, Yasothornsrikul S, Greenbaum D, Medzihradszky KF, Troutner K, Toneff T, Bundey R, Logrinova A, Reinheckel T, Peters C, Bogyo M. Cathepsin L and Arg/Lys aminopeptidase: a distinct prohormone processing pathway for the biosynthesis of peptide neurotransmitters and hormones. Biol Chem. 2004;385:473–80. doi: 10.1515/BC.2004.055. [DOI] [PubMed] [Google Scholar]
- 62.Hwang SR, Garza C, Mosier C, Toneff T, Wunderlich E, Goldsmith P, Hook V. Cathepsin L expression is directed to secretory vesicles for enkephalin neuropeptide biosynthesis and secretion. J Biol Chem. 2007;282:9556–63. doi: 10.1074/jbc.M605510200. [DOI] [PubMed] [Google Scholar]
- 63.Yasothornsrikul S, Greenbaum D, Medzihradszky KF, Toneff T, Bundey R, Miller R, Schilling B, Petermann I, Dehnert J, Logvinova A, Goldsmith P, Neveu JM, Lane WS, Gibson B, Reinheckel T, Peters C, Bogyo M, Hook V. Cathepsin L in secretory vesicles functions as a prohormone-processing enzyme for production of the enkephalin peptide neurotransmitter. Proc Natl Acad Sci U S A. 2003;100:9590–5. doi: 10.1073/pnas.1531542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Biswas N, Rodriguez-Flores JL, Courel M, Gayen JR, Vaingankar SM, Mahata M, Torpey JW, Taupenot L, O’Connor DT, Mahata SK. Cathepsin L Co-Localizes with Chromogranin a in Chromaffin Vesicles to Generate Active Peptides. Endocrinology. 2009;150:3547–57. doi: 10.1210/en.2008-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mahata SK, Mahata M, Wu H, Parmer RJ, O’Connor DT. Neurotrophin activation of catecholamine storage vesicle protein gene expression: signaling to chromogranin A biosynthesis. Neuroscience. 1999;88:405–24. doi: 10.1016/s0306-4522(98)00225-5. [DOI] [PubMed] [Google Scholar]
- 66.Livett BG, Boksa P, Dean DM, Mizobe F, Lindenbaum MH. Use of isolated chromaffin cells to study basic release mechanisms. J Auton Nerv Syst. 1983;7:59–86. doi: 10.1016/0165-1838(83)90069-3. [DOI] [PubMed] [Google Scholar]
- 67.De A, Krueger JM, Simasko SM. Tumor necrosis factor alpha increases cytosolic calcium responses to AMPA and KCl in primary cultures of rat hippocampal neurons. Brain Res. 2003;981:133–42. doi: 10.1016/s0006-8993(03)02997-4. [DOI] [PubMed] [Google Scholar]
- 68.Herrero CJ, Ales E, Pintado AJ, Lopez MG, Garcia-Palomero E, Mahata SK, O’Connor DT, Garcia AG, Montiel C. Modulatory mechanism of the endogenous peptide catestatin on neuronal nicotinic acetylcholine receptors and exocytosis. J Neurosci. 2002;22:377–88. doi: 10.1523/JNEUROSCI.22-02-00377.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mahata SK, Mahata M, Wakade AR, O’Connor DT. Primary structure and function of the catecholamine release inhibitory peptide catestatin (chromogranin A344-364): Identification of amino acid residues crucial for activity. Mol Endocrinol. 2000;14:1525–35. doi: 10.1210/mend.14.10.0531. [DOI] [PubMed] [Google Scholar]
- 70.Tsigelny I, Mahata SK, Taupenot L, Preece NE, Mahata M, Khan I, Parmer RJ, O’Connor DT. Mechanism of action of chromogranin A on catecholamine release: molecular modeling of the catestatin region reveals a β-strand/loop/β-strand structure secured by hydrophobic interactions and predictive of activity. Regul Peptides. 1998;77:43–53. doi: 10.1016/s0167-0115(98)00040-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Preece NE, Nguyen M, Mahata M, Mahata SK, Mahapatra NR, Tsigelny I, O’Connor DT. Conformational preferences and activities of peptides from the catecholamine release-inhibitory (catestatin) region of chromogranin A. Regul Pept. 2004;118:75–87. doi: 10.1016/j.regpep.2003.10.035. [DOI] [PubMed] [Google Scholar]
- 72.Wen G, Mahata SK, Cadman P, Mahata M, Ghosh S, Mahapatra NR, Rao F, Stridsberg M, Smith DW, Mahboubi P, Schork NJ, O’Connor DT, Hamilton BA. Both rare and common polymorphisms contribute functional variation at CHGA, a regulator of catecholamine physiology. Am J Hum Genet. 2004;74:197–207. doi: 10.1086/381399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mahata SK, Mahata M, Wen G, Wong WB, Mahapatra NR, Hamilton BA, O’Connor DT. The catecholamine release-inhibitory “catestatin” fragment of chromogranin a: naturally occurring human variants with different potencies for multiple chromaffin cell nicotinic cholinergic responses. Mol Pharmacol. 2004;66:1180–91. doi: 10.1124/mol.104.002139. [DOI] [PubMed] [Google Scholar]
- 74.Taupenot L, Mahata SK, Wu H, O’Connor DT. Peptidergic activation of transcription and secretion in chromaffin cells. cis and trans signaling determinants of pituitary adenylyl cyclase-activating polypeptide (PACAP) J Clin Invest. 1998;101:863–76. doi: 10.1172/JCI1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wakade AR. Studies on secretion of catecholamines evoked by acetylcholine or transmural stimulation of the rat adrenal gland. J Physiol. 1981;313:463–80. doi: 10.1113/jphysiol.1981.sp013676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wakade TD, Blank MA, Malhotra RK, Pourcho R, Wakade AR. The peptide VIP is a neurotransmitter in rat adrenal medulla: physiological role in controlling catecholamine secretion. J Physiol. 1991;444:349–62. doi: 10.1113/jphysiol.1991.sp018882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guo X, Wakade AR. Differential secretion of catecholamines in response to peptidergic and cholinergic transmitters in rat adrenals. J Physiol (Lond) 1994;475:539–45. doi: 10.1113/jphysiol.1994.sp020092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wakade AR, Wakade TD. Contribution of nicotinic and muscarinic receptors in the secretion of catecholamines evoked by endogenous and exogenous acetylcholine. Neuroscience. 1983;10:973–8. doi: 10.1016/0306-4522(83)90235-x. [DOI] [PubMed] [Google Scholar]
- 79.Mahata SK, Mahapatra NR, Mahata M, Wang TC, Kennedy BP, Ziegler MG, O’Connor DT. Catecholamine secretory vesicle stimulus-transcription coupling in vivo. Demonstration by a novel transgenic promoter/photoprotein reporter and inhibition of secretion and transcription by the chromogranin A fragment catestatin. J Biol Chem. 2003;278:32058–67. doi: 10.1074/jbc.M305545200. [DOI] [PubMed] [Google Scholar]
- 80.Tang K, Wu H, Mahata SK, Taupenot L, Rozansky DJ, Parmer RJ, O’Connor DT. Stimulus-transcription coupling in pheochromocytoma cells. Promoter region-specific activation of chromogranin A biosynthesis. J Biol Chem. 1996;271:28382–90. doi: 10.1074/jbc.271.45.28382. [DOI] [PubMed] [Google Scholar]
- 81.Tang K, Wu H, Mahata SK, Mahata M, Gill BM, Parmer RJ, O’Connor DT. Stimulus coupling to transcription versus secretion in pheochromocytoma cells. Convergent and divergent signal transduction pathways and the crucial roles for route of cytosolic calcium entry and protein kinase C. J Clin Invest. 1997;100:1180–92. doi: 10.1172/JCI119630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kruger PG, Mahata SK, Helle KB. Catestatin (CgA344-364) stimulates rat mast cell release of histamine in a manner comparable to mastoparan and other cationic charged neuropeptides. Regul Pept. 2003;114:29–35. doi: 10.1016/s0167-0115(03)00069-7. [DOI] [PubMed] [Google Scholar]
- 83.Munoz DG. Chromogranin A-like immunoreactive neurites are major constituents of senile plaques. Lab Invest. 1991;64:826–32. [PubMed] [Google Scholar]
- 84.Rangon CM, Haik S, Faucheux BA, Metz-Boutigue MH, Fierville F, Fuchs JP, Hauw JJ, Aunis D. Different chromogranin immunoreactivity between prion and a-beta amyloid plaque. Neuroreport. 2003;14:755–8. doi: 10.1097/00001756-200304150-00019. [DOI] [PubMed] [Google Scholar]
- 85.Taupenot L, Ciesielski-Treska J, Ulrich G, Chasserot-Golaz S, Aunis D, Bader MF. Chromogranin A triggers a phenotypic transformation and the generation of nitric oxide in brain microglial cells. Neuroscience. 1996;72:377–89. doi: 10.1016/0306-4522(96)83172-1. [DOI] [PubMed] [Google Scholar]
- 86.Ciesielski-Treska J, Ulrich G, Taupenot L, Chasserot-Golaz S, Corti A, Aunis D, Bader MF. Chromogranin A induces a neurotoxic phenotype in brain microglial cells. J Biol Chem. 1998;273:14339–46. doi: 10.1074/jbc.273.23.14339. [DOI] [PubMed] [Google Scholar]
- 87.Lechner T, Adlassnig C, Humpel C, Kaufmann WA, Maier H, Reinstadler-Kramer K, Hinterholzl J, Mahata SK, Jellinger KA, Marksteiner J. Chromogranin peptides in Alzheimer’s disease. Exp Gerontol. 2004;39:101–13. doi: 10.1016/j.exger.2003.09.018. [DOI] [PubMed] [Google Scholar]
- 88.Lugardon K, Raffner R, Goumon Y, Corti A, Delmas A, Bulet P, Aunis D, Metz-Boutigue MH. Antibacterial and antifungal activities of vasostatin-1, the N-terminal fragment of chromogranin A. J Biol Chem. 2000;275:10745–53. doi: 10.1074/jbc.275.15.10745. [DOI] [PubMed] [Google Scholar]
- 89.Metz-Boutigue MH, Goumon Y, Lugardon K, Strub JM, Aunis D. Antibacterial peptides are present in chromaffin cell secretory granules. Cell Mol Neurobiol. 1998;18:249–66. doi: 10.1023/A:1022573004910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gwadz RW, Kaslow D, Lee JY, Maloy WL, Zasloff M, Miller LH. Effects of magainins and cecropins on the sporogonic development of malaria parasites in mosquitoes. Infect Immun. 1989;57:2628–33. doi: 10.1128/iai.57.9.2628-2633.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shahabuddin M, Fields I, Bulet P, Hoffmann JA, Miller LH. Plasmodium gallinaceum: differential killing of some mosquito stages of the parasite by insect defensin. Exp Parasitol. 1998;89:103–12. doi: 10.1006/expr.1998.4212. [DOI] [PubMed] [Google Scholar]
- 92.Ghosh JK, Shaool D, Guillaud P, Ciceron L, Mazier D, Kustanovich I, Shai Y, Mor A. Selective cytotoxicity of dermaseptin S3 toward intraerythrocytic Plasmodium falciparum and the underlying molecular basis. J Biol Chem. 1997;272:31609–16. doi: 10.1074/jbc.272.50.31609. [DOI] [PubMed] [Google Scholar]
- 93.Conde R, Zamudio FZ, Rodriguez MH, Possani LD. Scorpine, an anti-malaria and antibacterial agent purified from scorpion venom. FEBS Lett. 2000;471:165–8. doi: 10.1016/s0014-5793(00)01384-3. [DOI] [PubMed] [Google Scholar]
- 94.Akaddar A, Doderer-Lang C, Marzahn MR, Delalande F, Mousli M, Helle K, Van Dorsselaer A, Aunis D, Dunn BM, Metz-Boutigue MH, Candolfi E. Catestatin, an endogenous Chromogranin A-derived peptide, inhibits in vitro growth of Plasmodium falciparum. Cell Mol Life Sci. 2009 doi: 10.1007/s00018-009-0235-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Goldberg DE, Slater AF, Cerami A, Henderson GB. Hemoglobin degradation in the malaria parasite Plasmodium falciparum: an ordered process in a unique organelle. Proc Natl Acad Sci U S A. 1990;87:2931–5. doi: 10.1073/pnas.87.8.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gaede AH, Lung MS, Pilowsky PM. Catestatin attenuates the effects of intrathecal nicotine and isoproterenol. Brain Res. 2009 doi: 10.1016/j.brainres.2009.09.088. [DOI] [PubMed] [Google Scholar]
- 97.Brutsaert DL, Fransen P, Andries LJ, De Keulenaer GW, Sys SU. Cardiac endothelium and myocardial function. Cardiovasc Res. 1998;38:281–90. doi: 10.1016/s0008-6363(98)00044-3. [DOI] [PubMed] [Google Scholar]
- 98.Gattuso A, Mazza R, Pellegrino D, Tota B. Endocardial endothelium mediates luminal Ach-NO signaling in isolated frog heart. Am J Physiol. 1999;276:H633–41. doi: 10.1152/ajpheart.1999.276.2.H633. [DOI] [PubMed] [Google Scholar]
- 99.Mazza R, Gattuso A, Mannarino C, Brar BK, Barbieri SF, Tota B, Mahata SK. Catestatin (chromogranin A344-364) is a novel cardiosuppressive agent: inhibition of isoproterenol and endothelin signaling in the frog heart. Am J Physiol Heart Circ Physiol. 2008;295:H113–22. doi: 10.1152/ajpheart.00172.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Aars H. Aortic baroreceptor activity in normal and hypertensive rabbits. Acta Physiol Scand. 1968;72:298–309. doi: 10.1111/j.1748-1716.1968.tb03851.x. [DOI] [PubMed] [Google Scholar]
- 101.West MJ, Korner PI. The baroreceptor-heart rate reflex in renal hypertension in the rabbit. Clin Exp Pharmacol Physiol. 1974;1:231–9. doi: 10.1111/j.1440-1681.1974.tb00545.x. [DOI] [PubMed] [Google Scholar]
- 102.Bristow JD, Honour AJ, Pickering GW, Sleight P, Smyth HS. Diminished baroreflex sensitivity in high blood pressure. Circulation. 1969;39:48–54. doi: 10.1161/01.cir.39.1.48. [DOI] [PubMed] [Google Scholar]
- 103.Goldstein DS. Arterial baroreflex sensitivity, plasma catecholamines, and pressor responsiveness in essential hypertension. Circulation. 1983;68:234–40. doi: 10.1161/01.cir.68.2.234. [DOI] [PubMed] [Google Scholar]
- 104.Grassi G, Cattaneo BM, Seravalle G, Lanfranchi A, Mancia G. Baroreflex control of sympathetic nerve activity in essential and secondary hypertension. Hypertension. 1998;31:68–72. doi: 10.1161/01.hyp.31.1.68. [DOI] [PubMed] [Google Scholar]
- 105.Parmer RJ, Cervenka JH, Stone RA. Baroreflex sensitivity and heredity in essential hypertension. Circulation. 1992;85:497–503. doi: 10.1161/01.cir.85.2.497. [DOI] [PubMed] [Google Scholar]