

Abstract
Ca2+ channels that underlie mitochondrial Ca2+ transport first reported decades ago have now just recently been precisely characterized electrophysiologically. Numerous data indicate that mitochondrial Ca2+ uptake via these channels regulates multiple intracellular processes by shaping cytosolic and mitochondrial Ca2+ transients, as well as altering the cellular metabolic and redox state. On the other hand, mitochondrial Ca2+ overload also initiates a cascade of events that leads to cell death. Thus, characterization of mitochondrial Ca2+ channels is central to a comprehensive understanding of cell signaling. Here, we discuss recent progresses in the biophysical and electrophysiological characterization of several distinct mitochondrial Ca2+ channels.
Keywords: Ca2+ channel, mitochondrial Ca2+ uniporter (MCU), mitochondrial ryanodine receptor (mRyR), rapid mode Ca2+ uptake (RaM), ruthenium red, Ru360, ryanodine
OVERVIEW
The identification of ion channels responsible for Ca2+ transport across the inner mitochondrial membrane has been a long and arduous journey. Following early findings that isolated mitochondria sequester cytosolic Ca2+ [1–3], extensive research focused on characterizing the different forms and properties that dictate mitochondrial Ca2+ uptake [4–7]. Mitochondrial Ca2+ uptake depends strongly on the mitochondrial inner membrane potential and is potently inhibited by both ruthenium red compounds and lanthanides [4,6]. The rate of mitochondrial Ca2+ uptake, measured in isolated mitochondria exhibits a sigmoidal dependence on extra-mitochondrial Ca2+ concentration that saturates at ~200 μM, a half maximal activation concentration at ~10 μM, and a Hill coefficient of ~2 [4,6]. Undoubtedly, these measurements are strongly influenced by both mitochondrial membrane potential and matrix Ca2+ accumulation [6]. Ca2+ uptake was initially considered to result from a single transport mechanism mediated by the mitochondrial Ca2+ uniporter (MCU), principally due to near complete inhibition by ruthenium red and lanthanides. However, subsequent studies have clearly identified additional Ca2+ uptake pathways (channels, Fig. 1), including the rapid mode of uptake (RaM) [8,9] and the mitochondrial ryanodine receptor (mRyR) [10–12]. These pathways exhibit kinetics, Ca2+ dependence (Fig. 2), and pharmacology that distinguish them from the MCU (Table 1).
Figure 1.

Mitochondrial Ca2+ channels/transporters and role in mitochondrial function. Mitochondrial Ca2+ uptake is determined by the mitochondrial Ca2+ uniporter (MCU), rapid mode of uptake (RaM), and ryanodine receptor (mRyR, or RyR1). The mitochondrial permeability transition pore (mPTP), Na+/Ca2+ exchanger (mNCX), H+/Ca2+ exchanger (mHCX, encoded by Letm1), and DAG activated cation channels (DCC) contribute to Ca2+ efflux. Mitochondrial Ca2+ uptake contributes to (a) shaping cytosolic Ca2+ signals and triggering metabolic coupling by enhancing mitochondrial ATP synthesis: (b) stimulation of Ca2+ dependent dehydrogenases of the TCA cycle [34] to increase NADH/FADH production used to feed electrons through the electron transport chain (ETC) and (c) activation of the ATP synthase [35]. However, mitochondrial Ca2+ overload can trigger (d) mPTP activation, (e) ROS generation, and cell death. Voltage dependent anion-selective channels (VDAC) provide a pathway for Ca2+ and metabolite transport across the mitochondrial outer membrane. MIM and MOM; mitochondrial inner and outer membranes, respectively.
Figure 2.
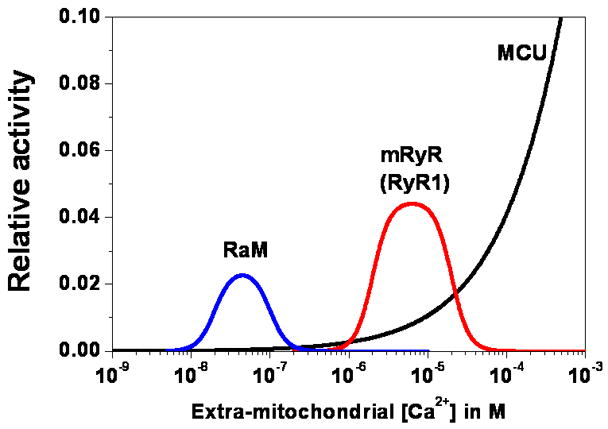
Ca2+ dependence of the major mitochondrial Ca2+ influx pathways. Relative activity of RaM (blue), mRyR (red), and the MCU (black) is estimated based on respective Ca2+ dependencies assuming a constant membrane potential and electrochemical gradient across the mitochondrial inner membrane. MCU is modeled according to patch clamp data of Kirichok et al. [43] and fitting with a Hill equation, 1/(1+(Km/x)n), where Km is the half-maximal concentration (19 mM) for activation, x is the extra-mitochondrial Ca2+ concentration, and n is the Hill coefficient (0.6). mRyR is modeled based on the Ca2+ dependent activation and inhibition of RyR1 channels using a modified Hill equation, c1*(1/(1+(Ka/x)n))*(1−1/(1+(Ki/x)n)), where c1 is a constant (0.0045) to enforce a 5-fold faster Ca2+ transport by mRyR compared to the MCU at 1 μM extra-mitochondrial Ca2+ according to the UV flash-induced mitochondrial Ca2+ uptake experiments of Beutner et al [10], Ka (2 μM) is the half-maximal concentration for Ca2+ dependent activation, Ki (20 μM) is the half-maximal concentration for Ca2+ dependent inhibition, x is the extra-mitochondrial Ca2+ concentration, and n is the Hill coefficient (4). RaM is modeled based on the same modified Hill’s equation, c2*(1/(1+(Ka/x)n))*(1−1/(1+(Ki/x)n)), where c2 is a constant (0.00049) to enforce a 50-fold faster Ca2+ transport of RaM compared to the MCU at 50 nM extra-mitochondrial Ca2+ according to the findings of Buntinas et al [8], Ka (20 nM) is the half-maximal concentration for Ca2+ dependent activation, Ki (100 nM) is half maximal concentration for Ca2+ dependent inhibition, x is the extra-mitochondrial Ca2+ concentration, and n is the Hill coefficient (4).
Table 1.
Biophysical properties of mitochondrial Ca2+ permeable channels
Comparison of mitochondrial Ca2+ channel biophysical properties. Properties of MiCa are taken from Kirichok et al [43], mCa1 and mCa2 from Michels et al [44], mRyR from Altschafl et al [12], DCC from Chinopoulos et al [63], and VDAC from [64,67]. Single channel conductance values of MiCa, mCa1, mCa2 were obtained in the presence of symmetrical 105 mM Ca2+, mRyR in symmetrical 300 mM Cs+ (225 pS in 150 mM Cs+ from our unpublished mitoplast patch clamp data), DCC and VDAC in symmetrical 150 mM KCl. Relative Ca2+ permeability value of mRyR is adapted from that of RyR1 in skeletal muscle [55]. Channel activity of mRyR exhibits long lasting subconductance openings in the presence of <10 μM ryanodine and is blocked at >100 μM ryanodine. RuR indicates ruthenium red.
| Single channel conductance | Ion selectivity | Voltage dependence | Molecular identity | Inhibitors | Activators | Other properties | ||
|---|---|---|---|---|---|---|---|---|
| MCU | MiCa | 2.6–5.2pS | Highly Ca2+ Selective | Inward rectifying | - | RuR, Ru360 | - | |
| mCa1 | 13.7 pS | Highly Ca2+ Selective | Inward rectifying | - | RuR, Ru360 | spermine | ||
| mCa2 | 7.67 pS | Highly Ca2+ Selective | Inward rectifying | - | Relatively insensitive to Ru360 | spermine | ||
| mRyR | 500–800 pS (225 pS) | Cation selective PCa/Pk =~6/1 | Linear | RyR1 | >100 μM Ryanodine | <10 μM Ryanodine Imperatoxin A | ||
| RaM | - | - | - | - | RuR | spermine | Only known as a kinetic mode | |
| DCC | 202 pS | Slightly cation selective Ca2+ Selectivity is not defined | Linear | - | 1 mM La3+ | DAG | ||
| VDAC | 700 pS | Anion or cation selective states PCa/PCl = 0.02–0.38 | Closed at > ±40 | VDAC 1,2,3 | DIDS, RuR, Ru360 | - | Outer membrane |
Substantial indirect evidence indicates that ion channels are involved in mitochondrial Ca2+ uptake. For example, mitochondrial Ca2+ uptake depends on the inner mitochondrial membrane potential and Ca2+ transport is not coupled to the movement of other ions [4,6,13,14]. In fact, the term “Ca2+ uniporter” was originally proposed as a counterpart to other known mitochondrial antiport systems such as the K+/H+, Na+/Ca2+, and H+/Ca2+ exchangers [4,14]. These original studies predicted the Ca2+ transport rate across the mitochondrial inner membrane to be low compared to that of conventional ion channels [4,6]. Thus, mitochondrial Ca2+ uptake mechanisms were collectively described under the guise of a “Ca2+ uniporter”, which eluded arguments of classification as either carrier or channel. However, recent innovative electrophysiological recordings more directly address this fundamental question and resulted in a detailed characterization of mitochondrial Ca2+ channels. Electrophysiological recordings using patch clamp techniques or lipid bilayer systems have advantages over conventional Ca2+ uptake measurements using fluorescence probes. Specifically, single channel recordings provide exquisite details regarding channel selectivity, conduction, and temporal resolution of channel gating (tens of microseconds) [15]. More importantly, the membrane voltage and concentration of Ca2+ ions on both sides of membrane, which affect Ca2+ transport rate, are tightly controlled. Thus, direct measurements of Ca2+ currents using these electrophysiological methods eliminates complications derived from Ca2+ flux itself or activation of other Ca2+ sensitive channels. Large conductance channels in the inner and outer mitochondrial membrane have been identified using electrophysiological methods [16]. Delays in definitive identification of native mitochondrial channels are primarily a result of significant technical challenges due to the small size of isolated mitochondria and mitoplasts (mitochondria in which the inner membrane is exposed). Furthermore, a low channel density, small unitary conductance, high Ca2+ selectivity, and low open probability of some mitochondrial channels provide additional technical challenges. Detailed consideration of the biophysical properties of these channels is discussed in subsequent sections of this review.
The Ca2+ concentration within the mitochondrial matrix is regulated by a variety of Ca2+ efflux mechanisms (Fig. 1) including the mitochondrial Na+/Ca2+ and H+/Ca2+ exchangers, neither of which have yet to be characterized electrophysiologically [7,14]. Depending on the concentration of Na+, H+ and Ca2+ across the mitochondrial inner membrane, these exchangers contribute to either Ca2+ uptake or release. Significant matrix Ca2+ efflux also occurs during opening of the mitochondrial permeability transition pore (mPTP) and the role of subconductance transient openings of the mPTP has also been discussed previously [17,18]. This review will focus the properties of mitochondrial Ca2+ uptake channels.
Mitochondrial Ca2+ channels and signaling
Mitochondrial Ca2+ channels play important roles in a myriad of intracellular signaling pathways in physiological and pathological conditions, which is more extensively reviewed elsewhere [7,17]. Mitochondrial Ca2+ uptake through these channels contributes to shaping beat-to-beat oscillations of cytosolic Ca2+ signals in heart cells [19–21]. More interestingly, mitochondrial Ca2+ uptake actively controls Ca2+ mobilizing mechanisms by regulating the Ca2+ concentration in the microdomain adjacent to the endoplasmic reticulum (ER) or plasma membrane in a variety of cell types. Ca2+ dependent activation or inactivation of plasma membrane and ER Ca2+ channels is strongly modulated by mitochondrial Ca2+ uptake [22,23]. Interestingly, privileged ER-mitochondrial communication is facilitated by electron dense tethering structures that physically connect mitochondria to the ER [24,25]. Subsarcolemmal mitochondria regulate the rate of sarcolemmal L-type Ca2+ channel inactivation in cardiomyocytes [26]. In addition, store operated Ca2+ channels, in which store depletion triggers oligomerization of ER Ca2+ sensor stromal interacting molecules (STIM1) and subsequent activation of Orai1/TRPC channels in the plasma membrane [27,28], are also regulated by mitochondrial Ca2+ uptake [29–32]. Finally, mitochondrial Ca2+ uptake and release modulates synaptic transmission by regulating presynaptic Ca2+ levels that underlie neuronal post-tetanic potentiation [33]. Thus, mitochondrial Ca2+ uptake channels play a critical role in regulating many essential cellular functions.
Mitochondrial Ca2+ channels are important regulators of cellular bioenergetics [34,35] (Fig. 1). During heart failure, dysregulation of mitochondrial Ca2+ handling is associated with contractile dysfunction [36]. Mitochondrial Ca2+ uptake activates several dehydrogenases [34] in the TCA cycle and ATP synthase [35]. In an elegant series of experiments, Brandes and Bers used real-time measurements of NADH auto-fluorescence to demonstrated Ca2+ activation of the TCA cycle during electrical stimulation of intact cardiac trabeculae [37]. When the frequency of electrical stimulation increased from 0.25 to 2 Hz, mitochondrial NADH levels initially decreased upon mitochondrial Ca2+ uptake, but then quickly recovered. Alternatively, when the frequency of stimulation was reduced from 2 to 0.25 Hz, NADH levels increased above control levels. A mathematical model was developed to validate “push” and “pull” models, which are based on Ca2+ activation of dehydrogenases and ADP-induced increase in respiration. Both models were required for the best mathematical reconstitution of time dependent changes in mitochondrial NADH dynamics [38]. Importantly, we found that inhibition of mitochondrial Ca2+ uptake inhibits ADP-induced state 3 respiration [11]. These findings indicate that mitochondrial Ca2+ channels act as metabolic transducers that operate by controlling Ca2+-dependent changes in mitochondrial ATP production required to meet dynamic changes in cytosolic energy needs [11]. Finally, mitochondrial Ca2+ is also a key regulator for the generation of reactive oxygen species (ROS), which intimately link to physiological redox signaling [39]. Under pathological conditions, however, mitochondrial Ca2+ overload contributes to excessive generation of ROS [17,39,40], activation of the mPTP [18,41], and initiation of cell death [42].
Mitochondrial Ca2+ uniporter (MCU)
The mechanisms of mitochondrial Ca2+ uptake are often studied in suspensions of isolated mitochondria where the control of membrane potential and ion concentrations is limited. Patch clamp studies overcome these limitations by directly controlling both membrane voltage and ion concentrations across the mitochondrial inner membrane. Mitoplasts prepared from COS-7 cells and macroscopic “whole–mitoplast” patch clamp recordings revealed a highly Ca2+ selective (<2 nM Ca2+ affinity) and inwardly rectifying current, named MiCa [43]. Inward MiCa currents exhibit partial Ca2+-independent inactivation at negative voltages. Macroscopic MiCa current density is large, reaching 55 pA/pF at −160 mV in the presence of 100 μM extra-mitochondrial [Ca2+], approximately the local Ca2+ concentration within ER-mitochondrial microdomains. Interestingly, inward MiCa currents saturate at >105 mM Ca2+ with a half maximal concentration of ~19 mM, and a Hill coefficient of ~0.6. These findings indicate that Ca2+ influx capacity through this pathway is enormous. Maximal Ca2+ flux through single MiCa channels with 105 mM extramitochondrial Ca2+ is ~5×106 ion/sec, which is comparable to that of most ion channels. Does this flux truly correspond to activity of the MCU? Indeed, the half maximal concentration and estimated MiCa flux rate is higher (~2×104 ions/sec, [4,6]) and the Hill coefficient lower compared to that previously reported for the MCU [6]. However, these differences could be due to the more uniform control of membrane potential and ion concentrations in patch-clamp experiments that are necessarily lacking in the biochemical measurements [6,43]. The permeability of MiCa to various divalent cations exhibits a similar rank order (Ca2+≈Sr2+≫Mn2+≈Ba2+, and Mg2+ being impermeable) as that previously reported for MCU. In addition, inhibition by nanomolar concentrations of ruthenium red, and the purified and more specific inhibitor Ru360, further support the notion that MiCa represents the electrophysiological correlate of the MCU. In inside-out patches, single channel MiCa activity exhibits characteristics remarkably similar to that observed macroscopically [43]. Specifically, channel open probability was high at negative voltages and decreased strongly with depolarization, reflecting the inwardly rectifying whole-mitoplast MiCa current. However, multichannel currents recorded from inside out patches do not exhibit the same inactivation properties as that observed in whole-mitoplast recordings. With symmetrical 105 mM CaCl2, single MiCa channel activity exhibits multiple subconductance states between 2.6–5.2 pS (Fig. 3A) with an estimated channel density of ~10–40 channels per μm2. Thus, the single channel recordings and estimated Ca2+ fluxes strongly support the notion that MiCa represents a bona fide Ca2+-permeable ion channel and not a carrier.
Figure 3.

Representative single channel current traces of different mitochondrial Ca2+ channels. A. Single channel current traces of MiCa recorded from a COS-7 cell mitoplast reproduced from the original recording reported by Kirichok et al. [43] (Nature, 2004, 427:360–364, supplemental figure 2a) with permission from Nature Publishing Group. The single channel activity of MiCa shows multiple conductance states ranging from 2.6–5.2 pS at −160 mV in symmetrical 105 mM CaCl2. At positive voltages (+140 mV), the single channel activity of MiCa shows fast flickering with very small subconductance states, an indication of conduction block by Ca2+. B. Single channel current trace of a mRyR channel purified from rat heart mitochondrial inner membrane and incorporated into an artificial lipid bilayer reproduced from the original recording reported by Altschafl et al. [12] (Biochimica et Biophysica Acta - Biomembranes, 2007, 1768:1784–1795, figure 7c) with permission from Elsevier. Peak unitary mRyR conductance is between 500–800 pS using a 300/50 mM, cis/trans, Cs-methanesulfonate gradient and 50 μM cytosolic Ca2+. Impera toxin A, a RyR channel modulator, induces subconductance opening of mRyR. C. Single channel current trace of a DAG activated cation channel (DCC) recorded from a brain mitoplast reproduced from the original report of Chinopoulos et al. [63] (Journal of Bioenergetics and Biomembranes, 2007, 37(4):237–247, figure 4d) with permission from Springer. The DCC current was recorded at −50 mV in symmetrical 150 mM KCl in the presence of 10 μM cytosolic Ca2+ and 100 μM 1-oleoyl-2-acetyl-sn-glycerol (OAG), a DAG analog. D. Single channel current trace of mPTP, previously referred to as the multi-conductance channel (MCC), recorded from rat heart mitoplast reproduced from the original report of Kinnally et al. [71] (Journal of Bioenergetics and Biomembranes, 24(1):99–110, figure 3) with permission from Springer. The mPTP activity was recorded at −60 mV in symmetrical 150 mM KCl. E. Single channel current traces of VDAC reproduced from the original report of Pavlov et al. [67] (Biochimica et Biophysica Acta - Bioenergetics, 2005, 1710:96–102, figure 2b & 2c) with permission from Elsevier. (a) Typical voltage dependent channel activity of VDAC with voltage ramps between ±40 mV shows anion- or cation-selective conductance states. (b) VDAC activity was recorded at 0 mV with 150/30 mM KCl gradient. Anion- and cation- selective states are shown with arrows. Solid line indicates the 0 current level. Scale bars are presented with some modification from the original article.
Recently, two voltage dependent Ca2+ channels in human heart mitoplasts, named mCa1 and mCa2, were characterized electrophysiologically in patch clamp experiments [44]. Both mCa1 and mCa2 exhibit high Ca2+ selectivity, maximal conductance at 105 mM Ca2+, and half saturation at 15.1 and 19.6 mM Ca2+, respectively. Like MiCa, mCa1 is inhibited by nanomolar Ru360 and exhibits increased open probability at negative voltages. However, mCa1 has a higher mean unitary conductance (13.7 pS) and exhibits multiple conductance states (10.1, 16.5, and 21.3 pS). Further, mCa1 exhibits completely distinct channel kinetics compared to MiCa. Specifically, mCa1 exhibits low channel open probability (PO=0.053) with long closed times and brief open times, whereas MiCa channels exhibit a high channel open probability (PO=0.9) due to short closed times and long-lived open times. These differences could be explained either by MiCa and mCa1 arising from distinct channels or to differences between experimental conditions including different species and cell types [44]. mCa2 shares the same voltage dependence with mCa1, but mCa2 channels exhibit a smaller unitary conductance (7.67 pS) and are insensitive to nanomolar concentrations and only partially reduced by micromolar concentration of Ru360. Spermine, which stimulates mitochondrial Ca2+ uptake [7], enhances both mCa1 and mCa2 channel activity. However, dantrolene, a type 1 ryanodine receptor inhibitor, does not alter mCa1 or mCa2 activity, suggesting that they are not associated with mRyR activity (see mRyR section for details). Importantly, mCa1 and mCa2 channel activity (decreased PO) and gating (prolonged closed times) are reduced in the failing heart, consistent with reduced mitochondrial Ca2+ uptake in heart failure.
Despite numerous attempts over many years, the molecular identity of the MCU remains elusive. Ca2+-binding glycoproteins were isolated from mitochondria in the 1970s [45,46]. Later, a 40 kDa glycoprotein was shown to form Ca2+ conducting channels in lipid bilayers and an antibody against this glycoprotein inhibits Ca2+ transport in liver mitoplasts [47]. Purification and reconstitution into lipid bilayers of a 2 kDa peptide of the 40 kDa glycoprotein shows a 20 pS ruthenium red-sensitive Ca2+ channel activity [48]. Antibodies against a different 20 kDa protein inhibit mitochondrial Ca2+ uptake [49] and 103Ru360 labels an 18 kDa protein purified from rat kidney mitochondria [50]. However, the molecular identities of these glycoproteins have not yet been identified. More recently, Trenker and colleagues reported that uncoupling protein 2 and 3 (UCP2/3) are fundamental for mitochondrial Ca2+ uptake based on experiments of mutants and following knockdown with small interference RNA [51,52]. However, it remains unclear and controversial as to whether or not UCP2/3 forms the MCU Ca2+ conducting pore or even regulates its activity. For example, Brookes et al found that mitochondrial Ca2+ uptake is unaffected by either UCP inhibitors, GDP and genipin, or UCP2/UCP3 deficiency, suggesting that UCPs do not function as the MCU [53]. Electrophysiological recordings of whole-mitoplast MiCa currents from normal and UCP2/3 knock-out mice together with mutational studies within putative pore regions of UCP2/3 are needed to more definitively resolve this issue.
A recent report has shown the promise of genome-wide high throughput RNA interference (RNAi) screens in the molecular identification of novel mitochondrial Ca2+ transport proteins [54]. Using this approach, the Drosophila homologue of mammalian Letm1 was identified as a mitochondrial H+/Ca2+ exchanger [54]. RNAi knockdown, overexpression, and liposome reconstitution of the purified protein demonstrate that Letm1 mediates pH dependent mitochondrial Ca2+ uptake and release. Although the authors did not report the identification of MCU in this initial report, this unbiased high throughput genome-wide screening approach could potentially lead to the molecular identification of the MCU and other mitochondrial ion channels and transporters in the future.
In summary, the biophysical analysis of MCU candidate channels confirms that they are highly Ca2+ selective and exhibit strongly inward rectifying properties. However, the different single channel conductance, gating, and pharmacological properties of these candidate channels indicate that MCU activity may result from a family of Ca2+ selective channels present in the mitochondrial inner membrane. Numerous questions regarding the MCU remain to be addressed. 1) What are the molecular identities of the MCU proteins? 2) Are there tissue and/or species differences in MCU activity and identity? 3) Is the MCU sufficient to account for mitochondrial Ca2+ uptake in different cell types? The plasma membrane expresses multiple subtypes of Ca2+ channels across a wide range of different cell types. Similarly, we envision that the cell type-specific differences in mitochondrial Ca2+ uptake likely reflect, in part, variations in MCU activity due to differences in the aggregate activity of multiple Ca2+ uptake channels (Fig. 2 and Table 1).
Rapid mode uptake (RaM)
A rapid mode of Ca2+ uptake (RaM), with kinetics hundreds of times faster than classical MCU activity, has been reported in isolated liver, heart, and brain mitochondria. Rapid mitochondrial Ca2+ uptake kinetics were revealed in experiments using fast Ca2+ pulses within a millisecond time scale [6–9]. Interestingly, unlike classical MCU activity, Ca2+ uptake by RaM is inhibited by increasing the extra-mitochondrial Ca2+ concentration (Fig. 2). Thus, to observe RaM, the basal extra-mitochondrial Ca2+ level between pulses needs to drop below ~100 nM for a period of time in order to permit removal of Ca2+ from a high-affinity external binding site [6]. RaM exhibits different characteristics between heart and liver mitochondria. Specifically, RaM-mediated Ca2+ uptake following a single pulse is significantly smaller in heart compared to liver mitochondria. Additionally, the reset time for the second Ca2+ pulse is longer in heart (>60 sec) than liver mitochondria (<0.3 sec) and RaM in heart mitochondria is less sensitive to blockade by ruthenium red. Spermine activates RaM in cardiac mitochondria but is less effective in liver. ATP and GTP activate RaM in liver but not in heart mitochondria. Finally, unlike in liver, RaM in heart mitochondria is activated by ADP and strongly inhibited by AMP.
Currently, RaM is only described as a kinetic mode of mitochondrial Ca2+ uptake. The protein(s) and molecular identity of RaM, and its relationship to the MCU, remain unknown [6]. Interestingly, MiCa currents measured in whole-mitoplast configuration exhibit transient inward currents followed by a persistent steady state current [43]. The relative amplitude of the transient current to the total peak current at −160 mV is about 21% with current decay exhibiting two time constants of 4 ms and 141 ms. This transient component of MiCa currents may be related to RaM. However, the extra-mitochondrial Ca2+ concentrations in these patch clamp recordings were high enough (20 μM to 105 mM) to completely inhibit RaM, which closes at concentrations above 100 nM Ca2+. Kirichok et al. showed that this transient component was not due to Ca2+ dependent inactivation since it was not affected by strong matrix Ca2+ buffering [43]. Thus, as it is unclear whether the transient component of MiCa current is related to RaM, it will be important to determine if AMP, which inhibits RaM in heart mitochondria [8], blocks the transient component of MiCa current.
Mitochondrial ryanodine receptor (mRyR)
A ryanodine-sensitive, rapid mitochondrial Ca2+ uptake mechanism in isolated heart mitochondria was firstly identified in 2001 by Beutner and colleagues [10]. Using [3H]ryanodine binding, immunogold labeling and Western blot analysis, a ryanodine receptor with a molecular mass of ~500 kDa was identified in the mitochondrial inner membrane. [3H]ryanodine binding to isolated heart mitochondria exhibits high affinity (Kd=9.8 nM), shows biphasic Ca2+ regulation, and is inhibited by Mg2+ (IC50=0.33 mM) and ruthenium red (IC50=105 nM). Interestingly, the bell-shaped Ca2+ dependence of [3H]ryanodine binding to purified heart mitochondria exhibits half-maximal activation at ~2 μM, peak binding between 10–40 μM, and is inactivated at higher Ca2+ concentrations. Considering that ryanodine binds only to the open channel [55], the bell shaped Ca2+ dependency of [3H]ryanodine binding demonstrates that mRyR is uniquely optimized for the physiological Ca2+ transport between the sarcoplasmic reticulum (SR) and mitochondria in the heart. mRyR activity in heart mitochondria is sensitive to dantrolene, consistent with mRyR being related to the skeletal muscle type 1 RyR isoform (RyR1), but not the cardiac isoform (RyR2) located in the SR. We further confirmed the subtype of mRyR in heart mitochondria being RyR1 using both subtype specific antibodies and by failure to detect mRyR in hearts from newborn RyR1 knockout mice [11].
Recently, single mRyR channel activity was recorded from purified mRyR proteins, extracted from mitochondrial inner membrane vesicles following incorporation in artificial planar lipid bilayers [12] (Fig. 3B). The inner mitochondrial membrane fractions used in these experiments exhibited robust labeling with RyR1-specific antibodies but were free of sarco-endoplasmic reticulum Ca2+ pump (SERCA), calsequestrin, and RyR2 reactivity, consistent with lack of SR contamination. The unitary conductance of mRyR is 500–800 pS with symmetrical 300 mM Cs+ solution. Changing the cis (cytosolic) Ca2+ concentration from 5 to 50 μM activates mRyR channels by increasing both bursting frequency and mean open time. Low micromolar concentrations of ryanodine lock mRyR channels into a long-lived subconductance state while higher ryanodine concentrations completely inhibit mRyR channel activity. These findings are classic characteristics of reconstituted ryanodine receptor channels [55,56]. Impera toxin A, a high affinity RyR1 modulator, also activated mRyR by promoting subconductance gating. The absence of effect of cyclosporine A and bongkrekic acid indicates that mRyR activity is not related to either the mPTP or adenine nucleotide translocator (ANT). We have also recorded ryanodine and ruthenium red sensitive single channel activities by directly patch clamping heart mitoplasts (unpublished data of Ryu, Kinnally, Dirksen, & Sheu). Together, these results indicate that functional mRyRs exist in the inner membrane of heart mitochondria.
Activation of a high conductance mRyR channel with relatively low Ca2+ selectivity could potentially depolarize the mitochondrial membrane potential, and thus, uncouple oxidative phosphorylation. However, Ca2+ and K+ transport through mRyR may actually serve to stabilize mitochondrial energy metabolism for several reasons. First, increased K+ permeability due to opening of large conductance Ca2+ activated K+ channels enhances mitochondrial energetic performance by inducing mitochondrial swelling with a maintained membrane potential [57]. Thus, increased mitochondrial K+ flux during mRyR activation may exert a similar enhancement of mitochondrial energetics. Second, Ca2+ dependent activation of several dehydrogenases in the TCA cycle and subsequent increase in NADH production will serve to counteract depolarization. Third, increased K+ flux and the bell-shaped Ca2+ dependence of mRyR activity may act as a built-in brake to regulate the electrochemical driving force or Ca2+ entry across the mitochondrial inner membrane. Fourth, the expected unitary mRyR conductance under physiological conditions will be smaller than that recorded experimentally in bilayers where high ionic strength solutions are used. In fact, the peak single channel conductance of native mRyR channel activity is only ~225 pS using symmetrical 150 mM Cs+ (our unpublished data). Furthermore, mRyR primarily opens to subconductance levels and physiological concentrations of cytosolic Mg2+ will decrease channel open probability, both of which serve to significantly limit net ion flux. Finally, maximal density of mRyR binding sites (Bmax) is 398.4 ± 12 fmol/mg of protein, only ~10% of the level in the SR, which will further limit the degree of mitochondrial depolarization.
Considering the electrochemical gradient for Ca2+ flux across the mitochondrial inner membrane, the properties and Ca2+ dependence of mRyR render these channels uniquely suited for mediating fast dynamic mitochondrial Ca2+ uptake. However, under certain situations (e.g. mitochondrial Ca2+ overload, reversal of Ca2+ electrochemical gradient), activation of mRyR channels could result in rapid mitochondrial Ca2+ efflux. In these conditions, mRyR-mediated Ca2+ efflux may serve an important protective role by reducing matrix Ca2+ and preventing activation of the more nonspecific pore mPTP and subsequent initiation of cell death.
Since our initial discovery of mRyR in 2001 [10], the idea that more than one Ca2+ influx mechanism exists in mitochondria has gradually gained wider recognition. Unfortunately, as of now we are still left with limited information regarding the molecular identities of these different Ca2+ influx mechanisms. This relatively slow progress is due to intrinsic difficulties in investigating mitochondrial ion channels, which is best exemplified by the fact that even 40 years after its discovery, the molecular identity of the MCU remains elusive. Indeed, the majority of mitochondrial ion channels have yet to be cloned, which lies in stark contrast with rapid progress in molecular identification and cloning of plasma membrane ion channels. Of particular interest, RyRs in the cardiac SR (RyR2) localize adjacent to mitochondria due to the physical tethering of these two organelles. Garcia-Perez et al showed that crude or Percoll-purified heavy mitochondrial fractions contain low levels of RyR2 and calsequestrin contamination [58]. In these mitochondrial preparations, caffeine plus thapsigargin induces mitochondrial Ca2+ uptake, consistent with intimate physical coupling between SR and mitochondria. Ru360, a presumed specific inhibitor of the MCU, blocked mitochondrial Ca2+ uptake induced by caffeine and thapsigargin. Thus, Garcia-Perez et al concluded that the observed mitochondrial Ca2+ uptake is not mediated by mRyR. For this reason, mRyR was suggested to either reflect an SR membrane “contaminant” in the purified mitochondrial preparation or to nonspecific binding of immunogold particles [59]. However, the concentration of Ru360 used in this study (10 μM) is also known to inhibit VDAC activity in the outer membrane [60], which would severely compromise Ca2+ transport across the outer membrane (see discussion below). In fact, addition of 10 μM Ca2+ in the presence of Ru360 induces a significant increase in mitochondrial Ca2+ [58], consistent with contributions of other Ca2+ transport mechanisms in the inner membrane. Moreover, several groups confirmed the presence of mRyR in mitochondrial cristae from electron micrographs using RyR antibody conjugated immunogold particles [10,61] and the absence of mRyR in purified mitochondria from RyR1 knock-out mice [11]. Resolution of these controversies will require further studies using RyR knock-out and knock-in mice to determine the molecular identity and functional significance of the mRyR.
Important future approaches/directions in mRyR research will include addressing several currently unresolved questions including: How is mRyR targeted to mitochondria? How do mRyR precursor proteins interact with protein import machineries in the outer and inner membrane? What are the important mRyR molecular regulators and binding partners? How is mRyR expression controlled? What are the implications of RyR1-linked diseases on mRyR and cardiac function? Does altered mRyR function contribute to cardiac abnormalities previously reported in individuals with RYR1 gene mutations linked to malignant hyperthermia [62]?
Other Ca2+-permeable mitochondrial channels
Diacylglycerol (DAG), which is generated by phospholipase C following PIP2 hydrolysis, induces Ca2+ release from Ca2+-loaded mitochondria. Patch clamp studies of brain mitoplasts demonstrate that DAG activates a novel La3+-sensitive cation channel in the mitochondrial inner membrane [63]. The single channel conductance of the novel DAG-activated cation-selective channel (DCC) is ~202 pS in symmetrical 150 mM KCl (Fig. 3C). DCC is not mediated by either the MCU, mRyR or mPTP since it is not inhibited by ruthenium red, bongkrekic acid, or cyclosporine A. Since DCC channel activity is not observed in membrane patches from pure lipid vesicles, DAG does not directly form these channels. The identification of mitochondrial DCCs is particularly intriguing since this data suggest mitochondrial Ca2+ handling is modulated by a second messenger, DAG, following activation of multiple G-protein coupled cell surface receptors.
Voltage dependent anion-selective channels (VDACs), located in the mitochondrial outer membrane, provide an access route into the intermembrane space for Ca2+ and various cellular metabolites including ATP and ADP [64,65]. The mitochondrial outer membrane was originally considered not to act as a significant barrier for the Ca2+ transport. However, over-expression of VDAC enhances ER-mitochondrial Ca2+ transport [66], consistent with the mitochondrial outer membrane serving a barrier function limiting mitochondrial Ca2+ transport. Furthermore, this finding suggests that VDAC provides a regulated pathway for Ca2+ transit across the outer membrane. VDAC is reported to exhibit both cation and anion selective conductance states [67] (Fig. 3E), with a cationic closed state paradoxically exhibiting greater Ca2+ permeability than the full open state [64,65]. The permeability ratio of VDAC for Ca2+ over Cl− (PCa2+/PCl−) is 0.02–0.38 [64]. Both VDAC cationic and anionic conductance states are blocked by ruthenium red and lanthanides [60,68]. Ruthenium compounds block by interacting with a Ca2+ binding site formed by two glutamate residues, E72 and E202, in the cytosolic loops of VDAC [69]. Ca2+ not only permeates but also regulates VDAC gating by inducing a prolonged fully open state that promotes increased metabolite exchange [70]. Thus, Ca2+ uptake is coordinated by a complex interplay between Ca2+-permeable channels in both the mitochondrial inner and outer membranes.
CONCLUSIONS
Mitochondrial Ca2+ handling plays a key physiological role in the control of many cellular functions. Dysfunction in proper mitochondrial Ca2+ homeostasis contributes to several pathological conditions. Mitochondrial Ca2+ channels provide the gateway for Ca2+ conduction across the mitochondrial outer and inner membranes (Fig. 1), enabling mitochondria to regulate the cytosolic Ca2+ signals, energy metabolism, ROS generation, and cell death. Remarkably, the unique Ca2+ dependence of MCU, RaM, and mRyR activities suggest their specific roles in various cytosolic Ca2+ environments (Fig. 2). Future studies are destined to provide new and exciting discoveries regarding the diversity in function, mechanisms of modulation/control, molecular identity/structure, and (patho)physiological roles of the mitochondrial Ca2+ channels. These findings will undoubtedly provide new insights into potential therapeutic targets for disorders as diverse as cancer, heart failure, myopathy, and neurodegenerative diseases.
Acknowledgments
The authors thank members of the Mitochondrial Research Innovation Group at the University of Rochester for critical discussions. We also thank Dr. Kirichok and Dr. Clapham for kindly allowing us to reproduce some of their work in this review. Fig 3A is reproduced by permission from Macmillan Publishers Ltd: [Nature] ([43]), copyright (2004). This work was supported by NIH grants HL-33333 to Sheu SS, AR44657 to Dirksen RT, GM57249 to Kinnally KW, and an American Heart Association Postdoctoral Fellowship grant 0525951T to Ryu SY.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Deluca HF, Engstrom GW. Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci U S A. 1961;47:1744–50. doi: 10.1073/pnas.47.11.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harris EJ. The uptake and release of calcium by heart mitochondria. Biochem J. 1977;168:447–56. doi: 10.1042/bj1680447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicholls DG. Calcium transport and porton electrochemical potential gradient in mitochondria from guinea-pig cerebral cortex and rat heart. Biochem J. 1978;170:511–22. doi: 10.1042/bj1700511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–86. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- 5.Gunter TE, Gunter KK, Sheu SS, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. Am J Physiol. 1994;267:C313–39. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- 6.Gunter TE, Yule DI, Gunter KK, Eliseev RA, Salter JD. Calcium and mitochondria. FEBS Lett. 2004;567:96–102. doi: 10.1016/j.febslet.2004.03.071. [DOI] [PubMed] [Google Scholar]
- 7.Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial Ca(2+) transport mechanisms. Biochim Biophys Acta. 2009;1787:1291–308. doi: 10.1016/j.bbabio.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Buntinas L, Gunter KK, Sparagna GC, Gunter TE. The rapid mode of calcium uptake into heart mitochondria (RaM): comparison to RaM in liver mitochondria. Biochim Biophys Acta. 2001;1504:248–61. doi: 10.1016/s0005-2728(00)00254-1. [DOI] [PubMed] [Google Scholar]
- 9.Sparagna GC, Gunter KK, Sheu SS, Gunter TE. Mitochondrial calcium uptake from physiological-type pulses of calcium. A description of the rapid uptake mode. J Biol Chem. 1995;270:27510–5. doi: 10.1074/jbc.270.46.27510. [DOI] [PubMed] [Google Scholar]
- 10.Beutner G, Sharma VK, Giovannucci DR, Yule DI, Sheu SS. Identification of a ryanodine receptor in rat heart mitochondria. J Biol Chem. 2001;276:21482–8. doi: 10.1074/jbc.M101486200. [DOI] [PubMed] [Google Scholar]
- 11.Beutner G, Sharma VK, Lin L, Ryu SY, Dirksen RT, Sheu SS. Type 1 ryanodine receptor in cardiac mitochondria: transducer of excitation-metabolism coupling. Biochim Biophys Acta. 2005;1717:1–10. doi: 10.1016/j.bbamem.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 12.Altschafl BA, Beutner G, Sharma VK, Sheu SS, Valdivia HH. The mitochondrial ryanodine receptor in rat heart: a pharmaco-kinetic profile. Biochim Biophys Acta. 2007;1768:1784–95. doi: 10.1016/j.bbamem.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 13.Kapus A, Szaszi K, Kaldi K, Ligeti E, Fonyo A. Is the mitochondrial Ca2+ uniporter a voltage-modulated transport pathway? FEBS Lett. 1991;282:61–4. doi: 10.1016/0014-5793(91)80444-8. [DOI] [PubMed] [Google Scholar]
- 14.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–55. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 15.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 16.Grigoriev SM, Muro C, Dejean LM, Campo ML, Martinez-Caballero S, Kinnally KW. Electrophysiological approaches to the study of protein translocation in mitochondria. Int Rev Cytol. 2004;238:227–74. doi: 10.1016/S0074-7696(04)38005-8. [DOI] [PubMed] [Google Scholar]
- 17.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. Am J Physiol Cell Physiol. 2004;287:C817–33. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 18.Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–31. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 19.Huser J, Blatter LA, Sheu SS. Mitochondrial calcium in heart cells: beat-to-beat oscillations or slow integration of cytosolic transients? J Bioenerg Biomembr. 2000;32:27–33. doi: 10.1023/a:1005556227425. [DOI] [PubMed] [Google Scholar]
- 20.Trollinger DR, Cascio WE, Lemasters JJ. Mitochondrial calcium transients in adult rabbit cardiac myocytes: inhibition by ruthenium red and artifacts caused by lysosomal loading of Ca(2+)-indicating fluorophores. Biophys J. 2000;79:39–50. doi: 10.1016/S0006-3495(00)76272-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robert V, Gurlini P, Tosello V, Nagai T, Miyawaki A, Di Lisa F, Pozzan T. Beat-to-beat oscillations of mitochondrial [Ca2+] in cardiac cells. EMBO J. 2001;20:4998–5007. doi: 10.1093/emboj/20.17.4998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parekh AB. Mitochondrial regulation of intracellular Ca2+ signaling: more than just simple Ca2+ buffers. News Physiol Sci. 2003;18:252–6. doi: 10.1152/nips.01458.2003. [DOI] [PubMed] [Google Scholar]
- 23.Duchen MR, Verkhratsky A, Muallem S. Mitochondria and calcium in health and disease. Cell Calcium. 2008;44:1–5. doi: 10.1016/j.ceca.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 24.Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science. 2009;325:477–81. doi: 10.1126/science.1175088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boncompagni S, Rossi AE, Micaroni M, Beznoussenko GV, Polishchuk RS, Dirksen RT, Protasi F. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol Biol Cell. 2009;20:1058–67. doi: 10.1091/mbc.E08-07-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sanchez JA, Garcia MC, Sharma VK, Young KC, Matlib MA, Sheu SS. Mitochondria regulate inactivation of L-type Ca2+ channels in rat heart. J Physiol. 2001;536:387–96. doi: 10.1111/j.1469-7793.2001.0387c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park CY, et al. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–90. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng W, Yuan JP, Kim MS, Choi YJ, Huang GN, Worley PF, Muallem S. STIM1 gates TRPC channels, but not Orai1, by electrostatic interaction. Mol Cell. 2008;32:439–48. doi: 10.1016/j.molcel.2008.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Glitsch MD, Bakowski D, Parekh AB. Store-operated Ca2+ entry depends on mitochondrial Ca2+ uptake. EMBO J. 2002;21:6744–54. doi: 10.1093/emboj/cdf675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. J Cell Biol. 1997;137:633–48. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zablocki K, Szczepanowska J, Duszynski J. Extracellular pH modifies mitochondrial control of capacitative calcium entry in Jurkat cells. J Biol Chem. 2005;280:3516–21. doi: 10.1074/jbc.M411507200. [DOI] [PubMed] [Google Scholar]
- 32.Ryu SY, Peixoto PM, Won JH, Yule DI, Kinnally KW. Extracellular ATP and P2Y2 receptors mediate intercellular Ca(2+) waves induced by mechanical stimulation in submandibular gland cells: Role of mitochondrial regulation of store operated Ca(2+) entry. Cell Calcium. 2010 doi: 10.1016/j.ceca.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JS, Kim MH, Ho WK, Lee SH. Presynaptic release probability and readily releasable pool size are regulated by two independent mechanisms during posttetanic potentiation at the calyx of Held synapse. J Neurosci. 2008;28:7945–53. doi: 10.1523/JNEUROSCI.2165-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta. 2009;1787:1309–16. doi: 10.1016/j.bbabio.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 35.Balaban RS. The role of Ca(2+) signaling in the coordination of mitochondrial ATP production with cardiac work. Biochim Biophys Acta. 2009;1787:1334–41. doi: 10.1016/j.bbabio.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maack C, O’Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res Cardiol. 2007;102:369–92. doi: 10.1007/s00395-007-0666-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brandes R, Bers DM. Simultaneous measurements of mitochondrial NADH and Ca(2+) during increased work in intact rat heart trabeculae. Biophys J. 2002;83:587–604. doi: 10.1016/S0006-3495(02)75194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cortassa S, Aon MA, Marban E, Winslow RL, O’Rourke B. An integrated model of cardiac mitochondrial energy metabolism and calcium dynamics. Biophys J. 2003;84:2734–55. doi: 10.1016/S0006-3495(03)75079-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feissner RF, Skalska J, Gaum WE, Sheu SS. Crosstalk signaling between mitochondrial Ca2+ and ROS. Front Biosci. 2009;14:1197–218. doi: 10.2741/3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duan Y, Gross RA, Sheu SS. Ca2+-dependent generation of mitochondrial reactive oxygen species serves as a signal for poly(ADP-ribose) polymerase-1 activation during glutamate excitotoxicity. J Physiol. 2007;585:741–58. doi: 10.1113/jphysiol.2007.145409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, Di Lisa F, Forte MA. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077–99. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- 42.Hajnoczky G, Csordas G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M. Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis. Cell Calcium. 2006;40:553–60. doi: 10.1016/j.ceca.2006.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–4. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- 44.Michels G, Khan IF, Endres-Becker J, Rottlaender D, Herzig S, Ruhparwar A, Wahlers T, Hoppe UC. Regulation of the human cardiac mitochondrial Ca2+ uptake by 2 different voltage-gated Ca2+ channels. Circulation. 2009;119:2435–43. doi: 10.1161/CIRCULATIONAHA.108.835389. [DOI] [PubMed] [Google Scholar]
- 45.Sottocasa G, Sandri G, Panfili E, De Bernard B, Gazzotti P, Vasington FD, Carafoli E. Isolation of a soluble Ca 2+ binding glycoprotein from ox liver mitochondria. Biochem Biophys Res Commun. 1972;47:808–13. doi: 10.1016/0006-291x(72)90564-5. [DOI] [PubMed] [Google Scholar]
- 46.Panfili E, Sandri G, Sottocasa GL, Lunazzi G, Liut G, Graziosi G. Specific inhibition of mitochondrial Ca2+ transport by antibodies directed to the Ca2+-binding glycoprotein. Nature. 1976;264:185–6. doi: 10.1038/264185a0. [DOI] [PubMed] [Google Scholar]
- 47.Saris NE, Sirota TV, Virtanen I, Niva K, Penttila T, Dolgachova LP, Mironova GD. Inhibition of the mitochondrial calcium uniporter by antibodies against a 40-kDa glycoproteinT. J Bioenerg Biomembr. 1993;25:307–12. doi: 10.1007/BF00762591. [DOI] [PubMed] [Google Scholar]
- 48.Mironova GD, Baumann M, Kolomytkin O, Krasichkova Z, Berdimuratov A, Sirota T, Virtanen I, Saris NE. Purification of the channel component of the mitochondrial calcium uniporter and its reconstitution into planar lipid bilayers. J Bioenerg Biomembr. 1994;26:231–8. doi: 10.1007/BF00763072. [DOI] [PubMed] [Google Scholar]
- 49.Zazueta C, et al. Identification of a 20-kDa protein with calcium uptake transport activity. Reconstitution in a membrane model. J Bioenerg Biomembr. 1994;26:555–62. doi: 10.1007/BF00762740. [DOI] [PubMed] [Google Scholar]
- 50.Zazueta C, Zafra G, Vera G, Sanchez C, Chavez E. Advances in the purification of the mitochondrial Ca2+ uniporter using the labeled inhibitor 103Ru360. J Bioenerg Biomembr. 1998;30:489–98. doi: 10.1023/a:1020546331217. [DOI] [PubMed] [Google Scholar]
- 51.Trenker M, Malli R, Fertschai I, Levak-Frank S, Graier WF. Uncoupling proteins 2 and 3 are fundamental for mitochondrial Ca2+ uniport. Nat Cell Biol. 2007;9:445–52. doi: 10.1038/ncb1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Graier WF, Trenker M, Malli R. Mitochondrial Ca2+, the secret behind the function of uncoupling proteins 2 and 3? Cell Calcium. 2008;44:36–50. doi: 10.1016/j.ceca.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brookes PS, et al. UCPs--unlikely calcium porters. Nat Cell Biol. 2008;10:1235–7. doi: 10.1038/ncb1108-1235. author reply 1237–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science. 2009;326:144–7. doi: 10.1126/science.1175145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- 56.Rousseau E, Smith JS, Meissner G. Ryanodine modifies conductance and gating behavior of single Ca2+ release channel. Am J Physiol. 1987;253:C364–8. doi: 10.1152/ajpcell.1987.253.3.C364. [DOI] [PubMed] [Google Scholar]
- 57.Aon MA, Cortassa S, Wei AC, Grunnet M, O’Rourke B. Energetic performance is improved by specific activation of K(+) fluxes through K(Ca) channels in heart mitochondria. Biochim Biophys Acta. 2009 doi: 10.1016/j.bbabio.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garcia-Perez C, Hajnoczky G, Csordas G. Physical coupling supports the local Ca2+ transfer between sarcoplasmic reticulum subdomains and the mitochondria in heart muscle. J Biol Chem. 2008;283:32771–80. doi: 10.1074/jbc.M803385200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Salnikov V, Lukyanenko YO, Lederer WJ, Lukyanenko V. Distribution of ryanodine receptors in rat ventricular myocytes. J Muscle Res Cell Motil. 2009;30:161–70. doi: 10.1007/s10974-009-9186-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gincel D, Vardi N, Shoshan-Barmatz V. Retinal voltage-dependent anion channel: characterization and cellular localization. Invest Ophthalmol Vis Sci. 2002;43:2097–104. [PubMed] [Google Scholar]
- 61.Norman JP, et al. HIV-1 Tat activates neuronal ryanodine receptors with rapid induction of the unfolded protein response and mitochondrial hyperpolarization. PLoS One. 2008;3:e3731. doi: 10.1371/journal.pone.0003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fenoglio JJ, Jr, Irey NS. Myocardial changes in malignant hyperthermia. Am J Pathol. 1977;89:51–8. [PMC free article] [PubMed] [Google Scholar]
- 63.Chinopoulos C, Starkov AA, Grigoriev S, Dejean LM, Kinnally KW, Liu X, Ambudkar IS, Fiskum G. Diacylglycerols activate mitochondrial cationic channel(s) and release sequestered Ca(2+) J Bioenerg Biomembr. 2005;37:237–47. doi: 10.1007/s10863-005-6634-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tan W, Colombini M. VDAC closure increases calcium ion flux. Biochim Biophys Acta. 2007;1768:2510–5. doi: 10.1016/j.bbamem.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mannella CA, Kinnally KW. Reflections on VDAC as a voltage-gated channel and a mitochondrial regulator. J Bioenerg Biomembr. 2008;40:149–55. doi: 10.1007/s10863-008-9143-0. [DOI] [PubMed] [Google Scholar]
- 66.Rapizzi E, et al. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J Cell Biol. 2002;159:613–24. doi: 10.1083/jcb.200205091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pavlov E, Grigoriev SM, Dejean LM, Zweihorn CL, Mannella CA, Kinnally KW. The mitochondrial channel VDAC has a cation-selective open state. Biochim Biophys Acta. 2005;1710:96–102. doi: 10.1016/j.bbabio.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 68.Gincel D, Zaid H, Shoshan-Barmatz V. Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem J. 2001;358:147–55. doi: 10.1042/0264-6021:3580147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Israelson A, Abu-Hamad S, Zaid H, Nahon E, Shoshan-Barmatz V. Localization of the voltage-dependent anion channel-1 Ca2+-binding sites. Cell Calcium. 2007;41:235–44. doi: 10.1016/j.ceca.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 70.Bathori G, Csordas G, Garcia-Perez C, Davies E, Hajnoczky G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC) J Biol Chem. 2006;281:17347–58. doi: 10.1074/jbc.M600906200. [DOI] [PubMed] [Google Scholar]
- 71.Kinnally KW, Antonenko YN, Zorov DB. Modulation of inner mitochondrial membrane channel activity. J Bioenerg Biomembr. 1992;24:99–110. doi: 10.1007/BF00769536. [DOI] [PubMed] [Google Scholar]