

Abstract
Background
Chronic use of long-acting β2-adrenergic receptor (β2AR) agonists (LABAs), resulting in β2AR desensitization, has been associated with increased asthma morbidity. When LABAs are used in combination with inhaled glucocorticoids (GCs), however, asthma control is improved, raising the question: Do GCs inhibit the proasthmatic mechanism that mediates altered contractility in LABA-exposed airway smooth muscle (ASM)?
Objective
This study aimed to identify the potential protective role and mechanism of action of GCs in mitigating the effects of prolonged LABA exposure on ASM constrictor and relaxation responsiveness.
Methods
Cultured human ASM (HASM) cells and isolated rabbit ASM tissues were examined for induced changes in agonist-mediated cAMP accumulation, constrictor and relaxation responsiveness, and expression of specific GC-regulated molecules following 24h exposure to the LABA, salmeterol, in the absence and presence of dexamethasone (DEX).
Results
Salmeterol-exposed ASM exhibited impaired cAMP and relaxation responses to isoproterenol and increased acetylcholine-induced contractility. These pro-asthmatic effects of prolonged LABA exposure were attributed to upregulated phosphodiesterase 4 (PDE4) activity, and ablated by pretreatment with DEX. Further studies demonstrated that: 1) DEX suppressed activation of the mitogen-activated protein kinase (MAPK), ERK1/2, which upregulates PDE4 expression in salmeterol-exposed ASM; and 2) the inhibitory actions of DEX on salmeterol-induced ERK1/2 activation and resultant PDE4-mediated changes in ASM responsiveness were prevented by gene silencing or pharmacological inhibition of DEX-induced expression of MAPK phosphatase-1 (MKP-1), an endogenous deactivator of ERK1/2 signaling.
Conclusion
GCs prevent the adverse proasthmatic effects of prolonged LABA exposure on airway responsiveness due to GC-induced upregulation of MKP-1, which inhibits proasthmatic ERK1/2 signaling in the LABA-exposed ASM.
Keywords: asthma, salmeterol, homologous β2-adrenergic receptor desensitization, airway smooth muscle, phosphodiesterase, ERK1/2 phosphorylation, siRNA, MAPK phosphatase-1
Introduction
While inhaled beta2-adrenergic receptor (ß2AR) agonists are commonly used to treat asthma, their chronic use in long-acting preparations can result in airway tolerance to ß2AR stimulation. This loss of bronchodilator effect is due to the development of homologous ß2AR desensitization, and has been associated with worsening of bronchial hyperreactivity and an increased incidence of asthma morbidity1-4. Conversely, when used in combination with inhaled glucocorticoids (GCs), long-acting ß2AR agonists (LABAs) have been shown to improve asthma control and alleviate airway inflammation5-9. Apart from reducing inflammation, the beneficial effects of this combinational therapy may reflect a complementary interaction between ß2AR and GC signaling in regulating airway smooth muscle (ASM) function under proasthmatic conditions10. Moreover, the therapeutic benefit of combined GC and LABA therapy may also relate to attenuation of the adverse effects of ß2AR desensitization, as previous studies demonstrated that GCs can reverse ß2AR tolerance in isolated human bronchi and small airways exposed to a short-acting ß2AR agonist11,12. To date, the role and mechanism by which GCs potentially mitigate the proasthmatic effects of homologous ß2AR desensitization on ASM constrictor and relaxation responsiveness remain unidentified.
We recently reported that prolonged exposure of cultured human ASM (HASM) cells and isolated rabbit ASM tissues to the LABA, salmeterol, leads to impaired acute ß2AR-induced cAMP accumulation and proasthmatic changes in ASM tissue responsiveness that are mediated by upregulated phosphodiesterase 4 (PDE4) activity in the ß2AR-desensitized ASM13. Moreover, the increase in PDE4 activity was attributed to ß2AR-induced Gi-βγ protein activation via cAMP-dependent PKA, which then activates the MAPK, ERK1/2, the latter leading to transcriptional upregulation of PDE4 expression13. Given this new information, the present study was undertaken to determine the potential role and mechanism of action of GCs in protecting ASM from the adverse effects of prolonged homologous ß2AR desensitization due to LABA exposure on ASM responsiveness. The results are the first to demonstrate that 1) GC-treated ASM is protected from the proasthmatic effects of prolonged exposure to a LABA on ASM constrictor and relaxation responsiveness; and 2) this protective action is attributed to GC-induced expression of mitogen-activated protein kinase phosphatase-1 (MKP-1), which suppresses ERK1/2 signaling and its consequent induction of PDE4 activity in the ß2AR desensitized state. These new findings highlight a heretofore-unidentified mechanism that may play a pivotal role in contributing to the known therapeutic efficacy of using GCs in combination with LABAs to treat asthma, and suggest that interventions targeted at this mechanism may yield new therapeutic approaches to improve asthma control.
Methods
Materials
Human ASM (HASM) cells were obtained from Bio Whittaker, Inc., and all chemicals were purchased from Sigma-Aldrich unless otherwise indicated.
Animals
Sixteen adult New Zealand White rabbits were used in this study, which was approved by the Biosafety and Animal Research Committee of the Research Institute at Children's Hospital of Philadelphia.
Culture and treatment of cells
HASM cells were cultured in growth medium supplemented with 10% FBS (Bio Whittaker), and maintained in an incubator containing 5% CO2 in air at 37°C. After attaining ∼95% conflue nce, the cells were starved in unsupplemented Ham's F12 media for 24 hr, subsequently treated with salmeterol or isoproterenol, and then examined for β2AR agonist-induced cAMP accumulation, PDE activity, ERK1/2 phosphorylation, and PDE4D5 mRNA expression under different experimental conditions.
cAMP accumulation and PDE activity
Confluent HASM cells were pretreated for 24 hr with either vehicle alone or salmeterol (10 μM), both in the absence and presence of a pre-determined maximally effective concentration of dexamethasone (DEX; 1 μM). In the continued presence of these treatments, cells were subsequently exposed for 5 min to isoproterenol (10 μM), and intracellular cAMP levels were detected using a cAMP enzyme immunoassay kit (R&D Systems)13,19. In separate studies, total cAMP PDE activity was measured in cell lysates using a colorimetric enzymatic assay (Biomol)13 following exposure of cells for 24 hr to either vehicle, salmeterol, or isoproterenol in the absence or presence of DEX under different co-treatment conditions, as described. The levels of cAMP and PDE activity were standardized to the cellular protein content.
Detection of PDE4D5 mRNA
As previously described13,14, PDE4D5 transcripts were detected following extraction of total RNA, and cDNAs were isolated by RT-PCR using the Superscript First Strand Synthesis System kit (Invitrogen), with the following oligonucleotide primer sets (Integrated DNA Technologies): for PDE4D5, 5′-TGCCAGCTGTACAAAGTTGACC-3′ (forward) and 5′TTCTCGGAGAGATCACTGGAGA-3′ (reverse); and for ß-actin, 5′-GAGAAGAGCTACGAGCTGCCTGAC-3′ (forward) and 5′-CGGAGTACTTGCGCTCAGGAGGAG-3′ (reverse). The reaction volume was 20 μl and cycling conditions used were 35 cycles of 30 sec denaturation at 95°C, followed by 30 sec annealing at 60°C and elongation at 72°C for 30 sec. Ex-Tag (Takara Biotechnology) was used as DNA polymerase.
Protein expression of ERK1/2 and MKP-1
Phosphorylated and total ERK1/2 and MKP-1 protein levels were detected by Western blotting in HASM cells before and after treatment with either salmeterol, isoproterenol, or DEX alone and in combination. Following protein extraction, lysates were loaded on a 10% SDS-PAGE gel for immunoblotting and, after transfer to a PVDF membrane, membranes were incubated overnight with monoclonal mouse anti-human primary antibodies directed against phospho-ERK1/2 and total ERK1/2, or a polyclonal goat anti-human primary antibody directed against MKP-1 (Imgenex Corp.). ERK levels were detected by ECL following incubation with a 1:2,000 dilution of HRP–conjugated rabbit anti-mouse secondary antibody, followed by exposure to autoradiography film. MKP-1 levels were detected with the Odyssey Infrared Imaging System (LI-COR Biosciences), using a 1:10,000 dilution of donkey anti-goat secondary antibody. The protein band intensities were quantified by densitometry.
siRNA knockdown of MKP-1
HASM cells were seeded into 6-well plates and, at ∼40% confluency, the medium was replaced with the reduced serum-containing medium, Opti-MEM (Invitrogen). As previously described13,14, cells were then transfected twice during a 24-hr interval with a pool of three siRNA duplexes targeted against human MKP-1 (Applied Biosystems: siRNA id # 5173,105846 and 146461), or with a non-targeted scrambled RNA (scRNA) duplex serving as a negative control, using Oligofectamine (Invitrogen) as the transfection agent. Each siRNA preparation was administered in a final concentration of 100 nM. In separate experiments, isolated rabbit ASM tissues (see below) were similarly transfected with the same siRNA preparations. Based on preliminary studies, this transfection approach was found to greatly enhance transfection efficiency, with DEX-induced MKP-1 protein expression inhibited by 78-91% in the siRNA-treated cells, and MKP-1 mRNA expression inhibited by 64-79% in the ASM tissues.
Pharmacodynamic studies of constrictor and relaxation responsiveness in rabbit ASM tissues
Following initial general anesthesia with intramuscular ketamine (50 mg/Kg), rabbits were sacrificed with an intravenous overdose of sodium pentobarbital (100 mg/kg). As previously described13,14, the tracheae were excised, loose connective tissue and epithelium were removed, and the tracheae were divided into ring segments that were incubated for 24 hr with either vehicle (control) or salmeterol, each administered under different pretreatment conditions, as described. The tissues were then placed in organ baths containing the same concentrations of their respective pharmacological treatments in modified Krebs-Ringer solution aerated with 5% CO2 in O2, and attached to force transducers to monitor isometric tension. Cholinergic contractility to cumulatively administered acetylcholine (ACh; 10-9 to 10-3 M) was assessed and, after rinsing with fresh buffer to remove the administered ACh, the pharmacological agents were immediately re-introduced into the organ baths and, ∼ 20 min thereafter, relaxation dose-response curves to isoproterenol (10-9 to 10-4 M) were generated after the tissues were initially half-maximally contracted with ACh. The constrictor and relaxation dose-response curves were analyzed with respect to each tissue's maximal isometric contractile force (Tmax) to ACh and maximal relaxation response (Rmax) to isoproterenol.
Statistical analyses
Results are expressed as mean ± SE values. Comparisons between groups were made using the Student's t-test (two-tailed) or ANOVA with Tukey's post-test analysis, where appropriate. A probability of <0.05 was considered statistically significant. Statistical analyses were conducted using the Prism computer program by Graph Pad Software Inc.
Results
GC prevention of homologous ß2AR desensitization in human ASM cells
To evaluate the role of GCs in modulating ASM function in the ß2AR desensitized state, we initially examined the effect of pretreatment with DEX on isoproterenol-induced cAMP accumulation in HASM cells following their prolonged homologous ß2AR desensitization. Accordingly, acute changes in intracellular cAMP accumulation, detected at 5 min following administration of a near half-maximal effective concentration of isoproterenol (ISO; 1.0 μM)13,14, were compared in HASM cells that were pretreated for 24 hr with either vehicle alone (control) or a maximally effective concentration of the LABA, salmeterol (10 μM)13, both in the absence and presence of co-treatment with DEX. As shown in Fig. 1, without DEX, salmeterol-exposed HASM cells exhibited homologous ß2AR desensitization, as evidenced by markedly suppressed cAMP responses to isoproterenol relative to those detected in control cells. Contrasting these observations, the cAMP responses to ISO were preserved in salmeterol-exposed cells that were co-treated with a pre-determined maximally effective concentration of DEX (1 μM), and similar to those detected in cells exposed to DEX alone.
Figure 1.

DEX prevents impaired acute ß2AR-induced cAMP accumulation in salmeterol-exposed HASM cells. In contrast to control cells, acute ISO induced cAMP accumulation is suppressed in HASM cells pre-exposed for 24 hr to salmeterol. By comparison, ISO-induced cAMP accumulation is preserved in salmeterol-exposed HASM cells that are co-treated with DEX. Data represent mean ± SE values from 4 paired experiments. **p<0.01 compared to untreated cells using paired t-test.
GCs prevent altered ASM responsiveness induced by prolonged ß2AR desensitization
We recently demonstrated that prolonged exposure of isolated rabbit ASM tissues to salmeterol elicits proasthmatic-like changes in ASM constrictor and relaxation responsiveness that are prevented by pretreating the tissues with the selective PDE4 inhibitor, rolipram13. To determine whether GCs exert a comparable protective action, agonist-induced constrictor and relaxation responses were compared in isolated rabbit ASM tissues that were exposed for 24 hr to either vehicle alone (control) or salmeterol (10 μM), both in the absence and presence of pretreatment with either DEX (1 μM) or rolipram (10 μM). Relative to their respective controls, the salmeterol-exposed ASM tissues exhibited significantly increased constrictor responsiveness to cumulative administration of ACh (Fig. 2A), yielding a mean ± SE maximal constrictor response (Tmax) of 136.0 ± 10.3 vs. 110.7 ± 7.2 g/g ASM wt. in the control tissues (p<0.05). Similar to the inhibitory effect of pretreatment with rolipram13, this enhanced constrictor responsiveness was also completely abrogated in salmeterol-exposed tissues that were pretreated with DEX.
Figure 2.

DEX prevents salmeterol-induced changes in agonist responsiveness in rabbit ASM tissues. Relative to controls, ASM tissues exposed to salmeterol exhibit significantly increased contractility to ACh (A) and impaired relaxation to isoproterenol (B). Pre-treatment with either rolipram or DEX prevents the salmeterol-induced changes in ASM responsiveness. Data represent mean ± SE values from 6-7 experiments. ANOVA used for multiple comparisons of mean Tmax values.
Under the same treatment conditions, cumulative administration of isoproterenol produced dose-dependent acute relaxation of pre-constricted ASM segments (Fig. 2B). Relative to controls, the relaxation responses were significantly attenuated in the salmeterol-exposed tissues, consistent with their development of homologous ß2AR desensitization. Accordingly, the mean ± SE maximal relaxation response (Rmax) in the salmeterol-exposed tissues amounted to 27.8 ± 4.9 % vs. 44.2 ± 5.5 % in the controls (p<0.05). This impaired relaxant responsiveness was also completely abrogated in salmeterol-exposed tissues that were pretreated with either rolipram or DEX. Of note, in separate experiments, relative to controls, ASM tissues that were treated with either DEX or rolipram alone showed no significant changes in responsiveness to either ACh or isoproterenol (data not shown).
GC suppression of PDE4 expression in ß2AR-desensitized ASM
Since transcriptional upregulation of PDE4 was found to mediate the above rolipram-sensitive changes in ASM responsiveness following its prolonged homologous or heterologous ß2AR desensitization13,14, we next examined whether DEX modulates PDE4 expression in the ß2AR-desensitized state. HASM cells were initially exposed to either vehicle, salmeterol (10 μM), or the shorter-acting ß2AR agonist, isoproterenol (10 μM), all in the absence and presence of co-treatment with DEX (1 μM), and then examined for induced changes in mRNA expression of PDE4D5, the functionally relevant PDE4 isoform in HASM15. As previously described13, relative to control cells (Fig. 3A: lane 1), HASM cells exposed to either isoproterenol or salmeterol for 6 hr (i.e., time to maximal response) exhibited upregulated expression of PDE4D5 mRNA transcripts (lanes 2 and 3). While constitutive transcript levels were essentially unaltered in cells exposed to DEX alone (lane 4), induction of PDE4D5 mRNA transcripts by the ß2AR agonists was completely suppressed in cells that were co-treated with DEX (lanes 5 and 6). As shown in Fig. 3B (open bars), comparable changes in total cAMP PDE activity were also detected under these experimental conditions. Accordingly, relative to control cells, HASM cells exposed for 24 hr to either isoproterenol or salmeterol exhibited similar significant increases in PDE activity that averaged approximately two-fold above control (p<0.01). Basal PDE activity was little affected in cells that were treated with DEX alone, whereas induction of PDE activity by the ß2AR agonists was completely abrogated in ASM cells that were co-treated with DEX (Fig. 3B; filled bars).
Figure 3.
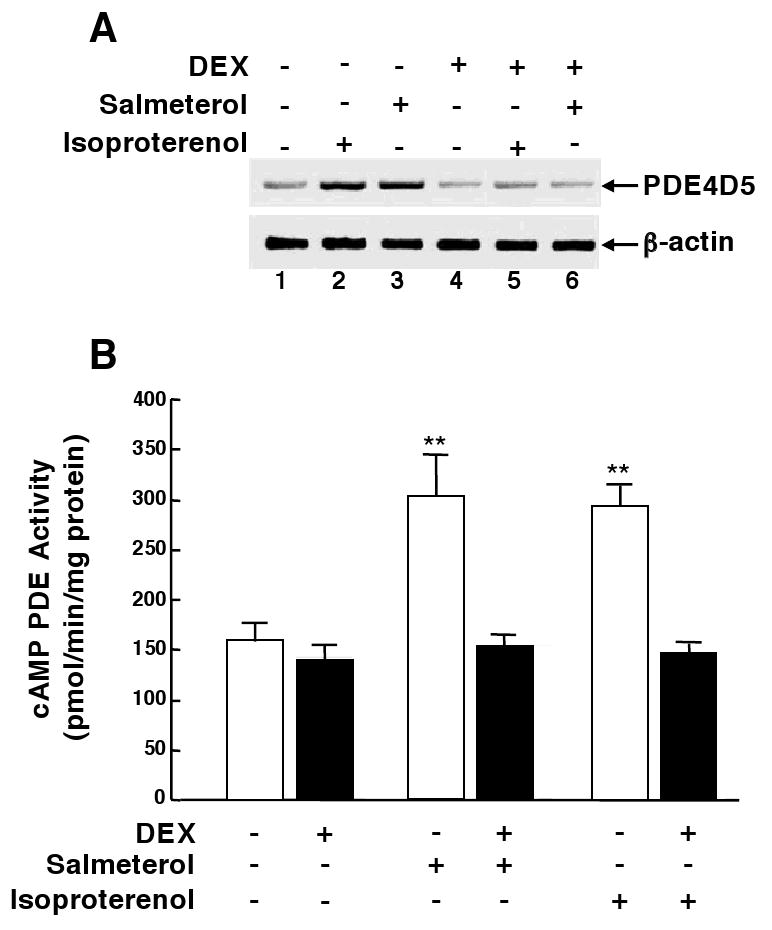
DEX prevents ß2AR agonist-induced upregulation of PDE4 in HASM cells. Relative to untreated controls, PDE4D5 mRNA expression (A) and cAMP PDE activity (B) are upregulated in HASM cells exposed to either salmeterol or isoproterenol. ß2AR agonist-induced upregulation of PDE4D5 transcripts and PDE activity is abrogated in cells pretreated with DEX. Data are mean ± SE values from 5 experiments. **p<0.01 compared to untreated cells using paired t-test.
GC suppression of ERK1/2 activation in ß2AR-desensitized ASM
In relation to the above observations, it should be noted that we previously demonstrated that the salmeterol-induced upregulation of PDE4 activity and associated PDE4-dependent changes in ASM constrictor and relaxation responsiveness are completely prevented by pretreating the ASM tissues with an inhibitor of either PKA or ERK1/2, and that these effects are reflective of PKA-dependent ERK1/2 activation in the ß2AR-desensitized ASM13. Given this evidence, we examined whether ß2AR agonist-induced activation of ERK1/2 is GC-sensitive. As previously described13, treatment of HASM cells with 10 μM of either isoproterenol or salmeterol acutely elicits increased phosphorylation of ERK1/2 proteins that peaks at 20 min and is sustained for at least up to 80 min. Accordingly, the effect of pretreatment with DEX (1 μM × 24 hr) on ERK1/2 phosphorylation was assessed in HASM cells at 60 min following exposure to 10 μM of salmeterol or isoproterenol. A representative immunoblot in Fig. 4A demonstrates that, relative to control cells (lane 1), ß2AR agonist-exposed cells exhibit increased ERK1/2 phosphorylation (lanes 2 and 3) and, while pretreatment with DEX alone has no effect (lane 4), the induction of ERK1/2 phosphorylation is suppressed in ß2AR agonist-exposed cells that are pretreated with DEX (lanes 5 and 6). The results obtained in 4 experiments demonstrated that isoproterenol and salmeterol elicited similar increases in ERK1/2 phosphorylation, expressed as fold-changes in the densitometric ratios of phosphorylated ERK1/2-to-total ERK1/2, that averaged ∼1.8-fold above that detected in control cells (p<0.01) and that, in contrast to the lack of effect of DEX alone, ß2AR agonist-induced phosphorylation of ERK1/2 was completely supressed in HASM cells that were pretreated with DEX (Fig. 4B). Thus, these data demonstrate that ß2AR agonist-induced ERK1/2 activation in HASM cells is GC-sensitive.
Figure 4.
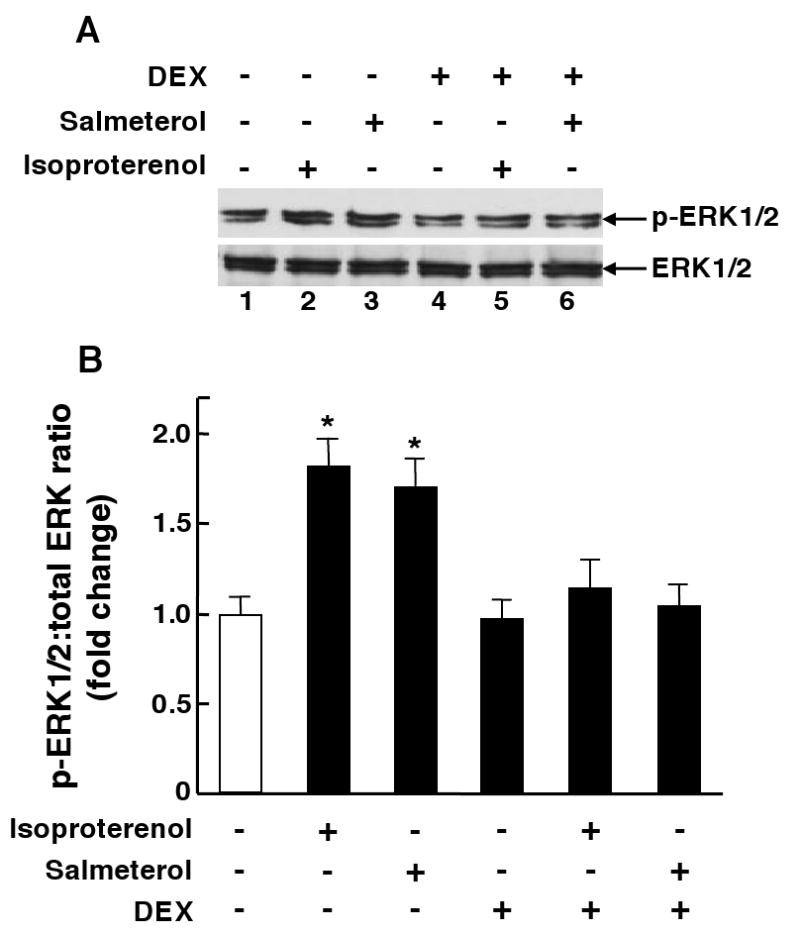
A: HASM cells exposed to either salmeterol or isoproterenol exhibit increased ERK1/2 phosphorylation that is prevented by pretreating the ß2AR agonist-exposed cells with DEX. B: Corresponding densitometric analysis demonstrates similar increases in ERK1/2 phosphorylation in salmeterol- and isoproterenol-exposed cells that are prevented by pretreatment with DEX. Data are mean ± SE values from 4 determinations under each treatment condition. *p<0.05 compared to untreated cells using paired t-test.
MKP-1 mediates GC suppression of ERK1/2 signaling and PDE4 expression in ß2AR-desensitized ASM
Since GCs evoke synthesis of the dual-specificity phosphatase, MKP-1, which acts as a potent endogenous deactivator of ERK1/2 signaling by dephosphorylating its tyrosine and threonine residues16, we next examined whether DEX-induced suppression of ERK1/2-coupled upregulation of PDE4 in ß2AR-desensitized HASM cells is mediated by altered MKP-1 expression. As previously documented17,18, our initial studies demonstrated that DEX (1 μM) acutely elicited increased MKP-1 protein expression in HASM cells that peaked at 2 hr and, as exemplified in Fig. 5A, this DEX-induced MKP-1 expression was markedly suppressed (i.e., between 78 and 91%) in cells that were transfected with a pool of three siRNA duplexes directed against human MKP-1. By comparison, transfection with a scrambled siRNA sequence (scRNA) serving as an inert negative control had no effect on DEX-induced MKP-1 expression (data not shown). Accordingly, these siRNA preparations were then used to ascertain the role of MKP-1 in mediating DEX-induced inhibition of ERK1/2 activation, PDE4D5 expression, and cAMP PDE activity in salmeterol-exposed HASM cells. Fig. 5B demonstrates that DEX-induced inhibition of salmeterol-evoked ERK1/2 activation was prevented in HASM cells that were pretreated with the MKP-1 siRNAs, or with the non-selective MKP-1 inhibitor, Ro-31-8220 (5 μM)19. Comparably, suppression by DEX of salmeterol-induced PDE4D5 mRNA expression was prevented in HASM cells transfected with the MKP-1 siRNAs, whereas transfection with scRNA had no effect (Figs. 5C). Finally, DEX-induced suppression of upregulated PDE activity in salmeterol-exposed cells was also abrogated either by transfection with the MKP-1 siRNAs (whereas scRNA transfection had no effect) or by pretreatment with Ro-31-8220 (Fig. 5D).
Figure 5.
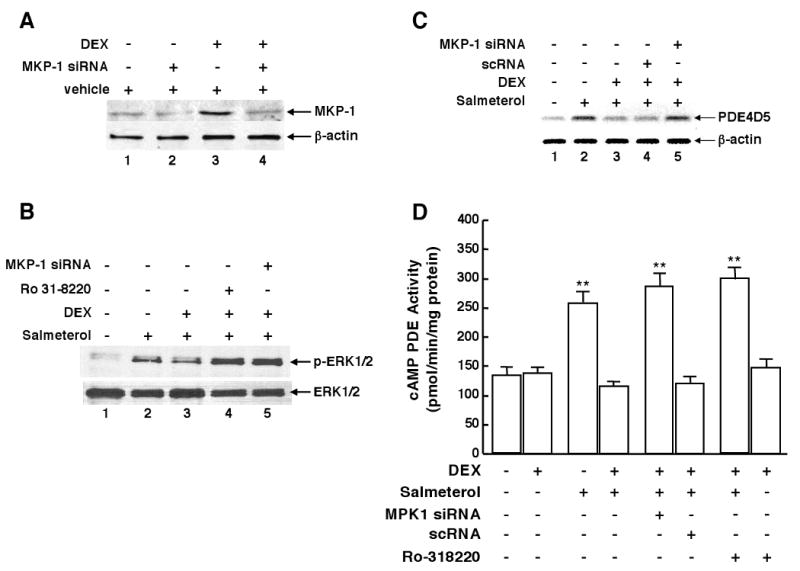
A: DEX-induced upregulation of MKP-1 protein is prevented in HASM cells transfected with MKP-1 siRNA duplexes. DEX-induced suppression of upregulated ERK1/2 phosphorylation (B) and PDE4D5 mRNA (C) expression in salmeterol-exposed cells is abrogated by transfection with MKP-1 siRNAs, whereas transfection with scRNA (negative control) has no effect. D: DEX-induced suppression of upregulated PDE activity in salmeterol-exposed cells is prevented by transfection with the MKP-1 siRNAs or by pretreatment with Ro-31-8220. Data are mean ± SE values from 5 experiments. **p<0.01 compared to untreated cells using paired t-test.
MKP-1 mediates GC-induced suppression of altered responsiveness in ß2AR-desensitized ASM
To ascertain the physiological relevance of the above observations, we next examined whether suppression of MKP-1 impairs the ability of DEX to prevent the PDE4-dependent changes in ASM responsiveness elicited in the ß2AR-desensitized state (as depicted in Fig. 2). In these studies, agonist-induced ASM constrictor and relaxation responses were compared in rabbit ASM tissues that were exposed to salmeterol (10 μM × 24 hr) following initial transfection with either scRNA (control) or the siRNA duplexes directed against MKP-1. Fig. 6 demonstrates that, relative to controls, the significantly increased constrictor responses to ACh (Fig. 6A) and impaired relaxation responses to isoproterenol (Fig. 6B) were abrogated in salmeterol-exposed ASM tissues that were pretreated with DEX (1 μM), and that the latter protective effects of DEX were ablated in salmeterol-exposed tissues that were transfected with the MKP-1 siRNA preparations. In relation to these observations, it should be noted that in separate studies conducted on control and salmeterol-exposed ASM tissues that were not pretreated with DEX, neither transfection with the MKP-1 siRNAs nor scRNA had a significant effect on the tissues' constrictor or relaxation responses (data not shown). Collectively, these data support the concept that prevention of the PDE4-mediated changes in ASM responsiveness elicited in the ß2AR-desensitized state by DEX is attributed to its induced upregulation of MKP-1, which suppresses ERK1/2 activation and its associated induction of PDE4 in the ß2AR-desensitized ASM.
Figure 6.

MKP-1 siRNA prevents DEX-induced suppression of the proasthmatic effects of salmeterol on ASM responsiveness. Rabbit ASM tissues exposed to salmeterol exhibit significantly increased contractility to ACh (A) and impaired relaxation responses to isoproterenol (B) that are prevented by pretreatment with DEX. The protective effects of DEX are abrogated in salmeterol-exposed tissues transfected with MKP-1 siRNAs. Data are mean ± SE values from 5-7 paired experiments. ANOVA used for multiple comparisons of mean Tmax values.
Discussion
Chronic use of LABAs to treat asthma has been associated with impaired bronchodilation, heightened bronchoconstriction and an increase in asthma morbidity1-3, presumably reflecting the adverse effects of prolonged homologous β2AR desensitization of the airways4. Although mechanisms implicated in mediating acute homologous desensitization of the β2AR in ASM are well described, including uncoupling of the β2AR from Gs protein due to its phosphorylation by G protein-coupled receptor kinases and cAMP-dependent PKA4,20,21, these mechanisms do not readily explain the worsening of asthma symptoms due to chronic use of LABAs. In this regard, we recently demonstrated that the induction of β2AR desensitization in ASM following its prolonged exposure to a LABA, as well as other cAMP-elevating agents, is associated with proasthmatic-like changes in ASM responsiveness that are mediated by upregulated PDE4 activity evoked by cross-talk between the PKA and ERK1/2 signaling pathways, the latter leading to transcriptional upregulation of PDE4D513,14. We now provide new evidence that identifies the role and mechanism of action of GCs in preventing the pro-asthmatic effects of LABA exposure on ASM function. Specifically, the present results demonstrate that: 1) DEX prevents the induction of PDE4-mediated changes in ASM responsiveness evoked by prolonged exposure to the LABA, salmeterol; and 2) this protective action of DEX is attributed to its induced upregulation of MKP-1, which suppresses ERK1/2 activation and its consequent upregulation of PDE4 activity. These findings are the first to highlight a heretofore unidentified mechanism of action of GCs in ASM that may fundamentally contribute to the reported improved asthma control and reduced risk of severe asthma morbidity in patients using LABAs in combination with inhaled GCs5-9.
While an earlier study suggested that reversal of ß2AR desensitization in human airways by GC administration may be related to up-regulation of ß2AR expression11, more recent studies have reported that the protective effects of GCs in ß2AR-desensitized rodent lung tissue22 and human small airways12 are not readily attributed to GC-induced changes in ß2AR expression, implying that GCs likely exert their protective action in ASM via an alternative mechanism. Our present observations demonstrated that: 1) pretreatment with DEX prevented the rolipram-sensitive changes in constrictor and relaxation responsiveness exhibited in homologous ß2AR-desensitized ASM (Fig. 2); and 2) the protective action of DEX was associated with its inhibition of upregulated PDE4D5 expression and cAMP PDE activity (Fig. 3), coupled to inhibition of ERK1/2 activation (Fig. 4), in the ß2AR-desensitized state. These observations raise certain relevant considerations. Accordingly, it should be noted that, although the assay used herein measured total PDE activity, we previously demonstrated that the upregulated PDE activity elicited in homologous ß2AR-desensitized ASM is specifically attributed to PDE4, as it is completely inhibited by pretreating salmeterol-exposed HASM cells with the PDE4 selective inhibitor, rolipram13. Moreover, the PDE4 activity most likely reflects that of PDE4D5 since it is the functionally relevant isoform in human ASM cells15. In relation to this evidence, it is noteworthy that recent studies have identified that PDE4 activity plays a critical role in regulating ASM contractility23, and that upregulated PDE4 activity mediates the constrictor hyperresponsiveness of the airways that accompanies allergen challenge in asthmatic subjects24 and in animal models of allergic asthma25-29. Moreover, in concert with our present findings, other studies have reported that GCs inhibit the upregulated PDE4 activity detected in the lungs of rodent models of allergic asthma27,28. Given these findings, together with those of the present study, it is reasonable to speculate that: 1) the induction of altered airway responsiveness following exposure to either a LABA or allergen in the sensitized asthmatic state is attributed to the same downstream mechanism that mediates upregulated PDE4 activity in ASM under these proasthmatic conditions; and 2) the bronchoprotective effect of GCs on airway function is due, at least in part, to the same mechanism that mediates GC-induced suppression of PDE4 activity in ASM under these conditions. These possibilities remain to be systematically investigated.
The anti-inflammatory actions of GCs, including in ASM10,30,31, are mediated by activation of the intracellular glucocorticoid receptor (GR), which serves to both suppress transactivation of various key proinflammatory transcription factors and promote transcription of genes that can acutely modulate the activities of various intracellular signalling cascades32,33. Acute GC induction of gene expression involves homodimerization of the activated GR complex, its nuclear translocation, and binding to palindromic glucocorticoid response elements (GREs) in the promotor region of various GC-responsive immediate-early response genes. The archetype of these GC-induced immediate-early response genes is MKP-1, which was found to have at least three putative GREs in its promotor region34. MKP-1 dephoshorylates activated MAPKs and, thereby, serves a crucial anti-inflammatory function16, including in ASM17,18,35. Our present results support the concept that GC-induced upregulation of MKP-1 and its consequent attenuation of ERK1/2 signaling plays a pivotal role in preventing the PDE4-dependent changes in ASM constrictor and relaxation responsiveness that accompany prolonged homologous ß2AR desensitization. Specifically, the results demonstrated that: 1) inhibition of MKP-1 activity by siRNA silencing of MKP-1 expression, or by the non-selective inhibitor, Ro-31-8220, abrogated DEX-induced suppression of ERK1/2 activation and its coupled upregulation of PDE4D5 expression and PDE4 activity in salmeterol-exposed HASM cells (Fig. 5); and 2) DEX-induced suppression of the PDE4-mediated changes in constrictor and relaxation responsiveness in salmeterol-exposed ASM tissues was also abrogated by siRNA silencing of MKP-1 (Fig. 6). Of note, these observations are consistent with those in previous studies reporting that ERK1/2 activation plays a crucial role in regulating the synthesis and activity of PDE4 in ASM13,14,36,37 and in mediating airway hyperreactivity in vivo and altered ASM responsiveness in vitro in response to different proasthmatic stimuli3841; and that GCs inhibit ERK1/2 activation by upregulating MKP-1 under various proinflammatory conditions in different cell systems16,42, including ASM18. Notwithstanding these considerations, it is important to indicate that the nature of LABA exposure and its effects examined under the ex vivo conditions herein may be significantly different from that related to LABA use in human asthmatics wherein, moreover, the presence of allergic airway inflammation may further modulate β2AR function.
In conclusion, this study is the first to report that: 1) GCs prevent the PDE4-dependent proasthmatic changes in ASM responsiveness that accompany ß2AR tolerance induced by prolonged exposure to a LABA; and 2) this protective action of GC is attributed to its induction of MKP-1 and, consequently, inhibition of ERK1/2-coupled upregulation of PDE4 activity in the ß2AR-desensitized ASM. These novel findings highlight a heretofore-unidentified mechanism by which GCs suppress the proasthmatic effects of prolonged airway exposure to a LABA on airway responsiveness. Thus, future interventions targeted at this mechanism may provide new therapeutic approaches to improve asthma control.
Acknowledgments
The authors thank David Kaltman for his technical assistance.
Declaration of funding sources: This work was supported by NIH grants HL-031467, HL-061038, and HL097739. The authors have no financial relationship with a biotechnology and/or pharmaceutical manufacturer that has an interest in the subject matter or materials presented in this manuscript.
Abbreviations
- β2AR
β2-adrenergic receptor
- LABA
long-acting β2-adrenergic receptor agonist
- GC
glucocorticoid
- DEX
dexamethasone
- ISO
isoproterenol
- HASM
human airway smooth muscle
- Tmax
maximal isometric contractile force
- Rmax
maximal relaxation response
- PDE4
phosphodiesertase 4
- MAPK
mitogen-activated protein kinase
- MKP-1
mitogen-activated protein kinase phosphatase-1
- ERK1/2
extracellular signal-regulated kinases 1 and 2
- siRNA
small interfering RNA
- GR
glucocorticoid receptor
- GRE
glucocorticoid response element
Footnotes
- GCs prevent the proasthmatic effects of prolonged homologous β2AR desensitization due to exposure to a LABA on ASM function.
- This bronchoprotective action is attributed to GC-induced upregulation of MKP-1 expression, which inhibits ERK1/2 activation and its consequent induction of PDE4-mediated changes in ASM responsiveness in the β2AR-desensitized state.
Implications: The above newly identified mechanism of GC action in preventing the proasthmatic effects of LABA exposure on ASM responsiveness may play a pivotal role in contributing to the therapeutic efficacy of using GCs in combination with LABAs for asthma control. Future interventions targeted at this mechanism may provide new approaches to treat asthma.
Capsule Summary: A mechanism of action of GCs in preventing the proasthmatic effects of a LABA on airway function is identified which may fundamentally contribute to the therapeutic efficacy of using GCs in combination with LABAs for asthma control.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Beasley R, Pearce N, Crane J, Burgess C. Beta-agonists: what is the evidence that their use increases the risk of asthma morbidity and mortality? J Allergy Clin Immunol. 1999;104:S18–30. doi: 10.1016/s0091-6749(99)70270-8. [DOI] [PubMed] [Google Scholar]
- 2.Cheung D, Timmers MC, Zwinderman AH, Bel EH, Dijkman JH, Sterk PJ. Longterm effects of a long-acting beta 2-adrenoceptor agonist, salmeterol, on airway hyperresponsiveness in patients with mild asthma. N Engl J Med. 1992;327:1198–203. doi: 10.1056/NEJM199210223271703. [DOI] [PubMed] [Google Scholar]
- 3.Nelson HS, Weiss ST, Bleecker ER, Yancey SW, Dorinsky PM. The Salmeterol Multicenter Asthma Research Trial: a comparison of usual pharmacotherapy for asthma or usual pharmacotherapy plus salmeterol. Chest. 2006;129:15–26. doi: 10.1378/chest.129.1.15. [DOI] [PubMed] [Google Scholar]
- 4.Giembycz MA, Newton R. Beyond the dogma: novel beta2-adrenoceptor signalling in the airways. Eur Respir J. 2006;27:1286–1306. doi: 10.1183/09031936.06.00112605. [DOI] [PubMed] [Google Scholar]
- 5.Woolcock A, Lundback B, Ringdahl N, Jaques LA. Comparison of addition of salmeterol to inhaled steroids with doubling of the dose of inhaled steroids. Am J Respir Crit Care Med. 1996;153:1481–8. doi: 10.1164/ajrccm.153.5.8630590. [DOI] [PubMed] [Google Scholar]
- 6.Pauwels RA, Lofdahl CG, Postma DS, Tattersfield AE, O'Byrne P, Barnes PJ, et al. Effect of inhaled formoterol and budesonide on exacerbations of asthma. N Engl J Med. 1997;337:1405–11. doi: 10.1056/NEJM199711133372001. [DOI] [PubMed] [Google Scholar]
- 7.Kips J, O'Connor B, Inman M, Svensson K, Pauwels RA, O'Byrne PM. A long-term study of the anti-inflammatory effect of low-dose budesonide plus formoterol versus high-dose budesonide in asthma. Am J Respir Crit Care Med. 2000;161:996–1001. doi: 10.1164/ajrccm.161.3.9812056. [DOI] [PubMed] [Google Scholar]
- 8.Bateman E, Nelson H, Bousquet J, Kral K, Sutton L, Ortega H, Yancey S. Meta-analysis: effects of adding salmeterol to inhaled corticosteroids on serious asthma-related events. Ann Int Med. 2008;149:33–42. doi: 10.7326/0003-4819-149-1-200807010-00229. [DOI] [PubMed] [Google Scholar]
- 9.Ni Chroinin M, Lasserson TJ, Greenstone I, Ducharme FM. Addition of long-acting beta-agonists to inhaled corticosteroids for chronic asthma in children. Cochrane Database of Systematic Reviews. 2009;(3):CD007949. doi: 10.1002/14651858.CD007949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giembycz MA, Kaur M, Leigh R, Newton R. A Holy Grail of asthma management: toward understanding how long-acting beta(2)-adrenoceptor agonists enhance the clinical efficacy of inhaled corticosteroids. Br J Pharmacol. 2008;153:1090–104. doi: 10.1038/sj.bjp.0707627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hauck RW, Harth M, Schulz C, Prauer H, Bohm M, Schomig A. Effects of β2-agonist- and dexamethasone-treatment on relaxation and regulation of β-adrenoceptors in human bronchi and lung tissue. Br J Pharmacol. 1997;121:1523–30. doi: 10.1038/sj.bjp.0701289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cooper PR, Panettieri RA., Jr Steroids completely reverse albuterol-induced β2-adrenergic receptor tolerance in human small airways. J Allergy Clin Immunol. 2008;122:734–40. doi: 10.1016/j.jaci.2008.07.040. [DOI] [PubMed] [Google Scholar]
- 13.Nino G, Hu A, Grunstein JS, Grunstein MM. Mechanism regulating proasthmatic effects of prolonged homologous β2-adrenergic receptor desensitization in airway smooth muscle. Am J Physiol Lung Cell Mol Physiol. 2009;297:L746–57. doi: 10.1152/ajplung.00079.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu A, Nino G, Grunstein JS, Fatma S, Grunstein MM. Prolonged heterologous β2-adrenoceptor desensitization promotes proasthmatic airway smooth muscle function via PKA/ERK1/2-mediated phosphodiesterase-4 induction. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1055–67. doi: 10.1152/ajplung.00021.2008. [DOI] [PubMed] [Google Scholar]
- 15.Billington CK, Le Jeune IR, Young KW, Hall IP. A major functional role for phosphodiesterase 4D5 in human airway smooth muscle cells. Am J Respir Cell Mol Biol. 2008;38:1–7. doi: 10.1165/rcmb.2007-0171OC. [DOI] [PubMed] [Google Scholar]
- 16.Abraham SM, Clark AR. Dual-specificity phosphatase 1: a critical regulator of innate immune responses. Biochem Soc Trans. 2006;34:1018–23. doi: 10.1042/BST0341018. [DOI] [PubMed] [Google Scholar]
- 17.Issa R, Xie S, Khorasani N, Sukkar M, Adcock IM, Lee KY, Chung KF. Corticosteroid inhibition of growth-related oncogene protein-alpha via mitogen-activated kinase phosphatase-1 in airway smooth muscle cells. J Immunol. 2007;178:7366–75. doi: 10.4049/jimmunol.178.11.7366. [DOI] [PubMed] [Google Scholar]
- 18.Kang BN, Jude JA, Panettieri RA, Jr, Walseth TF, Kannan MS. Glucocorticoid regulation of CD38 expression in human airway smooth muscle cells: role of dual specificity phosphatase 1. Am J Physiol Lung Cell Mol Physiol. 2008;295:L186–93. doi: 10.1152/ajplung.00352.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beltman J, McCormick F, Cook SJ. The selective protein kinase C inhibitor, Ro-31-8220, inhibits mitogen-activated protein kinase phosphatase-1 (MKP-1) expression, induces c-Jun expression, and activates Jun N-terminal kinase. J Biol Chem. 1996;271:27018–24. doi: 10.1074/jbc.271.43.27018. [DOI] [PubMed] [Google Scholar]
- 20.Penn RB, Panettieri RA, Jr, Benovic JL. Mechanisms of acute desensitization of the β2AR-adenylyl cyclase pathway in human airway smooth muscle. Am J Respir Cell Mol Biol. 1008;19:338–48. doi: 10.1165/ajrcmb.19.2.3025. [DOI] [PubMed] [Google Scholar]
- 21.Shore SA, Moore PE. Regulation of β-adrenergic responses in airway smooth muscle. Respir Physiol Neurobiol. 2003;137:179–95. doi: 10.1016/s1569-9048(03)00146-0. [DOI] [PubMed] [Google Scholar]
- 22.Mak JC, Hisada T, Salmon M, Barnes PJ, Chung KF. Glucocorticoids reverse IL-1β-induced impairment of β-adrenoceptor-mediated relaxation and up-regulation of G-protein-coupled receptor kinases. Br J Pharmacol. 2002;135:987–96. doi: 10.1038/sj.bjp.0704545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mehats C, Jin SL, Wahlstrom J, Law E, Umetsu DT, Conti M. PDE4D plays a critical role in the control of airway smooth muscle contraction. FASEB J. 2003;17:1831–41. doi: 10.1096/fj.03-0274com. [DOI] [PubMed] [Google Scholar]
- 24.van Schalkwyk E, Strydom K, Williams Z, Venter L, Leichtl S, Schmid-Wirlitsch C, Bredenbroker D, Bardin PG. Roflumilast, an oral, once-daily phosphodiesterase 4 inhibitor, attenuates allergen-induced asthmatic reactions. J Allergy Clin Immunol. 2005;116:292–8. doi: 10.1016/j.jaci.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 25.Hansen G, Jin S, Umetsu DT, Conti M. Absence of muscarinic cholinergic airway responses in mice deficient in the cyclic nucleotide phosphodiesterase PDE4D. Proc Natl Acad Sci U S A. 2000;97:6751–6. doi: 10.1073/pnas.97.12.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanehiro A, Ikemura T, Makela MJ, Lahn M, Joetham A, Dakhama A, Gelfand EW. Inhibition of phosphodiesterase 4 attenuates airway hyperresponsiveness and airway inflammation in a model of secondary allergen challenge. Am J Respir Crit Care Med. 2001;163:173–84. doi: 10.1164/ajrccm.163.1.2001118. [DOI] [PubMed] [Google Scholar]
- 27.Tang HF, Song YH, Chen JC, Chen JQ, Wang P. Upregulation of phosphodiesterase-4 in the lung of allergic rats. Am J Respir Crit Care Med. 2005;171:823–8. doi: 10.1164/rccm.200406-771OC. [DOI] [PubMed] [Google Scholar]
- 28.Sun JG, Deng YM, Wu X, Tang HF, Deng JF, Chen JQ, Yang SY, Xie QM. Inhibition of phosphodiesterase activity, airway inflammation and hyperresponsiveness by PDE4 inhibitor and glucocorticoid in a murine model of allergic asthma. Life Sci. 2006;79:2077–85. doi: 10.1016/j.lfs.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 29.Chapman RW, House A, Jones H, Richard J, Celly C, Prelusky D, et al. Effect of inhaled roflumilast on the prevention and resolution of allergen-induced late phase airflow obstruction in Brown Norway rats. Eur J Pharmacol. 2007;571:215–21. doi: 10.1016/j.ejphar.2007.05.074. [DOI] [PubMed] [Google Scholar]
- 30.Hirst SJ, Lee TH. Airway smooth muscle as a target of glucocorticoid action in the treatment of asthma. Am J Respir Crit Care Med. 1998;158:S201–6. doi: 10.1164/ajrccm.158.supplement_2.13tac190. [DOI] [PubMed] [Google Scholar]
- 31.Panettieri RA., Jr Effects of corticosteroids on structural cells in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2004;1:231–4. doi: 10.1513/pats.200402-021MS. [DOI] [PubMed] [Google Scholar]
- 32.Barnes PJ. Antiinfammatory action of glucocorticoids: molecular mechanisms. Clin Sci. 1998;94:557–72. doi: 10.1042/cs0940557. [DOI] [PubMed] [Google Scholar]
- 33.Hakonarson H, Halapi E, Whelan R, Gulcher J, Stefansson K, Grunstein MM. Association between IL-1β/TNF-α-induced glucocorticoid-sensitive changes in multiple gene expression and altered responsiveness in airway smooth muscle. Am J Respir Cell Mol Biol. 2001;6:761–71. doi: 10.1165/ajrcmb.25.6.4628. [DOI] [PubMed] [Google Scholar]
- 34.Noguchi T, Metz R, Chen L, Mattéi MG, Carrasco D, Bravo R. Structure, mapping and expression of erp, a growth factor-inducible gene encoding a nontransmembrane protein tyrosine phosphase and effect of erp on cell growth. Mol Cell Biol. 1993;13:5195–5205. doi: 10.1128/mcb.13.9.5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burgess JK, Lee JH, Ge Q, Ramsay EE, Poniris MH, Parmentier J, et al. Dual ERK and phosphatidylinositol 3-kinase pathways control airway smooth muscle proliferation: differences in asthma. J Cell Physiol. 2008;216:673–9. doi: 10.1002/jcp.21450. [DOI] [PubMed] [Google Scholar]
- 36.Liu H, Palmer D, Jimmo SL, Tilley DG, Dunkerley HA, Pang SC, et al. Expression of phosphodiesterase 4D (PDE4D) is regulated by both the cyclic AMP-dependent protein kinase and mitogen-activated protein kinase signaling pathways. A potential mechanism allowing for the coordinated regulation of PDE4D activity and expression in cells. J Biol Chem. 2000;275:26615–24. doi: 10.1074/jbc.M001634200. [DOI] [PubMed] [Google Scholar]
- 37.Houslay MD, Baillie GS. The role of ERK2 docking and phosphorylation of PDE4 cAMP phosphodiesterase isoforms in mediating cross-talk between the cAMP and ERK signalling pathways. Biochem Soc Trans. 2003;31:1186–90. doi: 10.1042/bst0311186. [DOI] [PubMed] [Google Scholar]
- 38.Duan W, Chan JH, Wong CH, Leung BP, Wong WS. Anti-inflammatory effects of mitogen-activated protein kinase kinase inhibitor U0126 in an asthma mouse model. J Immunol. 2009;172:7053–9. doi: 10.4049/jimmunol.172.11.7053. 4. [DOI] [PubMed] [Google Scholar]
- 39.Ohnishi H, Takeda K, Domenico J, Lucas JJ, Miyahara N, Swasey CH, et al. Mitogen-activated protein kinase/extracellular signal-regulated kinase 1/2-dependent pathways are essential for CD8+ T cell-mediated airway hyperresponsiveness and inflammation. J Allergy Clin Immunol. 2009;123:249–57. doi: 10.1016/j.jaci.2008.10.054. [DOI] [PubMed] [Google Scholar]
- 40.Grunstein MM, Veler H, Shan X, Larson J, Grunstein JS, Chuang S. Proasthmatic effects and mechanisms of action of the dust mite allergen, Der p 1, in airway smooth muscle. J Allergy Clin Immunol. 2005;116:94–101. doi: 10.1016/j.jaci.2005.03.046. [DOI] [PubMed] [Google Scholar]
- 41.Shan X, Hu A, Veler H, Fatma S, Grunstein JS, Chuang S, Grunstein MM. Regulation of toll-like receptor 4-induced pro-asthmatic changes in airway smooth muscle function by opposing actions of ERK1/2 and p38 MAPK signaling. Am J Physiol Lung Cell Mol Physiol. 2006;291:L324–33. doi: 10.1152/ajplung.00056.2006. [DOI] [PubMed] [Google Scholar]
- 42.Kassel O, Sancono A, Kratzschmar J, Kreft B, Stassen M, Cato AC. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. 2001;20:7108–16. doi: 10.1093/emboj/20.24.7108. [DOI] [PMC free article] [PubMed] [Google Scholar]