

Abstract
This review focuses on recent developments in the molecular mechanisms by which Ca activates cardiac sarcomeres and how these mechanisms play out in the cardiac cycle. I emphasize the role of mechanisms intrinsic to the sarcomeres as significant determinants of systolic elastance and ventricular stiffening during ejection. Data are presented supporting the idea that processes intrinsic to the thin filaments may promote cooperative activation of the sarcomeres and be an important factor in maintaining and modifying systolic elastance. Application of these ideas to translational medicine and rationale drug design forms an important rationale for detailed understanding of these processes.
1. Introduction
One of the significant challenges to investigations of control mechanisms in cardiac contraction and relaxation is the understanding of the relation between the molecular and cellular biology of working ventricular myocytes and the dynamics of the heart beat. There is wide acceptance that the transition from diastole to systole requires electrochemical coupling involving a membrane controlled release of Ca2+ into the sarcoplasmic space [1]. The rise in intracellular Ca2+ induces chemomechanical coupling in which Ca-binding to cardiac troponin C (cTnC) triggers a series of protein-protein interactions releasing the thin filaments from inhibition and promoting a force generating interaction between myosin cross-bridges and actin. The rise in intracellular Ca is transient as highly effective processes actively transport the Ca2+ back into cellular storage depots within the sarcoplasmic reticulum or back to the extra-cellular space. Thus, chemomechanical uncoupling has generally been considered to occur by a fall in intracellular Ca2+, release of Ca2+ from cTnC, waning of the actin-cross-bridge reaction, and a return of the myocytes to their diastolic state. In relating these molecular and cellular processes to the heart beat, there has been much focus on the idea that the dynamics are determined largely by the amounts and rates of movements of Ca2+ to and from the sarcomeres. A natural and understandable extension of this idea is that variations in the amounts of Ca2+ released to the sarcomeres largely determine contractility.
However, this perspective ignores the significant role of processes intrinsic to the sarcomeric proteins as determinants of the dynamics of the heart beat and as a controlled variable determining contractility. Early studies demonstrated that switches in isoform population of the myosin heavy chain have significant effects on dynamics of the heart beat independent of altered Ca2+ fluxes. More recent studies have demonstrated that isoform switches, posttranslational modifications, and mutations in sarcomeric proteins may also affect cardiac dynamics apart from membrane controlled alterations in cellular Ca2+ fluxes [2, 3]. It is also significant that the emphasis on Ca2+ as the main determinant of altered contractility is based largely on studies with unloaded cardiac myocytes in which intracellular Ca-transients and shortening are determined. The critical question, however, is how do these molecular events relate in time to the heart beat in which pressure first develops with no change in volume then pressure is sustained while ejection is ongoing and volume is decreasing, and finally pressure falls with no change in volume. What we need to know is how do the Ca2+ transients change in an ejecting beat, where is the Ca2+ bound during this time, and how does free and bound Ca2+ change with physiological, pathophysiological, and pharmacological manipulations of cardiac function. We need to fully integrate processes upstream and downstream to Ca-binding to cTnC and to know their relative significance in these manipulations.
In the present review, I focus on the molecular and cellular events in the ventricular heart beat that occur after Ca2+ binds to cTnC and induce conformational changes in the thin and the state of the thin and thick filaments after Ca2+ is released from cTnC. I discuss a current view of the molecular interactions among sarcomeric proteins responsible for the transition from diastole to systole and potential mechanisms by which activation of the actin-cross reaction may be sustained by cooperative processes in sarcomeric activation. I also present recent developments supporting the hypothesis that sarcomeric proteins are effective targets for inotropic agents useful in cardiac disorders including heart failure.
2. Promotion of the Actin-Cross-Bridge Reaction by On-Off Control of the Troponin Ca-Switch
Ca-binding to cTnC engages a remarkable and complex set of protein-protein interactions that result in the release of the thin filament from an inhibited state and in a force generating interaction between the myosin cross-bridge and actin. These interactions provide many potential control points for regulation of the sensitivity and steepness of the response to Ca2+, as well as force, ATPase rate, and cross-bridge kinetics at a given level of bound Ca2+. Recent studies [4–7] continue to illuminate these interactions and provide new perspectives.
Figure 1 illustrates a portion of a structural unit of the thin filament (7 actins in 1:1:1:1 complex with tropomyosin (Tm), troponin C (cTnC), troponin I (cTnI), and troponin T (cTnT). Figure 1 includes a current perception of these interactions based on evidence from the core crystal structure of cardiac troponin (Tn) [8], from elucidation of the structures by NMR [9], from biochemical investigations of protein-protein interactions [10–12], and from reconstructions and single particle analysis from electron micrographs of reconstituted myofilament preparations [5]. To clarify the relatively recent updates and new concepts in the structure functions relations, Figure 1(a) shows only the cTnI and cTnC (with Ca2+-bound to two C-terminal lobe structural sites) components binding to an actin-tropomyosin strand in the thin filament. Figure 1(b) adds cTnT and illustrates new data demonstrating that the N-terminal tail of cTnT binds to an actin strand opposite to that to which cTnI-cTnC binds. Figure 1(a) depicts cTnI and cTnC in the thin filament based on the core crystal structure of cTn. The crystal structure did not resolve significant regions including an inhibitory peptide (Ip), which tethers cTnI to actin, and the unique N-terminal peptide (~30 amino acids). The structure of these regions is based on biochemical evidence including determination of protein-protein interactions, florescence energy transfer, and NMR solution structure determination (reviewed in [10]). Figure 1(a) illustrates an off state with binding of cTnI to actin via two regions, a highly basic inhibitory peptide (Ip) and a second actin binding region. Importantly these regions flank a “switch peptide.” New data provide evidence for a role of the cTnI C-terminal mobile domain in the off state. This C-terminal domain has been demonstrated to bind to azimuthally localized actins with the far cTnI C-terminus lying across Tm [4]. Direct biochemical evidence for an interaction between this region of TnI and Tm has been reported by Mudalige et al. [13], who employed photochemical cross- linking studies with Tm labeled at positions 146 or 174. Their data indicate a cross-linking of Tm-146 with fast skeletal TnI (fsTnI) peptide 157-163 (DVGDWRK), which corresponds to a nearly identical C-terminal peptide in the cTnI (EVGDWRK). Importantly this study also demonstrated that the cross-linking between Tm and fsTnI occurred only under low Ca2+ conditions, whereas cross-linking of Tm-174 to TnT was not Ca2+-dependent. This interaction may be critical to control of thin filament activation state and provides a mechanism for earlier observations indicating a significant role for the far C-terminal peptide of cTnI [14, 15] in thin filament activation. Figure 1(b) illustrates the position of cTnT in the thin filament complex with the C-terminal regions interacting together with the near N-terminal region of cTnI and the C-terminal lobe of cTnC. cTnT is the main anchor site of the Tn complex to Tm. Sites of interaction occur between N-terminal regions of cTnT and Tm-175-190 and Tm-258-284 [10, 11, 16]. Figure 1(b) shows a new concept of the interaction of cTnT with the thin filament based on predictions reported by Galinska-Rakoczy et al. [4]. This study predicted that the N-terminal extension of cTnT binds to the strand of actins opposite to that binding the main core Tn complex. Thus, as described [4] and illustrated in Figures 1 and 2, Tm is wedged in the blocking position between the cTnI mobile domain on one side and the troponin core domain and TnT on the other. Thus the off state of the thin filament involves an immobilized Tm blocking the actin cross-bridge reaction. Immobilization of Tm involves anchoring of cTnI to actin as well as interactions between the N-terminal tail of TnT with Tm and possibly the C-terminal mobile domain of cTnI with Tm.
Figure 1.

Structure of a patch of cardiac thin filament demonstrating the position of troponin components with tropomyosin (Tm) in the “off” state. (a) Thin filament containing, only troponin I (cTnI) and troponin C (cTnC). cTnC is shown as a dumbbell shaped protein with the N-lobe containing a single regulatory Ca-binding site. cTnI is shown tethered to actin on an actin strand by an inhibitory peptide (Ip) and a second actin binding region flanking a switch peptide (Sw), which interacts with cTnC upon Ca-activation. The distal C-terminal end of cTnI drapes across azimuthal actins and may interact with Tm. cTnI has a unique stretch of N-terminal amino acids, which contain phosphorylation sites at S23, S24. The N-peptide interacts with the N-lobe of cTnC, but is released upon phosphorylation and may react with the Sw or Ip of cTnI. (b) Thin filament contains the complete Tn complex. Note that the Ip of cTnI binds to one actin strand, whereas the N-terminal tail of troponin T (cTnT) interacts with Tm on the adjacent actin strand. The consequence is that Tm is wedged and held in a blocking position.
Figure 2.

Alterations in thin filament structure and promotion of the actin-cross-bridge reaction induced by Ca2+-binding to cTnC. The left panel shows the diastolic state as in Figures 1 and 2. The right panel shows the systolic state and illustrates the release of cTnI from actin and binding of the switch peptide to cTnC most likely at a hydrophobic patch induced by Ca2+ -binding to the N-lobe of cTnC. cTnT from the Tn complex on the adjacent actin strand is shown with stripes. Activation is associated with release of Tm from an immobilized state by protein signaling to cTnT and possible release of Tm from an interaction with the C-terminal end of cTnI. See text for further description.
Figure 2 shows the transition in myofilament state with Ca2+ binding to cTnC. With its release into the intracellular space, Ca2+ binds to cTnC at a single site in the N-lobe and promotes the “on” state by inducing the generation of a hydrophobic patch, which draws the cTnI switch peptide to cTnC. This critical event in the on-off switching mechanisms releases the thin filament from inhibition by movements of the Ip and second actin binding site of cTnI from actin. The Ca-binding signal also induces a release of TnT from its interaction from Tm. These Ca-induced alterations in thin filament protein-protein interactions result in transition of Tm from a relatively immobilized state to a mobile state permitting the reaction of cross-bridges with actin. The propensity of interaction of the cross-bridges with actin is also governed by the relative radial position of the cross-bridge with respect to the thick filament proper. As discussed below there is compelling evidence that regulatory mechanisms control this radial movement of cross-bridges. These regulatory mechanisms involve thick filament associated proteins, myosin light chain 2 (MLC2) [17], myosin binding protein C (MyBPC) [18], and titin [19].
3. Modulation of the Troponin Ca2+ Switch
To illustrate modulation of the Ca2+ switch I discuss phosphorylation of cTnI. A major region of phosphorylation is the cTnI-N-terminal peptide, which is unique to the cardiac isoform. I have reviewed the multiple kinases controlling sarcomeric protein phosphorylation elsewhere [12]. Figure 1(a) indicates an interaction of the N-terminus of cTnI, which contains 2 prominent phosphorylation sites, which are substrates for PKA, PKG, PKD, and PKC-δ [12]. As discussed below this interaction is an important determinant of the affinity the cTnC N-lobe for Ca2+. Figure 1 also shows interactions of the unique N-terminal peptide of cTnI with the N-lobe of cTnC. This interaction is strong when the N-peptide is not phosphorylated at Ser 23, Ser 24, but with phosphorylation at these sites the N-peptide is released from cTnC [20]. The release of the peptide induces a significant depression in Ca-affinity of cTnC [21] and appears to promote an increase in kinetics of the actin-cross bridge reaction [22, 23]. These effects together with adrenergic control of Ca fluxes are important aspects of the tuning of the contraction relaxation cycle of the heart to altercations in heart rate. On the basis of NMR determination of the solution structure on the N-peptide of cTnI, we hypothesized that this peptide, which has negatively charged residues, might interact with the positively charged regions of the cTnI Ip [9, 24]. We tested this possibility employing a hetero-bifunctional cross linker linked to a Cys residue engineered into position 5 of cTnI normally containing Ser 5 and into position 19 normally containing an Iso residue [24]. Our data demonstrated an intermolecular interaction with positions Met-47 and Met-80 of cTnC and an intramolecular interaction between the N-peptide of cTnI and cTnI at Met-154 (I19C mutant) and Met-155 (S5C mutant). Met 154 and Met 155 are residues located in the cTnI switch peptide, and thus we hypothesized that this interaction may be a critical regulatory device and dependent on the charge state of the cTnI N-peptide. The hypothesis needs to be further tested by experiments employing other probes of conformation changes such as fluorescence resonance energy transfer and electron spin resonance. Other prominent and unique sites of phosphorylation of cTnI are Thr 144 in the Ip and Ser 43, Ser 45 in a near N-terminal region of cTnI, which interacts with the C-lobe of cTnC and C-terminal regions of cTnT [10, 12]. Phosphorylation of Thr 144 has been demonstrated to either enhance Ca2+ sensitivity or have little effect, whereas phosphorylation of Ser 43/Ser 45 has been reported to depress maximum tension and Ca2+ sensitivity of skinned fiber preparations. Phosphorylation of these sites has also been reported to slow down velocity of thin filament sliding in the motility assay, whereas phosphorylation of cTnI Ser 23/Ser 24 enhances sliding velocity [10].
4. The Time Course of Systolic Elastance and Rates of Isovolumic Relaxation Reflect Mechanisms at the Level of Sarcomeric Proteins
Relating the molecular mechanisms summarized in Figures 1 and 2 to the cardiac cycle especially in large animals remains a significant challenge, which is critical in translational studies. Figure 3 illustrates the cardiac cycle simply by the time course of the change in left ventricular pressure during an ejecting beat in basal conditions in a dog heart. The time course of the beat serves to constrain theories relating the cellular and molecular mechanisms of contraction and relaxation. Studies employing hearts loaded with Ca-sensitive dyes such as Rhod 2 and use of fiber optic directed excitation, and emission measurements have been carried out to determine the time course of cellular Ca2+ transients in a heartbeat [27, 28]. However, these studies have employed isovolumic rodent hearts. It is not clear therefore how the time course of the Ca2+ transient relates to the time course of systolic elastance in a beating ventricle. Even so, studies of Ca2+ transients and isometric force generation reveal that the duration of the Ca2+ transient is faster than the duration of force development [25, 29]. The critical dynamic is the time course of Ca2+-binding to cTnC during a beat of the heart. Unfortunately, we do not have measurements of the amounts of Ca2+ bound to the myofilaments during the heart beat. Early and more recent studies have demonstrated that the only site capable of exchanging Ca2+ during the beat is the single regulatory binding site on the N-lobe of cTnC (Figure 2). As illustrated in Figure 3, indirect estimations [30] and computations [25, 26] based on rate constants of Ca2+ exchange with cTnC indicate that, as expected, the Ca-binding increases with the transition from diastole to systole, but these same computations also indicate that cTnC bound Ca may decrease significantly during maintenance of systolic elastance and ahead of isovolumic relaxation. The data illustrated in Figure 3 have important consequences with regard to our understanding of physiological control of ejection, its modification in cardiac disorders, and in approaches to alterations in sarcomeric function by drug targeting. Moreover if Ca2+-binding and the Ca2+-transient are essentially over at the end of ejection and closure of the atrioventricular valves this means that isovolumic relaxation and re-establishment of the diastolic state depends on mechanisms intrinsic to the myofilaments. Hinken and Solaro [3] have previously discussed this disparity and potential mechanisms by which systolic elastance might be maintained in the face of a declining Ca2+ transient and cTnC bound Ca2+. The mechanisms involve a cooperative activation of the thin filaments in which Ca2+ binding to a cTnC on the thin filament promotes activation of near neighbor units. However, recent evidence has provided highly relevant and new perspectives on the cooperative processes, and on the modifications in the myofilaments that may modulate elastance [4–7]. These control processes intrinsic to the myofilaments are also of significance in preload (sarcomere length) sensing and engagement of immediate responses in terms of the Frank-Starling relations. Intrinsic myofilament mechanisms also are important in sensing the load in a negative feedback mechanism related to the force-velocity relation and in power generation [31, 32], and also in the relaxation kinetics related to rates of cross-bridge detachment and intersarcomere dynamics (as described by Pogessi et al. [33]).
Figure 3.

Relation between left ventricular pressure changes during the cardiac cyle and Ca2+-binding to cTnC in the myofilament lattice. The onset and end of ejection are indicated. Note that pressure (systolic stiffening or elastance) is maintained despite the fall in bound Ca2+. The Ca2+ binding kinetics are based on computations described Burkhoff and colleagues [25, 26]. See text for further discussion.
5. Thin and Thick Filament Mechanisms Related to Maintenance of Systolic Elastance
Cooperative activation of cardiac myofilaments has long been appreciated as a significance aspect of control of cardiac function including diastolic filling, the triggering and sustaining of developed pressure, and the dynamics of relaxation [10, 34]. Strong evidence for this cooperativity comes from measurements of the steady-state relation between free Ca2+ and force generation in detergent extracted single myocytes or bundles of myocytes. Despite control of activation by a single regulatory site on cTnC, the force-Ca2+ relations are steep and are commonly fit with Hill coefficients in the range of 3–5 [35]. Moreover as mentioned above in presteady state measurements of the relation between the duration force generation in fiber preparations, the dynamics of the Ca2+ transient show that the decline if the Ca2+ transient occurs well ahead of the Ca2+-transient.
Interactions among proteins within a functional unit and among the functional units of the sarcomere form the basis of the most prominent theories for the mechanism of the steep dependence of force on Ca2+. These interactions are illustrated in Figure 4. What promotes these interactions is under current debate. Detailed balance drives one of the theories in which the energies of interaction between cTnC and the actin-myosin interface flows in both directions. That is, the promotion of the actin-myosin interaction by Ca2+ signaling through cTnC in turn promotes Ca2+ binding to cTnC [10, 16, 34]. For example, in steady-state measurements of the Ca2+-force relation in skinned fibers, as more force generating cross-bridges react with the thin filament with increases in Ca2+, this theory predicts an increase in the affinity of cTnC also increases. Longitudinal spread of activation along the thin filament forms the basis for a second theory. The mechanism for the longitudinal spread remains a matter of debate. There are data demonstrating that strongly bound (rigor) cross-bridges are able to move Tm away from the cross-bridge binding sites on actin and closer to the groove in the actin helix [5]. Contiguous Tms overlap forming a continuous molecular strand believed to sensitive to this cross-bridge induced movement of Tm (Figure 4). Thus, in this theory activation spreads to a near neighbor functional unit without the need for Ca2+ binding to the cTnC in that unit. One of the issues with the theories described above is that they are based on measurements of Ca2+-binding and spread of activation in which nucleotide free (rigor) cross-bridges or cross-bridges modified with NEM are used as a surrogate for the strongly interacting and force generating cross-bridge. These cross-bridges strongly react with the thin filament but do not cycle and generate tension. Recent direct tests have failed to demonstrate that force generating, cycling cross-bridges promote molecular modifications in the thin filament in the same way as to rigor cross-bridges. One set of studies involved measurements of Ca2+ binding to the regulatory site of cTnC in reconstituted thin filaments alone or reacting with nucleotide-containing or nucleotide free cross-bridges in the form of myosin-S1 [36]. The data are summarized in Table 1. A significant findings in these data are that Ca2+ binding to the single regulatory site is cooperative (nH = 1.65) in the presence and absence of nucleotide bound myosin S-1. Moreover, reaction of the thin filaments with nucleotide-free S-1 increased Ca2+-affinity as predicted from previous studies, with a significant decrease in cooperativity (nH = 0.85). These data indicate that cooperative activation of myofilaments relies on mechanisms intrinsic to the thin filaments. Studies probing thin filament activation by use of fluorescent probes at specific sites on cTnC support this concept [6, 7]. cTnC labeled with probes sensing the “on” state and incorporated into skinned fiber preparations, tracked the Ca2+-force relation and show a similar cooperative response. The relation between the “on” states of cTnC remained the same when the cross-bridge interactions were completely blocked with blebbistatin. Thus these results indicate that the promotion of near-neighbor interactions along the thin filament do not require force generating cross-bridges. These results alter thinking with regard to the relative roles of cycling and rigor cross-bridges. There may be some effects of cycling cross-bridges on cooperative activation of the thin that are more apparent in presteady state measurements, but evidence of the relative low numbers of interacting cross-bridges during contractions suggests that this would be a minor influence. On the other hand in conditions of compromised generation of ATP and the elaboration of nucleotide free cross-bridges, there would be an influence on activation. Thus, the current evidence does not dismiss effects of rigor cross-bridges on myofilament activation, but relegates this mechanism to a patho-physiological role as in ischemia and cardiomyopathies rather than in physiological control mechanisms.
Figure 4.
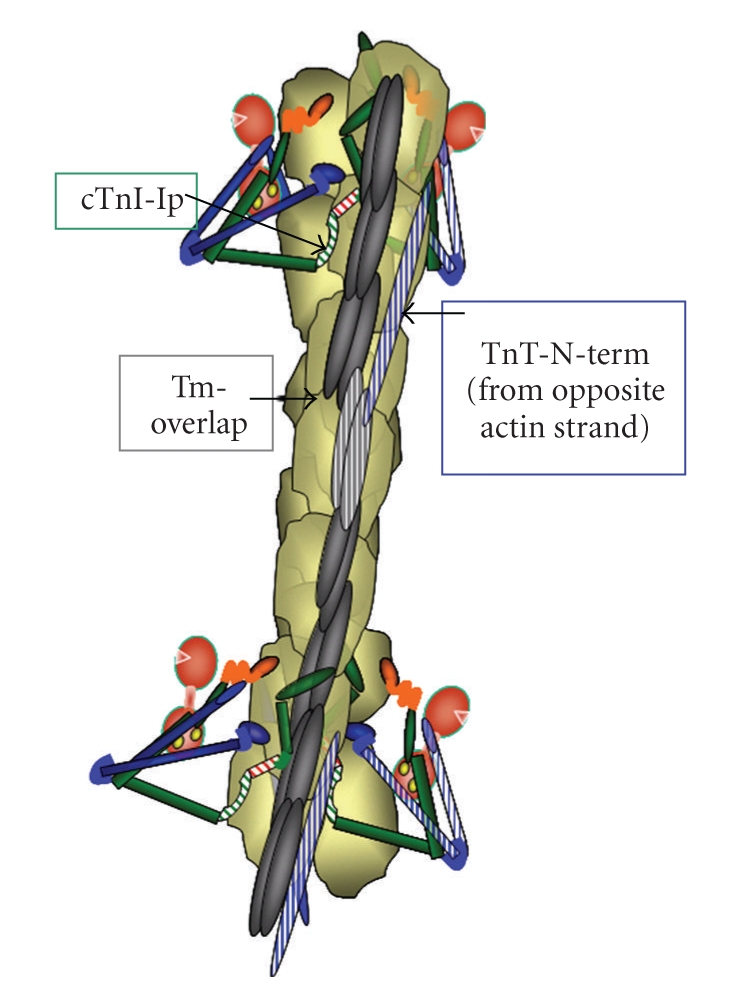
View of an extended region of a functional unit of the thin filament illustrating the position of the Tn complexes and the possible interactions among contiguous functional units. Cooperative spread of activation of one region of the thin filament may occur by actin-actin interactions or by spread via Tm overlap. Color coding of proteins as in Figure 2.
Table 1.
Strong binding rigor cross-bridges but no cycling cross-bridges affect Ca-binding to troponin C in reconstituted myofilament preparations. Data are from Davis et al. [36].
| System | Kd nM | Hill n | Koff(/s) |
|---|---|---|---|
| Thin Filament | 4810 ± 300 | 1.65 ± 0.04 | 105 + 1 |
| Thin Filament + S1 | 777 ± 30 | 0.81 ± 0.02 | 13 ± .1 |
| Thin Filament + S1 + ATP | 5040 ± 400 | 1.65 ± 0.08 | 110 ± 1 |
6. Signaling to Cardiac Sarcomeres and Control of Cooperative Activation
Modifications in thin filament proteins appear to affect cooperative spread of activation induced by force generating and rigor cross-bridges. Cross-bridge dependent thin filament activation depends on the isoform population of TnI. In myofilaments regulated by slow skeletal TnI (ssTnI), the ability of rigor and NEM-modified cross-bridges to activate force generation in the absence of Ca2+ was less than in the case of myofilaments controlled by cTnI [37]. This difference was particularly evident at acidic pH and evidence was developed to implicate a role. His residue at position 34 in ssTnI. When His-34 was replaced by Ala, which is the residue at the homologous position (Ala-66) in cTnI, activation by rigor cross-bridges was similar to that of myofilaments controlled by cTnI. These findings led us to compare cross-bridge dependent activation in cardiac myofilaments controlled either by wild type cTnI or by cTnI-A66H. When pH was reduced from pH 7.0 to pH 6.5, there was enhanced cross-bridge-dependent activation in cTnI myofilaments, but depressed activation in cTnI (A66H) myofilaments. These data may be of significance in our finding that compared to wild-type controls that adult hearts expressing ssTnI demonstrate protection against the stress of ischemia and reperfusion, where rigor cross-bridges are likely to be present [38]. In view of evidence that ischemia is associated with activation of protein kinase C (PKC), Engel et al. [39] also compared the ability of NEM-S1, a mimic of rigor cross-bridges to activate control cardiac myofilaments regulated by cTnI and myofilaments controlled by cTn complex containing either cTnI-(S43E/S45E) or cTnI-(T144E). These pseudo-phosphorylated sites are predominant PKC sites in Tn. Our data show that compared to controls, the myofilaments controlled by cTnI-(S43E/S45E) demonstrated a significant reduction in the ability of NEM-S1 to recruit cycling cross-bridges. Pseudo-phosphorylation of cTnI-(T144) had similar but less extensive effect. The functional significance of the modifications in Tn may be important in sparing energy consumption in ischemia.
Studies with transgenic models expressing cardiac specific and pseudo-phosphorylated mutants of cTnI (cTnI-S43E, S45E, T144E) also provide evidence of the significance of potential thin filament related cooperative mechanism [40]. In these studies extensive two-dimensional gel analysis demonstrated replacement of the endogenous cTnI with only 7.2 ± 0.5% of the mutant. Even with this modest increase in PKC-dependent phosphorylation, data from studies with perfused hearts, and intact and skinned myocardial preparations showed an induction of highly significant depressions in pressure and force generation with no change in Ca2+ transients. There were also significant depressions in relaxation rates. Mathematical modeling revealed that these effects are likely to be induced by a simultaneous reduction in the rate of cross-bridge entry into the force generating state, and by an increase in Ca2+ -independent persistence of the myofilament active state. Whatever the case, these data indicate that the small number of modified TnI proteins in the hearts of the transgenic mice expressing the mutant protein must be able to influence their neighbors on the thin filament. Effects of the expression of the pseudo-phosphorylated cTnI on systolic elastance and isovolumic relaxation have not as yet been carried out.
Modifications in thick filament proteins especially MyBP-C and MLC2 appear especially important in affecting systolic elastance. In comparing various transgenic models Palmer et al. [41] reported that only the hearts of the cMyPP-C t/t mice demonstrated a significant change in stiffening during ejection. Whereas the controls reached 42% of maximum elastance at the onset of ejection, the t/t mutant hears reached 77% of peak elastance. Moreover, elastance peaked earlier and was abbreviated in hearts of the cMyBP-C t/t mice compared to controls. Thus these data indicate that normal cardiac function and duration of systolic elastance rely on the integrity of MyBP-C. Scruggs et al. [17] have also generated evidence indicating the phosphorylation of MLC2 may be an important factor in determining the duration of systolic elastance. The states of both MyBP-C and MLC2 are likely to be important factors in determining radial disposition of the heads of myosin.
7. Sarcomeric Proteins as Targets for Inotropic Agents
Potential application to rational drug design is a significant aspect of the understanding of the relations between cellular and molecular mechanisms at the level of the sarcomeres. There has been a long standing effort to develop agents that directly modify the sarcomere response to Ca2+ with some success (see [42, 43] for reviews). Levosimendan and pimobendan are examples of such agents, which progressed into clinical use, and which are able to promote sarcomere responsiveness to Ca2+ and demonstrate some therapeutic benefits in acute and chronic heart failure. However, these agents have off target effects and there is a need for agents or approaches with more specific targeting to the sarcomeres [42]. For example, both Levosimendan and Pimobendan have been demonstrated to inhibit phosphor-diesterase III in addition to directly affecting Ca2+-signaling to TnC. The identification of the molecular processes involved in maintenance of systolic elastance and as determinants of isovolumic relaxation provides clues as to the location of these targets. A recent apparent success in the search for sarcomere activators has been the identification of a compound developed by Cytokinetics, Inc (CK-1827452) directly reacting with myosin and prolonging systolic elastance (reviewed in [44]). Evidence for factors maintaining systolic elastance summarized is thus significant in translational medicine, as these results with CK indicate that modification of systolic elastance is a viable pharmacological target. Modification of Tn, MLC2, MyBP-C or titin by drugs may also be an important road to success, but has not been explicitly investigated.
Acknowledgments
I am grateful to many colleagues for valuable interactions. Support from NIH grants PO1 HL 62426, RO1 HL 22231, and RO1 HL 64035 provide the bulk of support for experiments from our laboratory.
References
- 1.Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circulation Research. 2000;87(4):275–281. doi: 10.1161/01.res.87.4.275. [DOI] [PubMed] [Google Scholar]
- 2.Solaro RJ, de Tombe PP. Review focus series: sarcomeric proteins as key elements in integrated control of cardiac function. Cardiovascular Research. 2008;77(4):616–618. doi: 10.1093/cvr/cvn004. [DOI] [PubMed] [Google Scholar]
- 3.Hinken AC, Solaro RJ. A dominant role of cardiac molecular motors in the intrinsic regulation of ventricular ejection and relaxation. Physiology. 2007;22(2):73–80. doi: 10.1152/physiol.00043.2006. [DOI] [PubMed] [Google Scholar]
- 4.Galińska-Rakoczy A, Engel P, Xu C, et al. Structural basis for the regulation of muscle contraction by troponin and tropomyosin. Journal of Molecular Biology. 2008;379(5):929–935. doi: 10.1016/j.jmb.2008.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lehman W, Galińska-Rakoczy A, Hatch V, Tobacman LS, Craig R. Structural basis for the activation of muscle contraction by troponin and tropomyosin. Journal of Molecular Biology. 2009;388(4):673–681. doi: 10.1016/j.jmb.2009.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solaro RJ. Maintaining cooperation among cardiac myofilament proteins through thick and thin. The Journal of Physiology. 2009;587(1, article 3) doi: 10.1113/jphysiol.2008.166751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun Y-B, Lou F, Irving M. Calcium- and myosin-dependent changes in troponin structure during activation of heart muscle. The Journal of Physiology. 2009;587(1):155–163. doi: 10.1113/jphysiol.2008.164707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takeda S, Yamashita A, Maeda K, Maéda Y. Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature. 2003;424(6944):35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 9.Howarth JW, Meller J, Solaro RJ, Trewhella J, Rosevear PR. Phosphorylation-dependent conformational transition of the cardiac specific N-extension of troponin I in cardiac troponin. Journal of Molecular Biology. 2007;373(3):706–722. doi: 10.1016/j.jmb.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 10.Kobayashi T, Solaro RJ. Calcium, thin filaments, and the integrative biology of cardiac contractility. Annual Review of Physiology. 2005;67:39–67. doi: 10.1146/annurev.physiol.67.040403.114025. [DOI] [PubMed] [Google Scholar]
- 11.Tobacman LS. Thin filament-mediated regulation of cardiac contraction. Annual Review of Physiology. 1996;58:447–481. doi: 10.1146/annurev.ph.58.030196.002311. [DOI] [PubMed] [Google Scholar]
- 12.Solaro RJ. Multiplex kinase signaling modifies cardiac function at the level of sarcomeric proteins. Journal of Biological Chemistry. 2008;283(40):26829–26833. doi: 10.1074/jbc.R800037200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mudalige WAKA, Tao TC, Lehrer SS. Ca2+-dependent photocrosslinking of tropomyosin residue 146 to residues 157–163 in the C-terminal domain of troponin I in reconstituted skeletal muscle thin filaments. Journal of Molecular Biology. 2009;389(3):575–583. doi: 10.1016/j.jmb.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foster DB, Noguchi T, VanBuren P, Murphy AM, Van Eyk JE. C-terminal truncation of cardiac troponin I causes divergent effects on ATPase and force: implications for the pathophysiology of myocardial stunning. Circulation Research. 2003;93(10):917–924. doi: 10.1161/01.RES.0000099889.35340.6F. [DOI] [PubMed] [Google Scholar]
- 15.Rarick HM, Tu X-H, Solaro RJ, Martin AF. The C terminus of cardiac troponin I is essential for full inhibitory activity and Ca2+ sensitivity of rat myofibrils. Journal of Biological Chemistry. 1997;272(43):26887–26892. doi: 10.1074/jbc.272.43.26887. [DOI] [PubMed] [Google Scholar]
- 16.Tobacman LS, Sawyer D. Calcium binds cooperatively to the regulatory sites of the cardiac thin filament. Journal of Biological Chemistry. 1990;265(2):931–939. [PubMed] [Google Scholar]
- 17.Scruggs SB, Hinken AC, Thawornkaiwong A, et al. Ablation of ventricular myosin regulatory light chain phosphorylation in mice causes cardiac dysfunction in situ and affects neighboring myofilament protein phosphorylation. Journal of Biological Chemistry. 2009;284(8):5097–5106. doi: 10.1074/jbc.M807414200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sadayappan S, Gulick J, Osinska H, et al. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circulation Research. 2005;97(11):1156–1163. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fukuda N, Granzier HL, Ishiwata S, Kurihara S. Physiological functions of the giant elastic protein titin in mammalian striated muscle. Journal of Physiological Sciences. 2008;58(3):151–159. doi: 10.2170/physiolsci.RV005408. [DOI] [PubMed] [Google Scholar]
- 20.Ward DG, Brewer SM, Calvert MJ, Gallon CE, Gao Y, Trayer IP. Characterization of the interaction between the N-terminal extension of human cardiac troponin I and troponin C. Biochemistry. 2004;43(13):4020–4027. doi: 10.1021/bi036128l. [DOI] [PubMed] [Google Scholar]
- 21.Robertson SP, Johnson JD, Holroyde MJ. The effect of troponin I phosphorylation on the Ca2+-binding properties of the Ca2+-regulatory site of bovine cardiac troponin. Journal of Biological Chemistry. 1982;257(1):260–263. [PubMed] [Google Scholar]
- 22.Kentish JC, McCloskey DT, Layland J, et al. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circulation Research. 2001;88(10):1059–1065. doi: 10.1161/hh1001.091640. [DOI] [PubMed] [Google Scholar]
- 23.Biesiadecki BJ, Kobayashi T, Walker JS, Solaro RJ, de Tombe PP. The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circulation Research. 2007;100(10):1486–1493. doi: 10.1161/01.RES.0000267744.92677.7f. [DOI] [PubMed] [Google Scholar]
- 24.Warren CM, Kobayashi T, Solaro RJ. Sites of intra- and intermolecular cross-linking of the N-terminal extension of troponin I in human cardiac wholetroponin complex. Journal of Biological Chemistry. 2009;284(21):14258–14266. doi: 10.1074/jbc.M807621200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burkhoff D. Explaining load dependence of ventricular contractile properties with a model of excitation-contraction coupling. Journal of Molecular and Cellular Cardiology. 1994;26(8):959–978. doi: 10.1006/jmcc.1994.1117. [DOI] [PubMed] [Google Scholar]
- 26.Baran D, Ogino K, Stennett R, et al. Interrelating of ventricular pressure and intracellular calcium in intact hearts. American Journal of Physiology. 1997;273(3, part 2):H1509–H1522. doi: 10.1152/ajpheart.1997.273.3.H1509. [DOI] [PubMed] [Google Scholar]
- 27.Valverde CA, Mundiña-Weilenmann C, Reyes M, Kranias EG, Escobar AL, Mattiazzi A. Phospholamban phosphorylation sites enhance the recovery of intracellular Ca2+ after perfusion arrest in isolated, perfused mouse heart. Cardiovascular Research. 2006;70(2):335–345. doi: 10.1016/j.cardiores.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 28.Mejía-Alvarez R, Manno C, Villalba-Galea CA, et al. Pulsed local-field fluorescence microscopy: a new approach for measuring cellular signals in the beating heart. Pflugers Archiv European Journal of Physiology. 2003;445(6):747–758. doi: 10.1007/s00424-002-0963-1. [DOI] [PubMed] [Google Scholar]
- 29.Backx PH, Gao W-D, Azan-Backx MD, Marban E. The relationship between contractile force and intracellular [Ca2+] in intact rat cardiac trabeculae. Journal of General Physiology. 1995;105(1):1–19. doi: 10.1085/jgp.105.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Peterson JN, Hunter WC, Berman MR. Estimated time course of Ca2+ bound to troponin C during relaxation in isolated cardiac muscle. American Journal of Physiology. 1991;260(3, part 2):H1013–H1024. doi: 10.1152/ajpheart.1991.260.3.H1013. [DOI] [PubMed] [Google Scholar]
- 31.Landesberg A. End-systolic pressure-volume relationship and intracellular control of contraction. American Journal of Physiology. 1996;270(1, part 2):H338–H349. doi: 10.1152/ajpheart.1996.270.1.H338. [DOI] [PubMed] [Google Scholar]
- 32.McDonald KS, Herron TJ. It takes “heart” to win: what makes the heart powerful? News in Physiological Sciences. 2002;17(5):185–190. doi: 10.1152/nips.01396.2002. [DOI] [PubMed] [Google Scholar]
- 33.Poggesi C, Tesi C, Stehle R. Sarcomeric determinants of striated muscle relaxation kinetics. Pflugers Archiv European Journal of Physiology. 2005;449(6):505–517. doi: 10.1007/s00424-004-1363-5. [DOI] [PubMed] [Google Scholar]
- 34.Moss RL, Razumova M, Fitzsimons DP. Myosin crossbridge activation of cardiac thin filaments: implications for myocardial function in health and disease. Circulation Research. 2004;94(10):1290–1300. doi: 10.1161/01.RES.0000127125.61647.4F. [DOI] [PubMed] [Google Scholar]
- 35.Pan BS, Solaro RJ. Calcium-binding properties of troponin C in detergent-skinned heart muscle fibers. Journal of Biological Chemistry. 1987;262(16):7839–7849. [PubMed] [Google Scholar]
- 36.Davis JP, Norman C, Kobayashi T, Solaro RJ, Swartz DR, Tikunova SB. Effects of thin and thick filament proteins on calcium binding and exchange with cardiac troponin C. Biophysical Journal. 2007;92(9):3195–3206. doi: 10.1529/biophysj.106.095406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engel PL, Kobayashi T, Biesiadecki B, et al. Identification of a region of troponin I important in signaling cross-bridge-dependent activation of cardiac myofilaments. Journal of Biological Chemistry. 2007;282(1):183–193. doi: 10.1074/jbc.M512337200. [DOI] [PubMed] [Google Scholar]
- 38.Arteaga GM, Warren CM, Milutinovic S, Martin AF, Solaro RJ. Specific enhancement of sarcomeric response to Ca2+ protects murine myocardium against ischemia-reperfusion dysfunction. American Journal of Physiology. 2005;289(5):H2183–H2192. doi: 10.1152/ajpheart.00520.2005. [DOI] [PubMed] [Google Scholar]
- 39.Engel PL, Hinken A, Solaro RJ. Differential effects of phosphorylation of regions of troponin I in modifying cooperative activation of cardiac thin filaments. Journal of Molecular and Cellular Cardiology. 2009;47(3):359–364. doi: 10.1016/j.yjmcc.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirk JA, MacGowan GA, Evans C, et al. Left ventricular and myocardial function in mice expressing constitutively pseudophosphorylated cardiac troponin I. Circulation Research. 2009;105(12):1232–1239. doi: 10.1161/CIRCRESAHA.109.205427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palmer BM, Georgakopoulos D, Janssen PM, et al. Role of cardiac myosin binding protein C in sustaining left ventricular systolic stiffening. Circulation Research. 2004;94(9):1249–1255. doi: 10.1161/01.RES.0000126898.95550.31. [DOI] [PubMed] [Google Scholar]
- 42.Kass DA, Solaro RJ. Mechanisms and use of calcium-sensitizing agents in the failing heart. Circulation. 2006;113(2):305–315. doi: 10.1161/CIRCULATIONAHA.105.542407. [DOI] [PubMed] [Google Scholar]
- 43.Sorsa T, Pollesello P, Solaro RJ. The contractile apparatus as a target for drugs against heart failure: interaction of levosimendan, a calcium sensitiser, with cardiac troponin c. Molecular and Cellular Biochemistry. 2004;266(1-2):87–107. doi: 10.1023/b:mcbi.0000049141.37823.19. [DOI] [PubMed] [Google Scholar]
- 44.Solaro RJ. CK-1827452, a sarcomere-directed cardiac myosin activator for acute and chronic heart disease. IDrugs. 2009;12(4):243–251. [PubMed] [Google Scholar]