

Abstract
An overview is presented of some of the major insights that have come from studies of the structure, stability, and folding of T4 phage lysozyme. A major purpose of this review is to provide the reader with a complete tabulation of all of the variants that have been characterized, including melting temperatures, crystallographic data, Protein Data Bank access codes, and references to the original literature. The greatest increase in melting temperature (Tm) for any point mutant is 5.1°C for the mutant Ser 117 → Val. This is achieved in part not only by hydrophobic stabilization but also by eliminating an unusually short hydrogen bond of 2.48 Å that apparently has an unfavorable van der Waals contact. Increases in Tm of more than 3–4°C for point mutants are rare, whereas several different types of destabilizing substitutions decrease Tm by 20°C or thereabouts. The energetic cost of cavity creation and its relation to the hydrophobic effect, derived from early studies of “large-to-small” mutants in the core of T4 lysozyme, has recently been strongly supported by related studies of the intrinsic membrane protein bacteriorhodopsin. The L99A cavity in the C-terminal domain of the protein, which readily binds benzene and many other ligands, has been the subject of extensive study. Crystallographic evidence, together with recent NMR analysis, suggest that these ligands are admitted by a conformational change involving Helix F and its neighbors. A total of 43 nonisomorphous crystal forms of different monomeric lysozyme mutants were obtained plus three more for synthetically-engineered dimers. Among the 43 space groups, P212121 and P21 were observed most frequently, consistent with the prediction of Wukovitz and Yeates.
Keywords: T4 lysozyme, protein stability, crystal forms, protein cavities, thermophilic proteins
Introduction
The lysozyme from phage T4 (Fig. 1; PDB Code 3FA0) has served as a paradigm for studying the factors that determine the structure and stability of proteins. Also, the recent use of the protein to help crystallize and determine the first structure of a GPCR1,2 has renewed interest in its properties. The purpose of this review is to give a short overview of some of the major insights that have come from this system and to provide the interested reader with a tabular summary of all of the lysozyme mutants that have been made including key structural and stability information, and references to the original literature. Supporting Information Table S1 is such a summary of all of the 700 or so mutant lysozymes and lysozyme complexes that have been characterized. PDB codes are included where structures were determined. Table I is a short list of representative entries from Table S1. Because the emphasis of this review is on T4 lysozyme, we have not attempted to include parallel studies on other proteins.3
Figure 1.
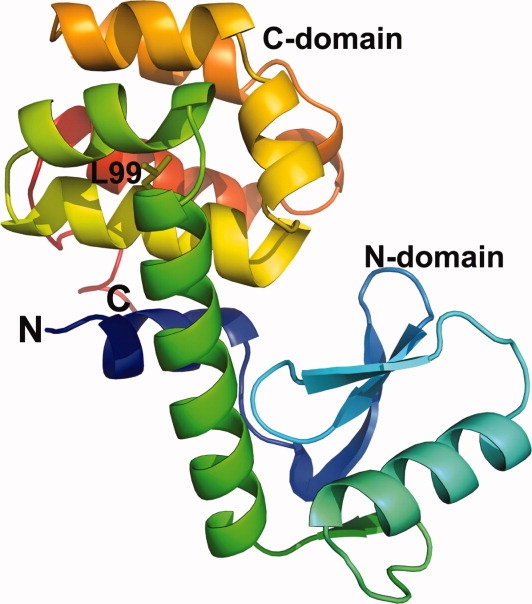
Backbone of T4 lysozyme showing its two-domain structure. The polypeptide chain is colored following the spectrum, from blue at the N-terminus to red at the opposite end.
Table I.
Thermodynamic, Crystallographic, and Other Key Information for Representative T4 Lysozyme Mutants and Complexes
| Thermodynamic data |
Crystallographic data |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Protein | PDB ID | Activity (%) | ΔTm (°C) | ΔΔG (kcal/mol) | pH | Space group | Resolution (Å) | Comment | Reference |
| WT (100K) | 3FA0 | P3221 | 0.98 | High resolution WT | 103,111 | ||||
| I3C/I9C/T21C/C54T/ T142C/L164C ox. | 152L | 0 | 23.4 | 2.0 | P212121 | 2.0 | Triple S-S bridge | 9,63 | |
| I3L/S38D/A41V/ A82P/N116D/ V131A/N144D | 189L | 2 | 8.32 | 3.57 | 5.42 | P212121 | 2.6 | Cumulative stabilization | 64 |
| T21C/S38D/L99A/ M102E/E108V/ S117V/T142C/N114D | 3GUI | −8.8 | −3.2 | 5.3 | P41212 | 1.45 | Buried charge in cavity | 112 | |
| S38D | 1L19 | 80 | 1.6 | 0.6 | 6.7 | P3221 | 1.7 | Helix dipole interaction | 15,16,64, 92 |
| S44F/WT* | 137L | ∼WT | 0.18 | 0.06 | 3.0 | P21 | 1.85 | Helix propensity analysis | 49,50 |
| S44W/WT* | 216L | ∼WT | 0.15 | 0.05 | 3.0 | C2 | 2.1 | Helix propensity analysis | 28,49,50 |
| K48-[HP]/WT* | 201L | ∼WT | −7.0 | −2.4 | 5.45 | P21 | 2.0 | Insertion “recovery” | 52,56 |
| A73-[AAA]/WT* | 209L | −15.9 | −5.0 | 5.4 | P6222 | 2.7 | Insertion | 72 | |
| R96H (100K) | 3F8V | −8.3 | −3.1 | 5.35 | P3221 | 1.08 | Site 96 survey | 103,104 | |
| L99A/WT* (200 MPa) | 2B6T | P3221 | 2.1 | Lysozyme cavity under pressure | 108,109 | ||||
| L99A/WT* + C6F5I | 3DN3 | P3221 | 1.8 | Halogenated benzene binding | 101 | ||||
| L99F/M102L/V111I/ F153L/WT* | 1L82 | 87 | −1.82 | −0.54 | 3.01 | P3221 | 2.1 | Designed core repacking | 34 |
| M102K/WT* | 1L54 | 35 | −20.3 | −6.9 | 5.3 | P3221 | 1.9 | Buried lysine | 35,77 |
| S117A/WT* | 165L | 3.64 | 1.27 | 3.01 | P3221 | 1.75 | Polyalanine helix 115–123 | 61 | |
This abbreviated list is excerpted from Table S1 in the supporting information, which also includes explanatory material and the full list of references.
Lysozyme is Tolerant of Changes in Amino Acid Sequence
One of the striking results to emerge from early studies of mutant lysozymes was that the protein is very tolerant of change. The most compelling demonstration came from Poteete and coworkers4 who used suppressor strains to introduce, in turn, 13 different amino acids at every one of the 164 sites in the protein, excluding the amino-terminal methionine. These mutant proteins were not purified, but their activity could be estimated from a convenient plate assay. Remarkably, at over 50% of the sites all 13 substitutions left the activity of the protein essentially equivalent to wild-type. These tolerant sites are in large part distributed over the surface of the protein (Fig. 2). This is consistent with the following first principle of protein folding: A residue that is on the surface of a folded protein is partially or fully solvent-exposed. Because such a residue will presumably remain solvated when the protein unfolds and therefore make similar interactions with its environment in the folded and unfolded form, it will contribute little to the stability of the protein. Conversely, it will make little difference if an alternative amino is placed at such a site. Poteete's survey also showed that many substitutions can be tolerated within the core of the protein.
Figure 2.
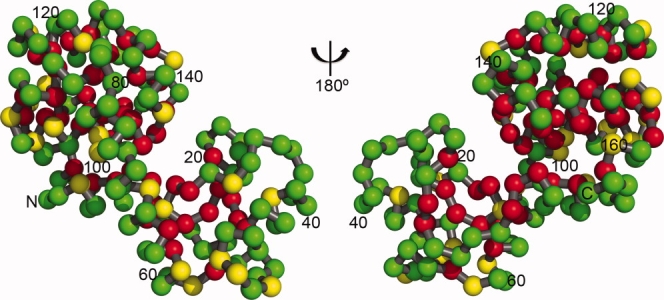
Figure illustrating the tolerance of T4 lysozyme to point mutation (following Ref. 4). Substitutions at the green-colored sites have little if any effect on folding or activity. One or more substitutions at the red sites compromise folding and/or activity. These locations, typically in the core or the active site cleft, are the least tolerant sites. The yellow sites are tolerant, but less so than the green ones (see text for details).
Figure 3a and b summarize the changes in melting temperature for all of the mutants of T4 lysozyme that have been characterized. Plots showing the change in the free energy of folding (ΔΔG) for these mutants are included in the Supporting Information. The plots for the single mutants are asymmetric, as also seen for other proteins.3 For T4 lysozyme, there are very few point mutations that increase the melting temperature by more than 3–4° (see later), but many that decrease stability by 3–4° or more. The most destabilizing point mutants reduce the melting temperature by about 20°C. These include M102K (1L54), which introduces a charged side chain into the hydrophobic core, A98F and A98W, which are “small-to-large” substitutions in the core, and L99G (1QUD), which is a “large-to-small” replacement in the core. R95A, which deletes multiple interactions made by the side-chain of Arg95, is also very destabilizing. As discussed in more detail later, a number of mutations increase stability slightly, but there is no single substitution that has a dramatic effect.
Figure 3.
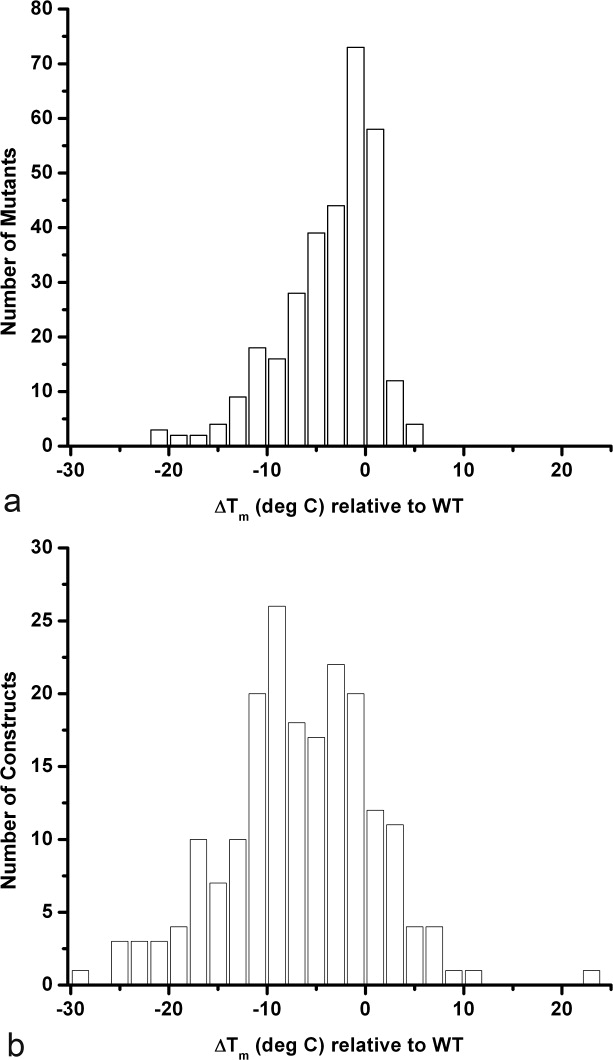
Histograms summarizing how known mutations in T4 lysozyme (Table S1) affect stability. (a) Histogram showing the distribution of ΔTm for 312 known point mutants. The most stabilizing and least stabilizing variants are discussed in the text. Roughly speaking, a change of 3°C in melting temperature corresponds to a change in the free energy of stabilization (ΔΔG) of about 1 kcal/mol. (b) Histogram summarizing the changes in melting temperature for all of the nonsingle site mutations in Table S1. This includes a total of 199 multiple mutants, insertion and deletion mutants and mutations that introduce disulfide bridges. Supporting Information Figures S1a and S1b are similar to Figure 3a and b but plot ΔΔG rather than ΔTm.
In addition to substitutions, lysozyme can also accommodate insertions and deletions.5 If, for example, a small number of amino acids are inserted into the middle of an α-helical region the protein might be expected to respond in one of two ways (Fig. 4): either (1) the inserted amino acids could form a local “bulge”, protruding from the side of the α-helix, or (2) the inserted amino acids could become part of the helix, and, in effect translate the insertion to one end or the other of the helix. Our experience suggests that the latter occurs in the overwhelming majority of cases.6 Presumably, bulges are avoided because they tend to introduce local strain, leave backbone carbonyls and amide groups with unsatisfied hydrogen bonds, and also disrupt hydrophobic contacts present in the parent structure.
Figure 4.
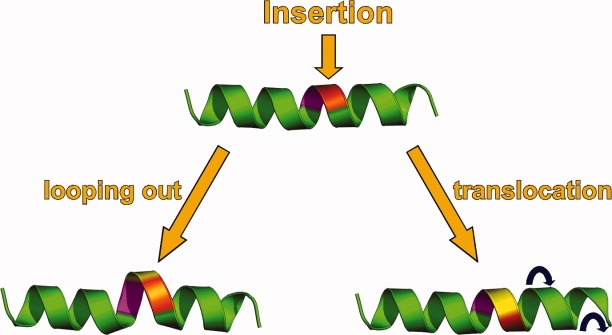
Sketch illustrating two different ways in which a protein could respond to an insertion within the body of an α-helix. The site at which the insertion is to be made is indicated at the top of the figure. The presence of the insertion can result in looping out (left), which is rare for T4 lysozyme, or translocation (right), which is common.
Mutations Cause Modest, Local Changes in Structure: Backbone Shifts are Common, Rotamer Changes are Rare
In the overwhelming number of cases, point mutations cause very modest, localized changes in the structure. Combinations of substitutions that are designed to create cavities or to repack the core also tend to result in relatively modest structural rearrangement.
Baldwin et al.7 used a genetic screen to select combinations of replacements that would repack the hydrophobic core of the protein. These alternative residues were accommodated in large part by adjustments in the local backbone, and only rarely by side-chains adopting different rotamers.
Hurley et al.8 used an in-house computational procedure based on the algorithm of Ponder and Richards9 to find alternative replacements of the buried residues Leu99, Met102, Val111, and Phe153. This culminated in the mutant L99F/M102L/V111I/F153L (1L82) whose structure had backbone shifts up to 0.6–1.0 Å not anticipated in the design procedure. Similarly, the computational procedure of Dahiyat and Mayo10 was used to predict alternative core packing sequences.11 Structure determination again showed that the designed sequence was accommodated primarily by backbone shifts (up to 2.8 Å) rather than changes in side-chain rotamers. Although the use of a fixed backbone template in protein redesign is computationally attractive, experience with lysozyme suggests that such a procedure is a poor approximation for the behavior of real proteins.
The lysozyme mutant in which the most extensive core-repacking was observed, A73-[AAA] (209L) has three alanine residues inserted in the long α-helix that connects the N- and C-terminal lobes of the molecule.6 This insertion caused the helix to increase in length by an extra turn (Fig. 5). In addition, residues 108–103 and 115–123, which form two short helices at right angles in the wild-type structure, rearrange to form a single, continuous helix (108–121) in the structure of the mutant. This major reorganization, which is unique among all the mutant lysozymes, presumably occurs because the lengthening of the long helix due to the insertion disrupts the contacts between the two short helices and the core of the protein.
Figure 5.
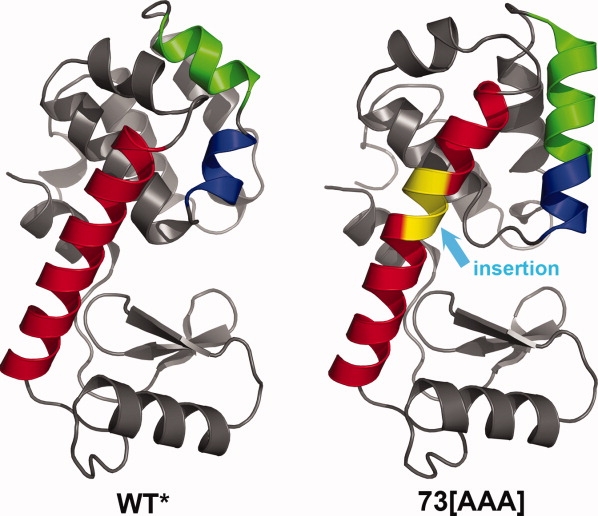
In mutant A73-[AAA] the insertion of three alanines (colored in yellow) within the long helix (red) connecting the lower N-terminal domain and the upper C-terminal domain causes large-scale reorganization that is highly unusual for any mutant structure. In particular, the blue and green helices, which are at an angle of 105° in wild-type (left) reorganize to form a single, straight helix in the mutant (right).
Different Crystal Forms: Hinge-Bending
The majority of the lysozyme variants crystallize isomorphously with wild-type in space group P3221 (Table S1). Not surprisingly, this includes many (but by no means all) of the point mutants. Multiple substitutions are more likely to yield novel crystals. As summarized in Table S2, a total of 43 different crystal forms have been observed for monomeric variants plus three more from mutants engineered to be synthetic disulfide-linked dimers.12 Usually, each of these 43 different packing arrangements was observed once, each for a different mutant. However, there were a few exceptions. For example, mutant S44F/WT* (137L) in space group P21 is isomorphous with the insertion mutant K48-[HP]/WT* (201L), and also with the tandem repeat mutant “L20/R63A” (1T97). Also R96A/WT* (175L) is isomorphous with S44W/WT* (216L) in space group C2 (Table S2).
In different crystal environments the lysozyme molecule displays a range of over 50° in the hinge-bending angle between the N- and C-terminal domains.13 This conformational flexibility appears to be an intrinsic property of the protein, not due to the effect of different mutations (i.e., large differences in the hinge-bending angle are still observed even when mutations are well away from the hinge region). During the hinge-bending motion the N- and C-terminal domains move essentially as rigid bodies, although there are backbone shifts of 0.2–0.5 Å that appear to be induced by differences in crystal contacts.
It could be that the ability of T4 lysozyme to undergo hinge-bending motion might have enhanced its adaptability for incorporation in, and crystallization of, the first GPCR structure.2 On the other hand, the N- and C-termini of T4 lysozyme are both located fairly close to the hinge-bending axis, so that the distance between these termini does not change much during hinge-bending. It might also be noted that by using a Gly-Gly-Ala connector to link the N- and C-terminal residues, it was possible to construct and determine the structure of a circular permutant.14 This permutant had a hinge-bending angle 11° larger than that of WT.
The frequency with which different space group symmetries are observed for the 43 nonisomorphous monomeric lysozyme crystals is summarized in Table II. In a classic article, Wukovitz and Yeates15 addressed the question “Why (do) protein crystals favor some space-groups over others?” They proposed that the favored space groups allow the protein molecules more rigid-body degrees of freedom and can therefore be realized in a greater number of ways. Based on the monomeric protein crystal structures available at the time, Wukovitz and Yeates found that the most commonly-occurring space groups do indeed have the largest number of rigid-body degrees of freedom, D. For comparison, the lysozyme crystal forms are summarized in Table II. The first point is that the two most common space groups for T4 lysozyme, namely P212121 and P21, are those that are most common for monomeric proteins at large. The second point is that the five most common crystal forms for T4 lysozyme have either the highest possible number of rigid-body degrees of freedom (D = 7) or the next-highest (D = 6). It provides further support for the rationalization of Wukovitz and Yeates.
Table II.
Space Group Frequency for Lysozyme Crystals and Proteins in General
| Frequency for monomeric proteins |
||||
|---|---|---|---|---|
| Lysozyme crystals |
Protein crystals in general |
|||
| Space group | D | Occurrences | % | % |
| P212121 | 7 | 10 | 23.3 | 36.1 |
| P21 | 6 | 9 | 20.5 | 11.1 |
| P21212, C2221 | 6 | 4 | 9.3 | 3.7 |
| C2 | 6 | 3 | 7.0 | 6.1 |
| P41212 | 6 | 3 | 7.0 | 2.0 |
| P65 | 5 | 2 | 4.7 | <1 |
| P6222 | 5 | 2 | 4.7 | 0 |
| P43212 | 6 | 0 | 0 | 5.7 |
| P3121 | 6 | 0 | 0 | 4.9 |
| Other | – | 10 | 23.5 | 30.4 |
The table shows the frequency with which the lysozyme crystals listed in supporting information Table S2 have distinct packing arrangements. “Distinct packing arrangement” is the same as “different space group symmetry” except in cases where crystals are isomorphous. For example, mutants R96A/WT* and S44W/WT* are isomorphous in space group C2. By definition, the molecules have the same packing arrangement and make similar contacts within the respective crystals. In the table this is only counted as a single occurrence of space group C2. Likewise, many mutants are isomorphous with wild-type in space group P3221, but this also is counted as a single occurrence. Because this table refers to monomeric proteins the three crystal forms in Table S2 that resulted from synthetically-engineered lysozyme dimers12 are not included. For lysozyme the category “other” includes space groups that occur only once. The table includes the space group frequency for the monomeric protein crystals tabulated by Wukovitz and Yeates.15D is the number of rigid-body degrees of freedom as defined by Wukovitz and Yeates and ranges from 4 to 7 for the 65 space groups possible for protein crystals.
Temperature-Sensitive Mutants
From an historical perspective, one of the main reasons for undertaking structural studies of T4 lysozyme was to take advantage of the genetic screen for temperature-sensitive (TS) mutants developed by George Streisinger.16 It was hoped that comparison of the structures and relative stabilities of such mutants would suggest how individual amino acids contributed to the stability of proteins. When the “lysozyme project” was started, methods for the generation of substitutions by site-directed mutagenesis were yet to be developed. Also the sequence changes in the first TS lysozymes had to be determined by classical protein chemistry rather than at the DNA level.
The five TS lysozymes characterized by these approaches were Arg96 → His (R96H), Met102 → Thr (M102T), Ala146 → Thr (A146T), Gly156 → Asp (G156D), and Thr157 → Ile (T157I).17 On average, these variants reduced the melting temperature of the protein by 10°C. In retrospect, it can be seen from Figure 3a that these TS mutants tend to be substantially more destabilizing than mutations in general. In the case of T157I, 12 other substitutions were made at site 157 and none destabilized the protein as much as the TS variant.18 At site 96 all possible substitutions were subsequently made.19 Thirteen were more stable than the TS mutant R96H and six were less stable at pH 5.35.
Even though the TS mutants tend to be relatively destabilizing, the reasons for the decrease in stability need not be obvious. This question has recently been probed in detail by high-resolution structure analysis of the variants with all possible substitutions at site 96.19,20 On one hand, there is some adaptability in achieving full stabilization at this site (i.e., the three variants of highest stability have Arg, Lys, or Gln at site 96). At the other extreme, to be maximally destabilizing at site 96, a mutation must not only eliminate favorable interactions but also introduce an unfavorable element such as steric strain or a hydrogen-bonding group that remains unsatisfied.
It is apparent that knowledge of the protein structure with high accuracy is essential to understand and to predict the stability of mutant proteins. For the same reason, it can be misleading to assume that conservative amino acid substitutions cause small changes in stability, and that large stability changes are associated with nonconservative replacements. For example, Asn and Gln are similar, but the lysozymes with these alternative amino acids at site 96 differ in melting temperature by 6.6°C. Conversely, Gly and Met are very different, but these substitutions at site 96 yield lysozymes with identical melting temperatures.
Mutations that Increase Stability
Many different methods have been tested to either select or design mutants that retain activity and have increased thermostability. These include the use of entropic effects,21 hydrophobic stabilization,22,23 helical propensity,24,25 salt-bridge interactions,26 metal binding,27 S-S bridges,28,29 methionine replacement,30 computational procedures,8,11 and genetic selection.31 In cases where disulfide bridges could be engineered without introducing strain into T4 lysozyme, they were very effective in increasing the stability of the protein. A good example is the Cys21-Cys142 bridge across the active-site cleft. In this case “hinge-bending” allowed the protein to adjust to accommodate the S-S bridge and the melting temperature was increased by 11°C at pH 2.32 Given this example, it was argued that it may be preferable to engineer disulfides into flexible rather than rigid parts of a protein of interest. By combining three disulfide bridges in the mutant I3C/I9C/T21C/C54T/T142C/L164C (PDB Code 152L), the melting temperature of the protein could be increased by 23.4°C.29
In terms of rationally designed point mutants, the approach most consistently beneficial was to introduce a negatively-charged side-chain at the N-terminus of an α-helix. Even though T4 lysozyme is a relatively small protein, the so-called “helix-dipole” mutants S38D, T109D, T115E, N116D, and N144D were all beneficial, increasing Tm, respectively, by 1.6, 1.5, 0.7, 1.6, and 1.4°C (PDB Codes 1L19, 1L62, 1L37, 1L57, 1L20).
Ignoring substitutions involved in the formation of disulfide bridges, the most stabilizing point mutation found to date is S117V, which increases Tm by 5.1°C.33 The structure of this mutant is not available, but in the related variant S117F/WT* (1TLA),34 the Phe117 side-chain is seen to occupy a preformed hydrophobic pocket on the surface of the protein. The resultant hydrophobic interaction accounted for some of the increase in stability, although not the whole amount. Adding to the puzzle, the mutant S117A/WT* (165L) was also substantially stabilizing (ΔTm = 3.6°C) even though this substitution cannot have the hydrophobic stabilization associated with S117V or S117F. Yet another puzzling observation was that the side-chain hydroxyl of Ser117 in WT lysozyme appeared to make a strong hydrogen bond to the side-chain of Asn132. The mutations S117A, S117V, and S117F all eliminated this hydrogen bond, yet increased stability. The answer to this conundrum was provided by the refinement of WT lysozyme at 1.06 Å resolution.35 This high-resolution analysis shows that the interaction distance between the side-chains of Ser117 and Asn132 is 2.48 ± 0.05 Å, substantially shorter than the lower bound of 2.7–2.8 Å or thereabouts expected for a typical H-bond. Thus, instead of this being a strong hydrogen bond, it is actually an unfavorable van der Waals contact. Consistent with this, when Asn132 is replaced by Phe, Ile or Met (i.e., in N132F, N132I, and N132M) the melting temperature is also increased (ΔTm = 3.0–3.6°C).33 In other words, the replacement of either Ser117 or Asn132 with a non-H-bonding substitute results in a significant increase in stability. [N132A has not been constructed as a single mutant but based on its beneficial effect on other variants,36 we anticipate that it also would be stabilizing (by about 1.5–2.0°C)].
Why is an apparent destabilizing interaction between Ser117 and Asn132 retained in the native lysozyme structure? Presumably because both of these residues are involved in the binding of the extended peptidoglycan that forms part of the cell-wall substrate of T4 lysozyme.37 It supports the hypothesis of Shoichet et al.33 that the residues in an enzyme that are involved in substrate binding and catalysis are not optimized for stability. Modifying these residues may yield an increase in stability but at the cost of catalytic activity. This is true for all the above replacements at sites 117 and 132. Likewise, Glu11 is thought to be the essential catalytic residue for T4 lysozyme and E11A, E11M, and E11F are all substantially stabilizing (ΔTm = 2.6–4.3°C), but having essentially no catalytic activity.
Thermophilic Proteins
In general, if multiple point mutations are located at sites that are not in contact, the effects on stability will be additive. Thus, by combining a series of stabilizing mutants, each having a modest benefit, it can be possible to substantially increase the overall stability of a protein.29,38,39 In the case of T4 lysozyme the engineered multiple mutant I3L/S38D/A41V/A82P/N116D/V131A/N144D (PDB Code 189L) increased Tm by 8.3°C.40
This finding explains how proteins from thermophiles typically have structures similar to their counterparts in mesophiles, but can have greatly enhanced thermostability. This thermotolerance can be achieved by a combination of different effects at different sites including hydrophobic stabilization, improved hydrogen bonding, metal ion binding and so on.41
Cavity-Creating Mutants
As is well known, the cores of proteins are tightly packed with little “nonprotein” space remaining. Occasionally water molecules can remain within the folded protein and will typically occupy cavities that are polar in character and up to about 150 Å3 in volume.42 Apolar cavities appear to be empty and typically have volumes up to about 50 Å3.42
If a bulky nonpolar residue in the core of a protein is replaced with a smaller one, a cavity may or may not be created. For T4 lysozyme a series of Leu→Ala, Ile→Ala, Phe→Ala, and Met→Ala created cavities ranging from 0 Å3 to 150 Å3.43,44 The Leu99→Ala (L99A) cavity is unusually large (150 Å3) in part because there was already a cavity of volume 41 Å3 at this site in the native protein, and also because the surrounding protein remains “rigid” and does not collapse into the cavity region. [Recent studies have also shown that this cavity retains its shape and volume even when the protein is under pressure of 200 MPa; under these conditions a cluster of approximately four water molecules move into the cavity45 (PDB Code 2B6T).] Experience with T4 lysozyme suggests that cavities engineered into well-buried, rigid (i.e., low B-factor) regions of the protein tend to retain their shape. In contrast, large-to-small substitutions made in more flexible regions of the protein, or close to the surface, tend to be associated with movement or “collapse” of the surrounding residues to reduce or eliminate the volume of the putative cavity.44
It was proposed that the reduction in protein stability associated with “large to small” substitutions of hydrophobic residues in the core is made up of two components. First, there is a constant term due to the reduced hydrophobicity (desolvation energy) of the smaller relative to the larger amino acid. Second, there is a variable term due to the loss in van der Waals (i.e., core packing) contacts. The variable term appeared to be proportional to the size of the cavity created (22 cal mol−1 Å−3).43,44 Efforts have been made to evaluate this relationship in other systems.46–48 The latter test48 is of special interest because it analyzed cavity-creating mutations in bacteriorhodopsin, an intrinsic membrane protein in a lipidic environment. The results, compared with those for T4 lysozyme43 are shown in Figure 6. As can be seen, the way in which the stabilities of bacteriorhodopsin and T4 lysozyme depend on cavity volume is strikingly similar. The slopes of the respective lines for the two proteins were considered by Joh et al.48 to be within experimental error. The obvious difference between the two proteins is the value of the intercept at zero change in cavity size. For lysozyme, this value is 1.9 kcal/mol and was attributed to the difference between the desolvation energy (hydrophobicity) of leucine and alanine. For lysozyme, a water-soluble protein, desolvation refers to the transfer of the amino acid in question from the nonpolar core of the protein to an aqueous environment. For bacteriorhodopsin, “desolvation” refers to transfer from the nonpolar protein core to a detergent micelle. Figure 6 suggests that this “solvophobic effect” contributes about 0.4–0.7 kcal/mol.
Figure 6.
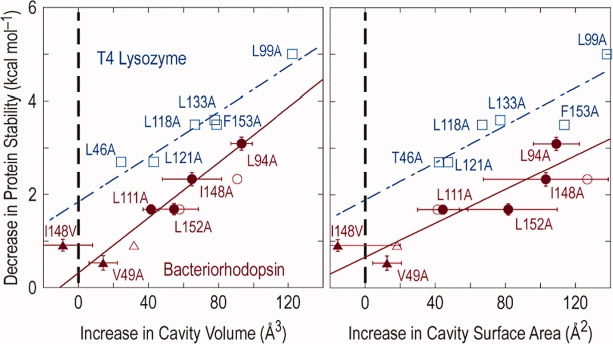
Comparison of the energetic effects of core mutations in T4 lysozyme (blue) and bacteriorhodopsin (red). Dependence of protein stability on cavity volume is shown on the left, and on surface area on the right. From Joh et al.48 with permission.
It might be noted that the calculation of cavity volume is not straightforward and can be sensitive to small changes in the coordinates. More important, for smaller cavities different algorithms can give volumes that vary by over a factor of two.44 For larger cavities these variations should become less important. On the other hand, the larger the volume of the cavity the greater the loss of protein stability. This limits the size of a nonpolar cavity that can be tolerated within a given protein.
Engineered Ligand Binding Sites
Although the L99A cavity in T4 lysozyme is internal and has no direct access to solvent it readily binds benzene (Fig. 7),49 which has also been shown by NMR to move in and out of the cavity on a microsecond timescale.50 The cavity binds nonpolar analogs of benzene, such as ethylbenzene and xylene.51 Recently, it has been shown that 1,2-azaborine and 1-ethyl-1,2-dihydro-1,2-azaborine also bind.52 These novel analogs provide a way to incorporate boron into compounds of biomedical interest. The L99A cavity also binds halogenated benzenes, such as C6F6, C6H5I, and C6F5I, among others.53 In the case of C6F5I, a “halogen bond” of length 3.0 Å was observed between the iodine atom and the sulfur of Met102 in the wall of the cavity (PDB Code 3DN3). This bond is 1.0 Å less than the sum of the van der Waals radii, but much larger than a covalent I-S bond (˜2.4 Å). Stability measurements, however, suggest that this interaction in the case of T4 lysozyme is quite weak (0.5–0.7 kcal/mol relative to a van der Waals contact).
Figure 7.
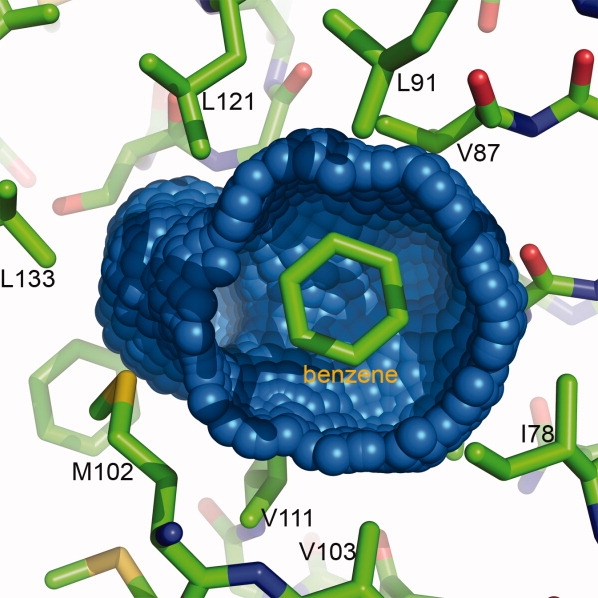
The blue-colored surface shows the cavity in the core of T4 lysozyme resulting from the mutation of leucine 99 to alanine. For clarity, part of the cavity surface has been removed to show the location occupied by benzene, one of many ligands that can bind within this cavity.
The L99A cavity in T4 lysozyme appears to be largely empty.54 In contrast to the active site of a typical enzyme where ligand binding is associated with extensive displacement of solvent, ligands binding to L99A enter a largely unoccupied site. This simplification makes L99A attractive as a test bed for in silico algorithms designed to screen libraries of compounds for their ability to bind to protein sites of interest.
The “original” L99A cavity (Fig. 6) is essentially apolar, and as noted above, binds various nonpolar ligands. Binding was not detected, however, for more polar ones including aniline and phenol.51 By making the supplemental mutation M102Q, a polar group was introduced into the wall of the cavity. This modified cavity does bind ligands, such as phenol and 2-fluoro-aniline and their binding conformations could be predicted quite well.55 To introduce a putatively-charged group into the cavity was more challenging because it was expected to be highly destabilizing. By engineering supplemental stabilizing substitutions elsewhere in the molecule (Table I, PDB Code 3GUI) it was, however, possible to change Met102 in the cavity wall into glutamic acid.56 In this L99A/M102E stabilized mutant the shape of the cavity remains essentially the same as in L99A, but a network of three water molecules bind near Glu102, which is at least partially charged under the conditions of the crystallographic analysis (pH ∼ 7.0). This variant not only binds the polar benzene analogs aniline and phenol but also nonpolar ones, such as p-xylene and ethylbenzene, even though the latter displaces all three water molecules from the vicinity of Glu102.
Access to the L99A Cavity
Recently Kay and coworkers57,58 have examined the L99A cavity mutant in more detail by NMR. They have characterized a transition between a highly populated (97%, 25°C) ground state conformation and a higher energy excited state. The transition involves a conformational rearrangement that includes the C-terminal portion of Helix E, Helix F, Helix I, and the loops connecting these elements to the rest of the protein. This is the same region, in particular Helix F (residues 108–113), which undergoes the largest conformational adjustments (∼1.5–2.5 Å) when larger ligands such as ethylbenzene and isobutylbenzene bind in the cavity59 (in Fig. 1, Helix F is yellow and runs essentially vertically at the right of the C-terminal domain). Helix F also has thermal factors substantially higher than those of the other helices that surround the L99A cavity. Furthermore, in mutant L99G, which could potentially create a cavity larger than that in L99A, part of Helix F moves 4–5 Å and creates a solvent-accessible declivity.60 In contrast, in the double mutant L99G/E108V Helix F returns to a position akin to that in WT and L99A. (Glu108 is on the surface of the protein 11 Å from Leu99. The mutation E108V appears to act by increasing the stability and rigidity of the protein.) Comparison of the L99G and L99G/E108V structures shows that Helix F can change between two conformations, one of which blocks access to the L99A cavity and another which leaves it open to solvent. These crystallographic observations are consistent with the suggestion of Mulder et al.57 that the excited state they observe could facilitate entry of ligands into the L99A cavity.
Lysozyme Folding
As has been discussed earlier, T4 lysozyme has a pronounced two-domain structure (Fig. 1). This was one of the most striking features of the original structure determination and it was remarked at the time that the enzyme would presumably have to undergo a fairly large conformational change to admit its peptidoglycan substrate.61 Consistent with this, there are many mutant crystal structures, which display large variations in the hinge-bending angle between the N- and C-terminal domain.13 NMR analysis also suggests hinge-bending in solution.62
The above observations might suggest that the N- and C-terminal domains of T4 lysozyme fold independently but careful early studies of Schellman and coworkers63 suggested that the protein folds in a strictly two-state manner, with no evidence for multistate behavior. Based on the use of pulsed hydrogen exchange, Lu and Dahlquist64 presented evidence for a folding intermediate that included parts of both the N- and C-terminal domains. Kato et al.65,66 further characterized this intermediate. They found it to be on the folding pathway and to exist after the rate-limiting step, consistent with folding akin to that of small proteins. This overall behavior is consistent with the fact that the N- and C-terminal domains of T4 lysozyme are energetically coupled.
Conclusions
Overall, the studies of T4 lysozyme mutants strongly support long-held beliefs regarding the nature of protein folding and stability. In particular, the stability and structure of every known lysozyme mutant can be rationalized using well-established principles of hydrophobicity, hydrogen bonding, electrostatics, and so on. Also reasonably good success can be achieved in using simple principles to design point mutants that increase protein stability. On the other hand, the relatively modest objective of accurately predicting the changes in stability and structure associated with small-to-large or large-to-small mutations within the core is yet to be met. Predicting the behavior of insertion or deletion mutants, or those involving major redesign remain as clear challenges for the future.
Acknowledgments
The results described here could not have been obtained without the dedication and help of a series of extraordinarily talented lab members.
References
- 1.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, Choi H-J, Yao X-J, Weis WI, Stevens RC, Kobilka BK. GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 2.Rosenbaum DM, Rasmussen SGF, Kobilka BK. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tokuriki N, Tawfik DS. Stability effects of mutations and protein evolvability. Curr Opin Struct Biol. 2009;19:596–604. doi: 10.1016/j.sbi.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Rennell D, Bouvier SE, Hardy LW, Poteete AR. Systematic mutation of bacteriophage T4 lysozyme. J Mol Biol. 1991;222:67–87. doi: 10.1016/0022-2836(91)90738-r. [DOI] [PubMed] [Google Scholar]
- 5.Heinz DW, Baase WA, Dahlquist FW, Matthews BW. How amino-acid insertions are allowed in an α-helix of T4 lysozyme. Nature. 1993;361:561–564. doi: 10.1038/361561a0. [DOI] [PubMed] [Google Scholar]
- 6.Vetter IR, Baase WA, Heinz DW, Xiong J-P, Snow S, Matthews BW. Protein structural plasticity exemplified by insertion and deletion mutants in T4 lysozyme. Protein Sci. 1996;5:2399–2415. doi: 10.1002/pro.5560051203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baldwin EP, Hajiseyedjavadi O, Baase WA, Matthews BW. The role of backbone flexibility in the accommodation of variants that repack the core of T4 lysozyme. Science. 1993;262:1715–1718. doi: 10.1126/science.8259514. [DOI] [PubMed] [Google Scholar]
- 8.Hurley JH, Baase WA, Matthews BW. Design and structural analysis of alternative hydrophobic core packing arrangements in bacteriophage T4 lysozyme. J Mol Biol. 1992;224:1143–1159. doi: 10.1016/0022-2836(92)90475-y. [DOI] [PubMed] [Google Scholar]
- 9.Ponder JW, Richards FM. Tertiary templates for proteins. Use of packing criteria in the enumeration of allowed sequences for different structural classes. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 10.Dahiyat BI, Mayo SL. De novo protein design: fully automated sequence selection. Science. 1997;278:82–87. doi: 10.1126/science.278.5335.82. [DOI] [PubMed] [Google Scholar]
- 11.Mooers BHM, Datta D, Baase WA, Zollars ES, Mayo SL, Matthews BW. Repacking the core of T4 lysozyme by automated design. J Mol Biol. 2003;332:741–756. doi: 10.1016/s0022-2836(03)00856-8. [DOI] [PubMed] [Google Scholar]
- 12.Banatao DR, Cascio D, Crowley CS, Fleissner MR, Tienson HL, Yeates TO. An approach to crystallizing proteins by synthetic symmetrization. Proc Natl Acad Sci USA. 2006;103:16230–16235. doi: 10.1073/pnas.0607674103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X-J, Wozniak JA, Matthews BW. Protein flexibility and adaptability seen in 25 crystal forms of T4 lysozyme. J Mol Biol. 1995;250:527–552. doi: 10.1006/jmbi.1995.0396. [DOI] [PubMed] [Google Scholar]
- 14.Sagermann M, Baase WA, Mooers BHM, Gay L, Matthews BW. Relocation or duplication of the Helix A sequence of T4 lysozyme causes only modest changes in structure but can increase or decrease the rate of folding. Biochemistry. 2004;43:1296–1301. doi: 10.1021/bi035702q. [DOI] [PubMed] [Google Scholar]
- 15.Wukovitz SW, Yeates TO. Why protein crystals favor some space-groups over others. Nature Struct Biol. 1995;2:1062–1067. doi: 10.1038/nsb1295-1062. [DOI] [PubMed] [Google Scholar]
- 16.Streisinger G, Okada Y, Emrich J, Newton J, Tsugita A, Terzaghi E, Inouye M. Frameshift mutations and the genetic code. Cold Spring Harb Symp Quant Biol. 1966;31:77–84. doi: 10.1101/sqb.1966.031.01.014. [DOI] [PubMed] [Google Scholar]
- 17.Alber T, Grütter MG, Gray TM, Wozniak J, Weaver LH, Chen B-L, Baker EN, Matthews BW. Structure and stability of mutant lysozymes from bacteriophage T4. In: Oxender DL, editor. Vol. 39. New York: Alan R. Liss; 1986. pp. 307–318. “UCLA Symposium on Molecular and Cellular Biology,” New Series. [Google Scholar]
- 18.Alber T, Dao-pin S, Wilson K, Wozniak JA, Cook SP, Matthews BW. Contributions of hydrogen bonds of threonine 157 to the thermodynamic stability of phage T4 lysozyme. Nature. 1987;330:41–46. doi: 10.1038/330041a0. [DOI] [PubMed] [Google Scholar]
- 19.Mooers BHM, Baase WA, Wray JW, Matthews BW. Contributions of all 20 amino acids at site 96 to the stability and structure of T4 lysozyme. Protein Sci. 2009;18:871–880. doi: 10.1002/pro.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mooers BHM, Tronrud DE, Matthews BW. Evaluation at atomic resolution of the role of strain in destabilizing the temperature-sensitive T4 lysozyme mutant Arg 96 → His. Protein Sci. 2009;18:863–870. doi: 10.1002/pro.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matthews BW, Nicholson H, Becktel WJ. Enhanced protein thermostability from site-directed mutations that decrease the entropy of unfolding. Proc Natl Acad Sci USA. 1987;84:6663–6667. doi: 10.1073/pnas.84.19.6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsumura M, Wozniak JA, Daopin S, Matthews BW. Structural studies of mutants of T4 lysozyme that alter hydrophobic stabilization. J Biol Chem. 1989;264:16059–16066. [PubMed] [Google Scholar]
- 23.Karpusas M, Baase WA, Matsumura M, Matthews BW. Hydrophobic packing in T4 lysozyme probed by cavity-filling mutants. Proc Natl Acad Sci USA. 1989;86:8237–8241. doi: 10.1073/pnas.86.21.8237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X-J, Baase WA, Matthews BW. Toward a simplification of the protein folding problem: a stabilizing polyalanine α-helix engineered in T4 lysozyme. Biochemistry. 1991;30:2012–2017. doi: 10.1021/bi00222a001. [DOI] [PubMed] [Google Scholar]
- 25.Heinz DW, Baase WA, Matthews BW. Folding and function of a T4 lysozyme containing 10 consecutive alanines illustrate the redundancy of information in an amino acid sequence. Proc Natl Acad Sci USA. 1992;89:3751–3755. doi: 10.1073/pnas.89.9.3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dao-pin S, Sauer U, Nicholson H, Matthews BW. Contributions of surface salt bridges to the stability of bacteriophage T4 lysozyme determined by directed mutagenesis. Biochemistry. 1991;30:7142–7153. doi: 10.1021/bi00243a015. [DOI] [PubMed] [Google Scholar]
- 27.Wray JW, Baase WA, Ostheimer GJ, Zhang X-J, Matthews BW. Use of a non-rigid region in T4 lysozyme to design an adequate metal-binding site. Protein Eng. 2000;13:313–321. doi: 10.1093/protein/13.5.313. [DOI] [PubMed] [Google Scholar]
- 28.Matsumura M, Becktel WJ, Levitt M, Matthews BW. Stabilization of phage T4 lysozyme by engineered disulfide bonds. Proc Natl Acad Sci USA. 1989;86:6562–6566. doi: 10.1073/pnas.86.17.6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsumura M, Signor G, Matthews BW. Substantial increase of protein stability by multiple disulfide bonds. Nature. 1989;342:291–293. doi: 10.1038/342291a0. [DOI] [PubMed] [Google Scholar]
- 30.Lipscomb LA, Gassner NC, Snow SD, Eldridge AM, Baase WA, Drew DL, Matthews BW. Context-dependent protein stabilization by methionine-to-leucine substitution shown in T4 lysozyme. Protein Sci. 1998;7:765–773. doi: 10.1002/pro.5560070326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pjura P, Matsumura M, Baase WA, Matthews BW. Development of an in vivo method to identify mutants of phage T4 lysozyme of enhanced thermostability. Protein Sci. 1993;2:2217–2225. doi: 10.1002/pro.5560021221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jacobson R, Matsumura M, Faber HR, Matthews BW. Structure of a stabilizing disulfide bridge mutant that closes the active-site cleft of T4 lysozyme. Protein Sci. 1992;1:46–57. doi: 10.1002/pro.5560010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shoichet BK, Baase WA, Kuroki R, Matthews BW. A relationship between protein stability and protein function. Proc Natl Acad Sci USA. 1995;92:452–456. doi: 10.1073/pnas.92.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson DE, Hurley JH, Nicholson H, Baase WA, Matthews BW. Hydrophobic core repacking and aromatic-aromatic interaction in the thermostable mutant of T4 lysozyme Ser 117 → Phe. Protein Sci. 1993;2:1285–1290. doi: 10.1002/pro.5560020811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mooers BHM, Matthews BW. Use of an ion-binding site to bypass the 1000-atom limit to structure determination by direct methods. Acta Cryst. 2004;D60:1726–1737. doi: 10.1107/S0907444904017020. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X-J, Baase WA, Matthews BW. Multiple alanine replacements within α-helix 126–134 of T4 lysozyme have independent, additive effects on both structure and stability. Protein Sci. 1992;1:761–776. doi: 10.1002/pro.5560010608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuroki R, Weaver LH, Matthews BW. A covalent enzyme-substrate intermediate with saccharide distortion in a mutant T4 lysozyme. Science. 1993;262:2030–2033. doi: 10.1126/science.8266098. [DOI] [PubMed] [Google Scholar]
- 38.Pantoliano MW, Whitlow M, Wood JF, Dodd SW, Hardman KD, Rollence ML, Bryan PN. Large increases in general stability for subtilisin BPN' through incremental changes in the free energy of unfolding. Biochemistry. 1989;28:7205–7213. doi: 10.1021/bi00444a012. [DOI] [PubMed] [Google Scholar]
- 39.Serrano L, Day AG, Fersht AR. Step-wise mutation of barnase to binase. A procedure for engineering increased stability of proteins and an experimental analysis of the evolution of protein stability. J Mol Biol. 1993;233:305–312. doi: 10.1006/jmbi.1993.1508. [DOI] [PubMed] [Google Scholar]
- 40.Zhang X-J, Baase WA, Shoichet BK, Wilson KP, Matthews BW. Enhancement of protein stability by the combination of point mutations in T4 lysozyme is additive. Protein Engin. 1995;8:1017–1022. doi: 10.1093/protein/8.10.1017. [DOI] [PubMed] [Google Scholar]
- 41.Matthews BW, Weaver LH, Kester WR. The conformation of thermolysin. J Biol Chem. 1974;249:8030–8044. [PubMed] [Google Scholar]
- 42.Hubbard SJ, Gross K-H, Argos P. Intramolecular cavities in globular proteins. Protein Eng. 1994;7:613–626. doi: 10.1093/protein/7.5.613. [DOI] [PubMed] [Google Scholar]
- 43.Eriksson AE, Baase WA, Zhang X-J, Heinz DW, Blaber M, Baldwin EP, Matthews BW. Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science. 1992;255:178–183. doi: 10.1126/science.1553543. [DOI] [PubMed] [Google Scholar]
- 44.Xu J, Baase WA, Baldwin E, Matthews BW. The response of T4 lysozyme to large-to-small substitutions within the core and its relation to the hydrophobic effect. Protein Sci. 1998;7:158–177. doi: 10.1002/pro.5560070117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Collins MD, Hummer G, Quillin ML, Matthews BW, Gruner SM. Cooperative water filling of a nonpolar protein cavity observed by high-pressure crystallography and simulation. Proc Natl Acad Sci USA. 2005;102:16668–16671. doi: 10.1073/pnas.0508224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buckle AM, Cramer P, Fersht AR. Structural and energetic responses to cavity-creating mutations in hydrophobic cores: observation of a buried water molecule and the hydrophilic nature of such hydrophobic cavities. Biochemistry. 1996;35:4298–4305. doi: 10.1021/bi9524676. [DOI] [PubMed] [Google Scholar]
- 47.Kadonosono T, Chatani E, Hayashi R, Moriyama H, Ueki T. Minimization of cavity size ensures protein stability and folding: structures of Phe46-replaced bovine pancreatic RNase A. Biochemistry. 2003;42:10651–10658. doi: 10.1021/bi034499w. [DOI] [PubMed] [Google Scholar]
- 48.Joh NH, Oberai A, Yang D, Whitelegge JP, Bowie JU. Similar energetic contributions of pacing in the core of membrane and water-soluble proteins. J Am Chem Soc. 2009;131:10846–10847. doi: 10.1021/ja904711k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eriksson AE, Baase WA, Wozniak JA, Matthews BW. A cavity-containing mutant of T4 lysozyme is stabilized by buried benzene. Nature. 1992;355:371–373. doi: 10.1038/355371a0. [DOI] [PubMed] [Google Scholar]
- 50.Feher VA, Baldwin EP, Dahlquist FW. Access of ligands to cavities within the core of a protein is rapid. Nature Struct Biol. 1996;3:516–521. doi: 10.1038/nsb0696-516. [DOI] [PubMed] [Google Scholar]
- 51.Morton A, Baase WA, Matthews BW. Energetic origins of specificity of ligand binding in an interior nonpolar cavity of T4 lysozyme. Biochemistry. 1995;34:8564–8575. doi: 10.1021/bi00027a006. [DOI] [PubMed] [Google Scholar]
- 52.Liu L, Marwitz AJV, Matthews BW, Liu S-Y. Boron in disguise: 1-ethyl-1,2-dihydro-1,2-azaborine binds in a nonpolar cavity of T4 lysozyme similarly to ethylbenzene. Angew Chem Int Ed. 2009;48:6817–6819. doi: 10.1002/anie.200903390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu L, Baase WA, Matthews BW. Halogenated benzenes bound within a nonpolar cavity in T4 lysozyme provide examples of I…S and I…Se halogen-binding. J Mol Biol. 2009;385:595–605. doi: 10.1016/j.jmb.2008.10.086. [DOI] [PubMed] [Google Scholar]
- 54.Liu L, Quillin ML, Matthews BW. Use of experimental crystallographic phases to examine the hydration of polar and nonpolar cavities in T4 lysozyme. Proc Natl Acad Sci USA. 2008;105:14406–14411. doi: 10.1073/pnas.0806307105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wei BQ, Baase WA, Weaver LH, Matthews BW, Shoichet BK. A model binding site for testing scoring functions in molecular docking. J Mol Biol. 2002;322:339–355. doi: 10.1016/s0022-2836(02)00777-5. [DOI] [PubMed] [Google Scholar]
- 56.Liu L, Baase WA, Matthews BW. Use of stabilizing mutations to engineer a charged group within a ligand-binding hydrophobic cavity in T4 lysozyme. Biochemistry. 2009;48:8842–8851. doi: 10.1021/bi900685j. [DOI] [PubMed] [Google Scholar]
- 57.Mulder FAA, Mittermaier A, Hon B, Dahlquist FW, Kay LE. Studying excited states of proteins by NMR spectroscopy. Nat Struct Biol. 2001;8:932–935. doi: 10.1038/nsb1101-932. [DOI] [PubMed] [Google Scholar]
- 58.Mulder FAA, Hon B, Mittermaier A, Dahlquist FW, Kay LE. Slow internal dynamics in proteins: application of NMR relaxation dispersion spectroscopy to methyl groups in a cavity mutant of T4 lysozyme. J Am Chem Soc. 2002;124:1443–1451. doi: 10.1021/ja0119806. [DOI] [PubMed] [Google Scholar]
- 59.Morton A, Matthews BW. Specificity of ligand binding in a buried nonpolar cavity of T4 lysozyme: linkage of dynamics and structural plasticity. Biochemistry. 1995;34:8576–8588. doi: 10.1021/bi00027a007. [DOI] [PubMed] [Google Scholar]
- 60.Wray JW, Baase WA, Lindstrom JD, Weaver LH, Poteete AR, Matthews BW. Structural analysis of a noncontiguous second-site revertant in T4 lysozyme shows that increasing the rigidity of a protein can enhance its stability. J Mol Biol. 1999;292:1111–1120. doi: 10.1006/jmbi.1999.3102. [DOI] [PubMed] [Google Scholar]
- 61.Matthews BW, Remington SJ. The three-dimensional structure of the lysozyme from bacteriophage T4. Proc Natl Acad Sci USA. 1974;71:178–4182. doi: 10.1073/pnas.71.10.4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goto NK, Skyrnnikov NR, Dahlquist FW, Kay LE. What is the average conformation of bacteriophage T4 lysozyme in solution? A domain orientation study using dipolar couplings measured by NMR. J Mol Biol. 2001;308:745–764. doi: 10.1006/jmbi.2001.4614. [DOI] [PubMed] [Google Scholar]
- 63.Hawkes R, Grütter MG, Schellman JA. Thermodynamic stability and point mutations of bacteriophage T4 lysozyme. J Mol Biol. 1984;175:195–212. doi: 10.1016/0022-2836(84)90474-1. [DOI] [PubMed] [Google Scholar]
- 64.Lu J, Dahlquist FW. Detection and characterization of an early folding intermediate of T4 lysozyme using pulsed hydrogen exchange and two-dimensional NMR. Biochemistry. 1992;31:4749–4756. doi: 10.1021/bi00135a002. [DOI] [PubMed] [Google Scholar]
- 65.Kato H, Feng H, Bai Y. The folding pathway of T4 lysozyme: the high-resolution structure and folding of a hidden intermediate. J Mol Biol. 2007;365:870–880. doi: 10.1016/j.jmb.2006.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kato H, Vu N, Feng H, Zhou Z, Bai Y. The folding pathway of T4 lysozyme: an on-pathway hidden folding intermediate. J Mol Biol. 2007;365:881–891. doi: 10.1016/j.jmb.2006.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]