

Abstract
Point mutations in proteins can have different effects on protein stability depending on the mechanism of unfolding. In the most interesting case of I27, the Ig-like module of the muscle protein titin, one point mutation (Y9P) yields opposite effects on protein stability during denaturant-induced “global unfolding” versus “vectorial unfolding” by mechanical pulling force or cellular unfolding systems. Here, we assessed the reason for the different effects of the Y9P mutation of I27 on the overall molecular stability and N-terminal unraveling by NMR. We found that the Y9P mutation causes a conformational change that is transmitted through β-sheet structures to reach the central hydrophobic core in the interior and alters its accessibility to bulk solvent, which leads to destabilization of the hydrophobic core. On the other hand, the Y9P mutation causes a bend in the backbone structure, which leads to the formation of a more stable N-terminal structure probably through enhanced hydrophobic interactions.
Keywords: NMR, protein unfolding, titin, mitochondrial protein import, AFM
Introduction
Proteins are generally programed to attain well-defined three-dimensional structures to perform their functions. In cells, however, large conformational changes or even global unfolding are often called to respond to the demand of cellular homeostasis. Protein unfolding can be achieved both in vitro and in vivo by several means. Unfolding pathways could differ depending on the unfolding mechanism, reflecting differences in the regions where unfolding factors or systems directly act.1 In chemical unfolding, denaturants such as urea or guanidinium chloride (GdHCl) have global effects on the protein to destabilize folded states. In mechanical-forced unfolding, atomic force microscopy (AFM) acts on two different regions of the protein to exert stretching forces in opposite directions.1,2 Cellular unfolding systems often achieve vectorial unfolding by grabbing a specific region (e.g., the N-terminus) of a substrate protein and threading it through a narrow pore, which causes mechanical unfolding of the rest of the molecule.1 Such narrow pores are provided by protein-conducting channels of protein translocators in membranes and by ATP-dependent proteases including proteasome and Clp proteases.
The I27 domain (the 27th immunoglobulin domain of human cardiac titin) is a model protein whose unfolding by denaturants, AFM, and mitochondrial protein import has been studied extensively.2–4 The I27 domain (89 amino-acids long) consists of seven β-strands (strands A–G) that fold into two layers (strands A, B, E, and D in one layer and strands A′, G, F, and C in the other) of β-sheets through backbone hydrogen bonds and hydrophobic side-chain interactions [Fig. 1(A)].5,6 In denaturant-induced unfolding, the I27 domain becomes unfolded without a stable intermediate; instead, I27 undergoes unfolding through a distinct transition state, in which only the A′ and G strands are completely unstructured while all other β-strands are structured to some extent.6 On the other hand, during the forced unfolding by AFM, I27 becomes populated by an unfolding intermediate, whereby β-strand A is completely detached from its β-sheet partner (β-strand B) while the rest of the molecule remains structurally intact7,8; higher forces are then required for the detachment of β-strand A′ from strand G, leading to the collapse of hydrophobic cores and global unfolding.
Figure 1.
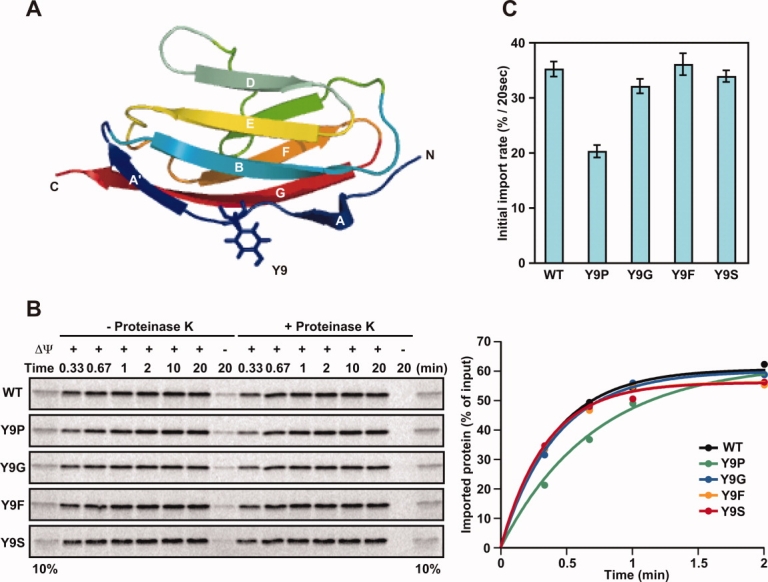
Structure of I27 and import of its Y9 mutants. (A) Cartoon diagram showing the β-sandwich structure of the I27 molecule (PDB ID, 1TIT). Tyr9 is shown in stick form. (B) Effects of mutations at residue 9 of the I27 domain on the import of radiolabeled pb2(80)-I27 fusion proteins. Mitochondria were isolated from yeast strain D273-10B. The indicated radiolabeled proteins were incubated with mitochondria at 25°C for indicated times in the absence (+ΔΨ) or presence (−ΔΨ) of valinomycin, which dissipates the membrane potential (ΔΨ) across the inner membrane. The mitochondria were treated with (+Proteinase K) or without (−Proteinase K) proteinase K, and radioactive proteins were analyzed by SDS-PAGE and radioimaging (left panels). The amounts of radiolabeled proteins added to each reaction were set to 100% and imported proteins were plotted against incubation times (right panels). 10%, ten percent of the radiolabled proteins added to each reaction. (C) Imported, proteinase-K-protected fractions in (B) were quantified, and import rates (initial slopes of the import reactions) were plotted. Values are presented as mean ± SD.
In mitochondrial protein import, an N-terminally attached mitochondrial targeting signal can direct nonmitochondrial “passenger” proteins to mitochondria both in in vitro and in vivo.9,10 Mitochondrial protein import is mediated by the membrane protein complexes called translocators in the outer and inner mitochondrial membranes.10–12 Protein translocators contain receptor(s) that recognize the targeting and/or sorting signals encoded in the presequence or mature domain of substrate proteins and a protein-conducting channel through which mitochondrial proteins traverse the hydrophobic barrier of the mitochondrial membranes. When incubated with isolated mitochondria, the I27 domain fused to a mitochondrial targeting signal can be unfolded to move through the narrow pores of the translocator complexes in the outer and inner mitochondrial membranes.4,13 In this “import-coupled unfolding,” β-strand A is easily detached from β-strand B, as in the case of mechanical unfolding, yet the intermediate with detached segment A is not resistant against further disruption of the β-strand pair of strands A′ and G, which is clearly different from the case of mechanical unfolding by AFM.
It has been recognized that point mutations can have different effects on protein stability depending on the mechanism of unfolding.4,7,13–15 However, much of the structural basis of those mutational effects on different unfolding processes remains unclear, especially for vectorial unfolding.2 Amino acid residues in the N-terminal region (β-strand A, linker segment, and β-strand A′) of the I27 domain have been systematically replaced with Pro, and analyzed with respect to their effects on mechanical unfolding and mitochondrial import rates.4,7,13,14 As Pro is incapable of forming a backbone hydrogen bond as a donor, proline mutagenesis in a β-segment would likely destabilize the β-sheet structure. This prediction was indeed consistent with the denaturant-induced unfolding of I27, but not always consistent with the mechanical unfolding by AFM or unfolding upon mitochondrial protein import. For example, introduction of a proline mutation at residue 6 in β-strand A13 or at residue 9 in the linker between β-strands A and A′ (this study) destabilizes the I27 domain during unfolding by denaturants. However, the K6P mutation appeared to eliminate the unfolding intermediate with only β-strand A, not β-strand A′, detached from β-strand B during AFM-mediated unfolding.7 This observation was initially interpreted in such a way that the K6P mutation ruptured the hydrogen bonds bridging the AB β-strands, thereby destabilizing the N-terminal AB β-strand pair.7 However, later interpretation indicated that the K6P mutation stabilized the N-terminal structure, rendering β-strand A more resistant (to the level of β-strand A′) against detachment from β-strand B at low forces by AFM.4 Similarly, the I27 domain bearing the Y9P mutation required increased pulling forces for AFM-mediated unfolding as compared with wild-type.14 In mitochondrial protein import, the presence of K6P or Y9P mutations on chimeric I27 constructs fused to presequences (targeting sequences) decreased import rates, suggesting that these mutations rendered the N-terminal region resistant against unraveling by the mitochondrial import systems.4
In this study, we addressed the question of why the Y9P mutation stabilizes the I27 domain against unfolding by AFM and by mitochondrial import systems while it renders the I27 domain sensitive to denaturant-induced unfolding. Our work revealed that the proline mutation not only caused global effects on the I27 molecule, including the central hydrophobic core which destabilizes the overall molecular stability, but also leads to reorganization of the hydrophobic side chains near the N-terminus and stabilization of the N-terminal region.
Results
Proline substitution at residue 9 of I27 decreases the mitochondrial import rate
Because mitochondrial proteins are synthesized in the cytosol as precursor forms and imported into mitochondria posttranslationally, folded mature domains of precursor proteins often have to become unfolded again to pass through the protein-import channels. As unfolding of the mature domains is usually a rate-limiting step of the import process, analyses of the import rates in vitro offer information on the stability of the mature domains against vectorial unfolding exerted by the mitochondrial import system. We previously analyzed the effects of proline substitution at residues 4, 5, 6, 9, 11, 13, and 15 in the N-terminal segment of the I27 domain on the mitochondrial import rates of their presequence-attached fusion proteins, and found that the K6P and Y9P mutations decreased the import rates while the other N-terminal mutations did not.4 In this study, we first asked whether substitutions at residue 9 with residues other than proline might similarly lower import rates [Fig. 1(A)].
Using an in vitro reticulocyte lysate system, we synthesized radiolabeled fusion constructs consisting of pb2(80), the entire 80-residue presequence of yeast cytochrome b2, fused to the I27 domain (wild-type (WT) or with mutations Y9P, Y9G, Y9F, or Y9S). We then incubated them with isolated yeast mitochondria in the presence of ATP and ΔΨ, the membrane potential across the inner membrane, and quantified the imported fractions, which were protected against externally added protease (proteinase K) [Fig. 1(B), left panel]. As a control, the fusion proteins were not imported into mitochondria in the presence of valinomycin, which dissipates the ΔΨ [Fig. 1(B), left panel, rightmost lanes]. Imported fractions were plotted against incubation time, and import rates were obtained as initial slopes of each curve [Fig. 1(B), right panel]. Proline substitution of residue 9 decreased the import rates as observed previously, yet other substitutions at this site (Gly, Phe, or Ser) did not affect import rates [Fig. 1(C)]. Therefore, of the amino acid substitutions assayed, the N-terminal stabilization measured by the import rates is specific for proline.
Proline substitution at residue 9 leads to global destabilization of I27
To assess the basis for the specific effect of proline substitution at residue 9, we prepared recombinant I27 domains (WT and those bearing Y9P, Y9G, Y9F, or Y9S mutations) from E. coli cells. The far-UV CD spectra of these four I27 domains are nearly the same, suggesting that those four mutations did not cause any obvious changes in secondary structures [Fig. 2(A)].
Figure 2.
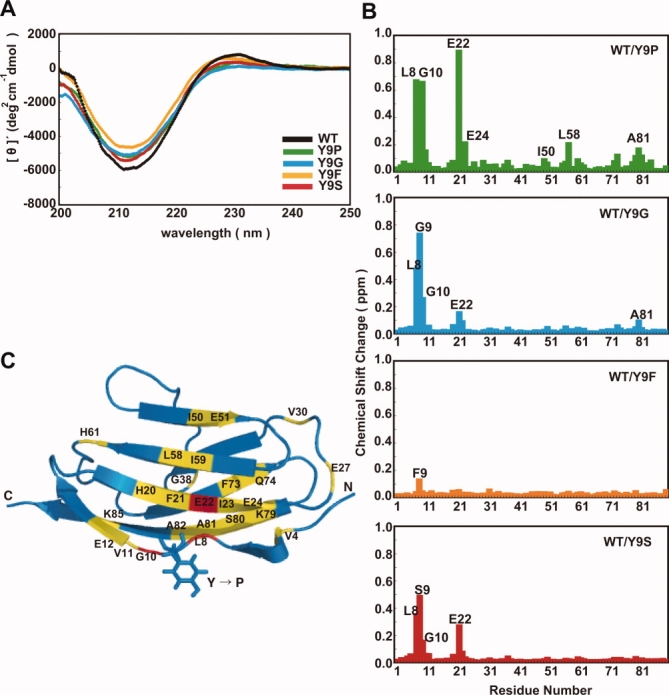
CD and NMR spectra and H-D exchange of I27 and Y9 mutants. (A) CD spectra of the I27 mutants. Far-UV CD spectra of 10 μM I27 mutants were recorded in 50 mM KPi (pH 7.4) at 25°C. (B) Chemical shift changes of backbone amides of I27 derivatives as compared with wild-type (WT) I27. Chemical shift changes were calculated in [1H, 15N]-HSQC spectra according to the equation, [Δδ(1H)2 + (Δδ(15N)/7)2]1/2. (C) Residues of the I27 derivatives were colored in ribbon form according to chemical shift changes as compared with WT I27: >0.50 ppm (red), 0.25–0.50 ppm (orange), 0.05–0.25 ppm (yellow) and 0-0.05 ppm (blue).
We then recorded [1H, 15N]-HSQC NMR spectra of 15N-labeled WT and mutant (Y9P, Y9G, Y9F, or Y9S) I27 domains and plotted chemical shift differences for backbone amide protons with WT I27 as a reference [Fig. 2(B)]. Replacement of Tyr9 with Phe hardly caused spectral perturbation of I27, indicating that the hydrophobic phenyl group, not the hydroxyl group, of Tyr9 contributes to its structural role. Spectral perturbation arising from the substitution of Tyr9 with Gly or Ser was also small and limited to the region around Tyr9 and Glu22 in β-strand B, which is in the vicinity of Tyr9, and remote residues were not affected by Y9G or Y9S mutations at all. Therefore, glycine and serine substitutions of Tyr9 induce only local structural perturbation in I27.
The Y9P mutant produced larger chemical-shift perturbations around both Tyr9 in the linker and Glu22 in β-strand B than did the other three I27 domain mutants (Y9G, Y9F, and Y9S) [Fig. 2(B)]. Moreover, Y9P-induced chemical shift perturbations were detected for the residues in the adjacent β-strand G, which forms a β-sheet pair with strand A′, and also in the more remote β-strands E and F [Fig. 2(C)]. This indicates that structural perturbation was transmitted from residue 9 to remote regions in both upper and lower β-sheet layers of the molecule, traversing several β-sheet strands.
Because the Y9P mutation affects the entire I27 molecule, we compared its global stability against unfolding by denaturants with those of WT and other mutant I27 domains. The free energy of unfolding (ΔGD) was determined using GdHCl-induced denaturation as monitored by CD measurements (Table I). Because the values of m did not differ significantly among wild-type and mutant I27 domains, ΔGD was calculated by fitting the data to ΔG = ΔGD − m[GdHCl], where m for WT I27 was used for the other I27 mutants. The obtained values showed that, although the Y9P, Y9G, Y9F, and Y9S mutants exhibited nearly the same CD spectra as WT I27, only the Y9P mutant is destabilized during unfolding by GdHCl.
Table I.
The Effects of Mutations on the Overall Stability of I27
| [GdHCl]50%a | ΔGDb (kcal mol−1) | |
|---|---|---|
| WT | 2.7 ± 0.1 | 5.9 ± 0.3 |
| Y9P | 1.7 ± 0.1 | 3.7 ± 0.2 |
| Y9G | 2.3 ± 0.1 | 5.1 ± 0.2 |
| Y9F | 2.5 ± 0.1 | 5.5 ± 0.5 |
| Y9S | 2.6 ± 0.1 | 5.7 ± 0.3 |
[GdHCl] at which 50% of the protein is denatured.
Calculated with m = 2.2 for WT was used for both WT and mutant I27 domains.
Although hydrogen-deuterium (H-D) exchange of buried NHs in proteins is not necessarily correlated with overall protein stability, buried backbone NHs and Trp indole NHs of the immunoglobulin-fold domain were found to exchange with solvent deuterons through global unfolding under physiological conditions.16 We thus compared the H-D exchange of backbone NHs between WT and Y9P I27 domains. We first confirmed that NMR chemical shift indexes that reflect secondary structures17 were similar for WT and Y9P I27 domains [Fig. 3(A)], as suggested by their similar CD spectra [Fig. 2(A)]. The residues involved in backbone hydrogen bonding also appeared similar for the WT and Y9P I27 domains because we observed nearly the same set of slowly exchanging NHs except for Val11 NH in [1H, 15N]-HSQC NMR spectra ∼300 min after H-D exchange [Fig. 3(B)]. We then measured H-D exchange of slowly exchanging backbone NHs in WT and Y9P I27 domains [Fig. 3(C)] and mapped the protection factors (PFs) against H-D exchange onto the I27 structure [Fig. 3(D)]. It is evident that the decreased stability of the entire Y9P mutant is also reflected in the accelerated H-D exchange of backbone NHs.
Figure 3.
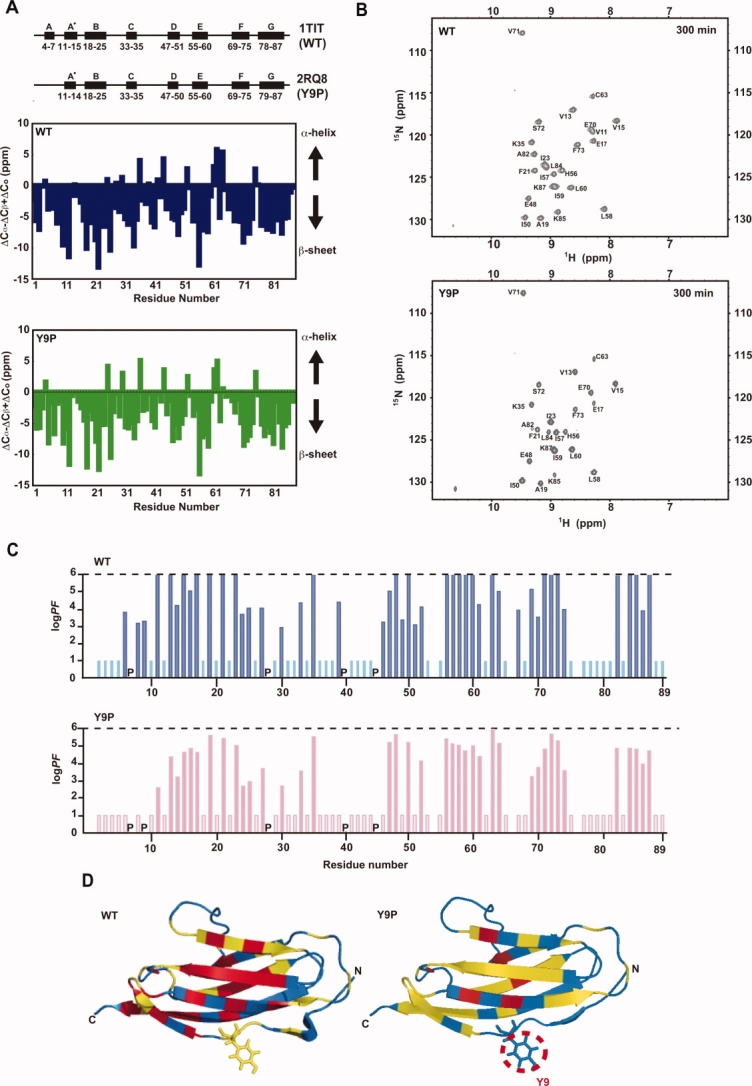
Secondary structures and H-D exchange of I27 and Y9P. (A) Upper panel: secondary structure diagrams of WT I275 and Y9P (estimated from the NMR structure [Table II] by PROCHECK). PDB ID codes for each structure are shown. Central and lower panels: NMR chemical shift indexes17 for WT5 and Y9P (this study) reflecting secondary structures. (B) Slowly exchanging NH signals for WT I27 (upper panel) and Y9P I27 (lower panel) observed at 300 min after H-D exchange. Note that the NH signal of V11 is observed only for WT I27, but not for Y9P. (C) PFs for H-D exchange of backbone NHs in WT and Y9P I27. Bars reaching logPF = 6, exchange too fast (log PF ≥ 6); bars with log PF = 1, exchange too slow (log PF ≤ 1). P, Pro residue. (D) PF (protection factor) values of H-D exchange of backbone NHs of each residue are mapped onto the molecular structure of I27. Protection factors are calculated from the equation, PF = kex(U)/kex(N), where kex(N) and kex(U) represent the rates of exchange for a given NH under folded and unfolded conditions, respectively.18 The exchange rates of fully solvent-exposed NHs, kex(U), were estimated on the basis of the primary structure, pH, and temperature using a program SPHERE (http://www.fccc.edu/research/labs/roder/sphere). Residues are colored according to PF values; log PF > 5.5 (red), log PF = 2.5–5.5 (yellow), and log PF < 2.5 (blue). Tyr9 is shown in stick form.
The central hydrophobic core region that contributes to the entire molecular stability of the I27 domain was identified previously.6,19 The core region consists of Phe21, Ile23, Trp34, His56, Leu58, Val71, and Phe73 [Fig. 4(A)]. The residues whose chemical shifts were affected by the Y9P mutations included some of those hydrophobic core residues, that is, Phe21, Ile23, Leu58, and Phe73 [Fig. 4(B,C)]. Therefore, as discussed later, the Y9P mutation likely causes structural perturbation of the central core region, thereby lowering the entire molecular stability.
Figure 4.
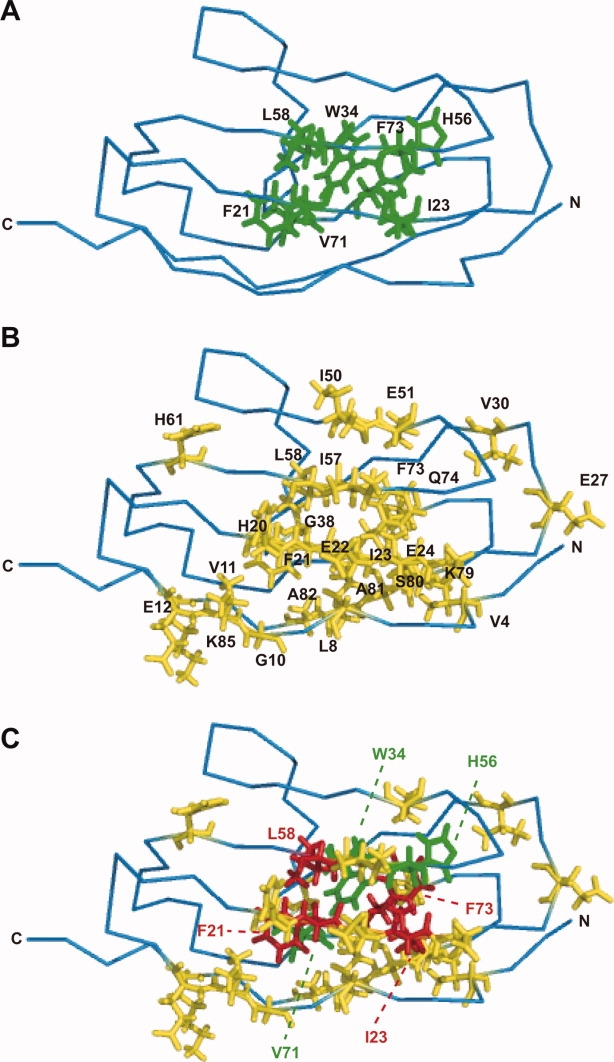
Mapping of the residues affected by the Y9P mutation. (A) The residues proposed to form a central hydrophobic core of the I27 domain are shown in stick form (green): F21, I23, W34, H56, L58, V71, and F73. (B) The residues that showed large chemical shift changes (>0.05 ppm) are shown in stick form (yellow): V4, L8, G10, V11, E12, H20, F21, E22, I23, E24, E27, V30, G38, I50, E51, I57, L58, H61, F73, Q74, K79, S80, A81, A82, and K85. (C) The residues included in (A) and (B) are shown in red: F21, I23, L58, and F73.
The Y9P mutation stabilizes the N-terminal labile region of I27
The aforementioned results suggest that the Y9P mutation in the I27 domain significantly destabilizes the global structure during unfolding by denaturants. However, AFM and mitochondrial protein import experiments suggest that the N-terminal region of Y9P is more resistant against unfolding by locally endowed mechanisms.4,14 What is the structural basis for those contrasting effects (i.e., N-terminal stabilization against local unfolding and global destabilization during unfolding by denaturants)?
To address this question, we determined the NMR structure of Y9P I27 on the basis of 1232 NOEs (Table II) and compared it with the previously reported structure of WT I27.5 At first glance, the overall main-chain folding of the determined NMR structure of Y9P is close to that of wild-type I27, with the RMSD between the two structures being 2.04 Å [Fig. 5(A)].
Table II.
Structural Statistics for 20 Final NMR Structures
| Distance restraints | |
| Total NOE | 1232 |
| Short-range | 320 |
| Medium-range | 411 |
| Long-range | 501 |
| Hydrogen bond restraintsa | 36 |
| Dihedral angle constraintsb | |
| φ | 44 |
| ψ | 44 |
| Mean CYANA target function | 0.98 |
| Ramachandran plot statistics (%) | |
| Most favored regions | 78.5 |
| Additionally allowed regions | 20.3 |
| Generously allowed regions | 1.0 |
| Disallowed regions | 0.2 |
| RMSDs to mean structure (residues) Å | |
| Backbone atoms (N, Cα, C′, O) | 0.48 ± 0.15 |
| All heavy atoms | 0.99 ± 0.15 |
Hydrogen-bond constraints were derived from H-D exchange experiments [Fig. 3(C)] and implemented as two upper (HN-O 2.5 Å, N-O 3.6 Å) and two lower (HN-O 1.5 Å, N-O 2.4 Å) distance constraints for each hydrogen bond.
Backbone φ and ψ dihedral constraints determined with TALOS.
Figure 5.
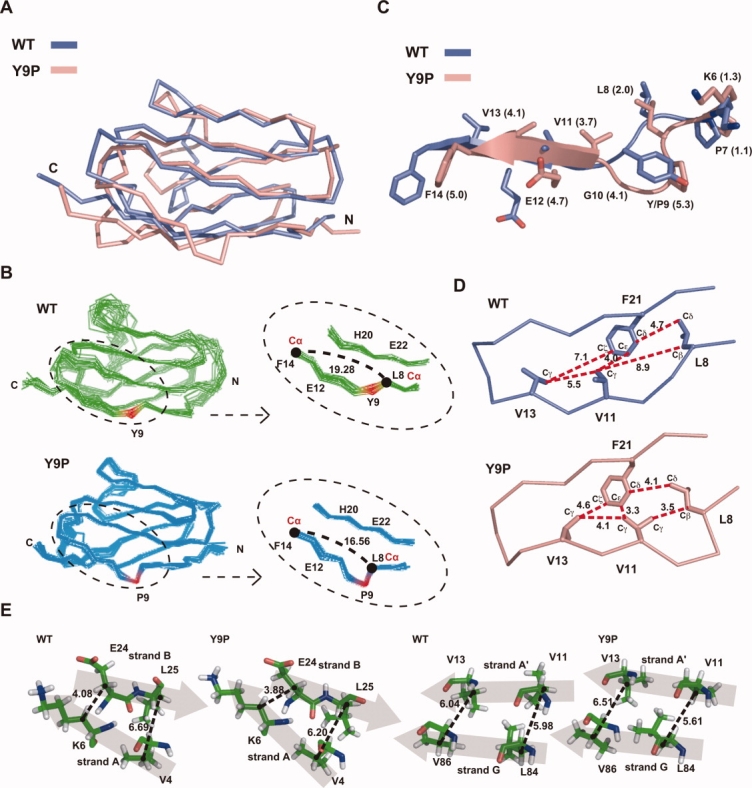
Effects of the Y9P mutation on the N-terminal structure of the I27 molecule. (A) Main-chain structures of WT (blue) and Y9P (pink) I27 are superimposed on the basis of secondary structure matching using a program CCP4 suite.20 RMSD of the two structures is 2.04 Å. (B) Overlay of the 24 (WT) and 20 (Y9P) NMR structures (left panels) and their close-up around residue 9 (right panels). Residue 9 is shown in red and distances between the Cα atoms of L8 and F14 are indicated. (C) The N-terminal structures around residue 9 of WT (blue) and Y9P (pink) I27 are superimposed as in (A). Distances between Cα atoms in the two structures are shown in parentheses. (D) Relative geometry of hydrophobic residues (L8, V11, V13, and F21) in the N-terminal region for the NMR structures of WT I27 and Y9P I27. Distances (Å) between the C atom pairs specified with broken lines are indicated. (E) Comparison of the relative geometry of the β-strand pairs A-B and A′-G between WT I27 and Y9P I27. Distances (Å) for V4Cβ–L25Cβ, K6Cβ–E24Cβ, V11Cβ– L84Cβ, and V13Cβ–V86Cβ are indicated. β-strands are shown with shaded arrows, and Cα atoms are indicated by dots.
However, closer examination of the structures of the N-terminal regions of WT and Y9P I27 domains revealed distinct structural differences between the two proteins. The Y9P mutation induced a backbone bend around residue 9 [Fig. 5(B), left panels, red], reducing the distance between the remote residue pair, Leu8 and Phe14 from 19.3 Å in WT I27 to 16.6 Å in the Y9P mutant [Fig. 5(B), right panels]. This structural bend pulls strand A′ toward the N-terminus by around 5 Å, which is illustrated in superposition of the N-terminal structures of WT and Y9P I27 [Fig. 5(C)]. As a result, the hydrophobic residues near Phe21 become closer to each other in the Y9P mutant compared with the WT I27 domain: distances between residues Leu8-Val11, Val11-Phe21, and Phe21-Val13 are 8.0, 4.0, and 7.1 Å, respectively, in wild-type I27, while those between the same residues become 3.5, 3.3, and 4.6 Å, respectively, in the Y9P mutant [Fig. 5(D)]. Therefore, the structural bend due to the Y9P mutation likely causes tighter packing of these residues in the hydrophobic core in the N-terminal region, which may contribute well to stabilization of the N-terminal region in Y9P I27 against vectorial unfolding.
Finally, we addressed the structural basis for the destabilization of the overall structure in the Y9P mutant. Although the NMR chemical-shift changes suggested that the Y9P mutation caused structural perturbations that destabilized the central core region, we could not detect a significant difference in the central core region structures (Phe21, Ile23, Trp34, Ile49, His56, Leu58, Val71, and Phe73) between WT and Y9P I27 [Fig. 6(A)]. However, when we viewed the WT and Y9P I27 molecules from different angles, we could detect a water-penetratable channel leading to the hydrophobic core in the interior of the molecules [Fig. 6(B), yellow]. The residues constituting the channels or cavities leading to the hydrophobic cores were identified as Gln33, Trp34, Lys35, Leu36, Pro40, Leu41, Thr42, Ala43, Cys47, Ile49, Leu58, Leu60, and Val71 for WT I27, and Phe21, Leu36, Lys37, Gly38, Gln39, Asp46, Leu58, Leu60, His61, Asn62, Cys63, Gln64, Leu65, Gly69, Glu70, Val71, Leu84, and Val86 for Y9P I27 [Fig. 6(B), cyan] by the program for identification and size characterization of surface pockets and occluded cavities, CASTp.21,22 The volume of the cavity was indeed expanded from 209.1 Å3 in WT I27 to 382.3 Å3 in Y9P I27. This expansion of the cavity was brought about by significant rearrangement of the channel-forming residues. In particular, the entrance of the channel is half-closed by the hydrophobic side chains of Leu36, Leu41, and Met67 in WT I27 [Fig. 6(C), left panel, red]. On the other hand, in Y9P I27, the side chains of Leu36 and Leu41 change their orientations away from the channel and Met67 is pulled apart from those two residues so that the channel interior becomes more accessible to bulk solvent [Fig. 6(C), right panel, red]. Is this structural perturbation directly caused by the Y9P mutation? As we described earlier, the Y9P mutation causes a shift of β-strand A′ toward the N-terminus. Because β-strand A′ is hydrogen bonded with the β-strand G, which is hydrogen bonded with another β-strand F, substantial movement of β-strand A′ causes structural distortion around the β-strands A′, G, and F. This distortion causes popping out of the loop N-terminal to β-strand F, so that Met67 in the loop moves away from the channel [Fig. 6(D)].
Figure 6.

Effects of the Y9P mutation on the central core domain of the I27 molecule. (A) Side chains of the central core residues are shown for WT (blue) and Y9P (pink) I27. Orientations of the side chains of F21 in both structures are adjusted for the best match. (B) The WT (blue) and Y9P (pink) I27 structures are drawn in space-filling form, showing the channels leading to the central hydrophobic cores (F21, I23, W34, H56, L58, V71, and F73; yellow) in the interiors. The residues constituting the channels or cavities leading to the hydrophobic cores are shown in cyan and the volumes of the cavities were calculated by the program CASTp21 available on the Internet (http://sts-fw.bioengr.uic.edu/castp/index.php). (C) L36, L41, and M67, which form the entrance of the channel in WT I27, are shown in red in the WT and Y9P I27 structures drawn as in (B). (D) The same view of the WT (blue) and Y9P (pink) I27 structures as in (B) are shown in ribbon form. Side chains of the central hydrophobic core residues (yellow) and channel forming residues (L36, L41, and M67; red) are indicated in stick form.
Discussion
In this study, we have revealed the structural basis for the opposite effects of the point mutation, Y9P, on the stability of the I27 domain against global unfolding caused by denaturants and N-terminal unraveling by AFM and mitochondrial import systems. Substitution of Pro for Tyr at position 9 caused a bend in the backbone structure, although the peptide bond of Pro9 remained in the trans-conformation. This conformational change induced a structural change that was transmitted through the β-sheet organization and side-chain interactions, reaching the core region around Trp34, as shown by NMR chemical shift perturbation [Fig. 2(C)]. This implicated conformational change was not obvious in the determined NMR structure of Y9P I27 [Fig. 5(A)]; however, the conformational change around residue 9 did cause structural distortion around the β-strands A′, G, and F, which apparently increased the solvent accessibility of the channel leading to the central hydrophobic core [Fig. 6(B,C)]. The resultant destabilization of the overall structure was reflected in chemical-denaturant induced unfolding (Table I) and H-D exchange of backbone amide protons [Fig. 3].
On the other hand, the bend of the backbone structure around residue 9 caused local rearrangement of the hydrophobic side-chain packing in the N-terminal region through the shift of β-strand A′ [Fig. 5(B–D)]. This rearrangement would likely enhance hydrophobic interactions involving the residues in β-strand A′ (Val11 and Val13) and β-strand B (Phe21), which belong to different β-sheet layers. In contrast, relative geometry of the β-strand pair A-B in the same β-sheet layer and that of the β-strand pair A′-G in the other β-sheet layer did not differ significantly between WT I27 and Y9P I27 [Fig. 5(E)]. Therefore, the possible stabilized N-terminal hydrophobic interactions of Y9P I27 may well resist against disruption of the inter-layer interactions, thereby stabilizing the Y9P mutant against N-terminal unraveling by AFM or the mitochondrial import machinery. Of course, the revealed N-terminal structural rearrangement could be compatible with, if any, other structural mechanisms that contribute to the enhanced local stability in the N-terminal region.
The previous studies indicated that the K6P mutation also stabilized the I27 domain against N-terminal unraveling associated with mitochondrial import and mechanical unfolding by AFM.4,14 Our preliminary analyses suggested that the K6P mutation, like the Y9P mutation, both stabilizes the N-terminal region and destabilizes the overall structure against unfolding by denaturants (Oguro et al., unpublished). However, in contrast with the Y9P mutation, the K6P mutation generates two distinct sets of NMR signals, indicating that the K6P mutant undergoes a slow conformational equilibrium on the NMR time scale, hampering the detailed NMR structural analyses as conducted in this study. Determination of the X-ray crystal structures of both conformations of the K6P mutant will reveal if the K6P mutation stabilizes the N-terminal region by the mechanism similar to the case of Y9P mutant.In conclusion, this study revealed the structural basis for the opposite effects of a single mutation (Y9P) on the stabilities of the I27 domain, that is, N-terminal stabilization against local vectorial unfolding and global destabilization during unfolding by denaturants. There may be different mechanisms to cause opposite effects on local and global stabilities, which should be studied in different mutant proteins in future.
Materials and Methods
I27 mutants and their fusion proteins
The genes for I27 mutants and I27 mutant fusion proteins with the N-terminal 80 residues of the cytochrome b2 precursor were derived from those used in the previous study.4 The Y9G, Y9F, and Y9S mutations were introduced into the gene for I27 or pb2(80)-I27 by standard PCR-directed mutagenesis. The genes for pb2(80)-I27 mutant fusion proteins in the plasmid pGEM-4Z were used for in vitro translation and those for I27 mutants in the plasmid pET21a(+) were used for protein purification from E. coli cells. The recombinant Y9P, Y9G, Y9F, and Y9S proteins were purified from E. coli cells as described previously.13
NMR measurements
NMR spectra were recorded at 298K on a Bruker AVANCE600 NMR spectrometer. A series of three-dimensional double and triple resonance (CBCANH, CBCA(CO)NH, HNCO, HN(CA)CO) experiments were performed for spectral assignments of the Y9P protein. NMR samples were either in 90% H2O/10%D2O and contained 50 mM KPi, pH 7.4, and the protein concentration was 0.4 mM. Distance constraints for the structure determination were obtained from 3D 15N-edited [1H, 1H]-NOESY (H2O), 15N-NOESY-HSQC, and 13C-NOESY-HSQC spectra of Y9P. Slowly exchanging amide protons were identified by acquiring a series of [1H, 15N]-HSQC spectra after dissolving the lyophilized protein in 99.9% D2O. Structure calculations were performed using the program CYANA.23 Details of NMR structural determination of the Y9P protein are described in Table II. The coordinates of ensembles of the 20 conformers have been deposited in the Protein Data Bank (http://www.pdb.org; PDB ID code 2RQ8).
CD measurements
Circular dichroism (CD) spectra were recorded on a JASCO J-720 spectropolarimeter at 25°C, using a 0.2 cm path-length cell.
Import assay
pb2(80)-I27 fusion proteins were synthesized in a rabbit reticulocyte lysate in the presence of [35S]methionine and subjected to in vitro import into isolated yeast mitochondria as described previously.4
Acknowledgments
The authors thank J. M. Fernandez for the I27 gene, Nathan N. Alder for careful reading of the manuscript, and the members of the Endo laboratory for discussions and comments.
Glossary
Abbreviations:
- AFM
atomic force microscopy
- CD
circular dichroism
- GdHCl
guanidinium hydrochloride
- NOE
nuclear Overhauser effect
- PF
protection factor.
References
- 1.Prakash S, Matouschek A. Protein folding in the cell. Trends Biochem Sci. 2004;29:593–600. doi: 10.1016/j.tibs.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Forman JR, Clarke J. Mechanical unfolding of proteins: insights into biology, structure and folding. Curr Opin Struct Biol. 2007;17:58–66. doi: 10.1016/j.sbi.2007.01.006. [DOI] [PubMed] [Google Scholar]
- 3.Carrion-Vazquez M, Oberhauser AF, Fisher TE, Marszalek PE, Li H, Fernandex JM. Mechanical design of proteins studied by single-molecule force spectroscopy and protein engineering. Prog Biophys Mol Biol. 2000;74:63–91. doi: 10.1016/s0079-6107(00)00017-1. [DOI] [PubMed] [Google Scholar]
- 4.Sato T, Esaki M, Fernandez JM, Endo T. Comparison of the protein unfolding pathways between mitochondrial protein import and atomic force microscopy measurements. Proc Natl Acad Sci USA. 2005;102:17999–18004. doi: 10.1073/pnas.0504495102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Improta S, Politou AS, Pastore A. Immunoglobulin-like modules from titin I-band: extensible components of muscle elasticity. Structure. 1996;4:323–337. doi: 10.1016/s0969-2126(96)00036-6. [DOI] [PubMed] [Google Scholar]
- 6.Fowler SB, Clarke J. Mapping the folding pathway of an immunoglobulin domastructural detail from phi value analysis and movement of the transition state. Structure. 2001;9:355–366. doi: 10.1016/s0969-2126(01)00596-2. [DOI] [PubMed] [Google Scholar]
- 7.Marszalek PE, Lu H, Li H, Carrion-Vazquez M, Oberhauser AF, Schulten K, Fernandez JM. Mechanical unfolding intermediates in titin modules. Nature. 1999;402:100–103. doi: 10.1038/47083. [DOI] [PubMed] [Google Scholar]
- 8.Fowler SB, Best RB, Toca-Herrera JL, Rutherford TJ, Steward A, Paci E, Karplus M, Clarke J. Mechanical unfolding of a titin Ig domastructure of unfolding intermediate revealed by combining AFM, molecular dynamics simulations, NMR and protein engineering. J Mol Biol. 2002;322:841–849. doi: 10.1016/s0022-2836(02)00805-7. [DOI] [PubMed] [Google Scholar]
- 9.Schatz G, Dobberstein B. Common principles of protein translocation across membranes. Science. 1996;271:1519–1526. doi: 10.1126/science.271.5255.1519. [DOI] [PubMed] [Google Scholar]
- 10.Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Neupert W, Herrmann JM. Translocation of proteins into mitochondria. Ann Rev Biochem. 2007;76:723–749. doi: 10.1146/annurev.biochem.76.052705.163409. [DOI] [PubMed] [Google Scholar]
- 12.Endo T, Yamano K. Multiple pathways for mitochondrial protein traffic. Biol Chem. 2009;390:723–730. doi: 10.1515/BC.2009.087. [DOI] [PubMed] [Google Scholar]
- 13.Oguro T, Yagawa K, Momose T, Sato T, Yamano K, Endo T. Structural stabilities of different regions of the titin I27 domain contribute differently to unfolding upon mitochondrial protein import. J Mol Biol. 2009;385:811–819. doi: 10.1016/j.jmb.2008.10.076. [DOI] [PubMed] [Google Scholar]
- 14.Li H, Carrion-Vazquez M, Oberhauser AF, Marszalek PE, Fernandez JM. Point mutations alter the mechanical stability of immunoglobulin modules. Nat Struct Biol. 2000;7:1117–1120. doi: 10.1038/81964. [DOI] [PubMed] [Google Scholar]
- 15.Huang S, Ratliff KS, Schwartz MP, Spenner JM, Matouschek A. Mitochondria unfold precursor proteins by unraveling from their N-termini. Nat Struct Biol. 1999;6:1132–1138. doi: 10.1038/70073. [DOI] [PubMed] [Google Scholar]
- 16.Kawata Y, Goto Y, Hamaguchi K, Hayashi F, Kobayashi Y, Kyogoku Y. Hydrogen-exchange kinetics of the indole NH proton of the buried tryptophan in the constant fragment of the immunoglobulin light chain. Biochemistry. 1988;27:346–350. doi: 10.1021/bi00401a052. [DOI] [PubMed] [Google Scholar]
- 17.Wishart DS, Sykes BD. The 13C chemical shift index: a simple method for the identification of protein secondary structure using 13C chemical-shift data. J Biomol NMR. 1994;4:171–180. doi: 10.1007/BF00175245. [DOI] [PubMed] [Google Scholar]
- 18.Marmorino JL, Auld DS, Betz SF, Doyle DF, Young GB, Pielak GJ. Amide proton exchange rates of oxidized and reduced Saccharomyces cerevisiae iso-1-cytochrome c. Protein Sci. 1993;2:1966–1974. doi: 10.1002/pro.5560021118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geierhaas CD, Paci E, Verdruscolo M, Clarke J. Comparison of the transition states for folding of two Ig-like proteins from different superfamilies. J Mol Biol. 2004;343:1111–1123. doi: 10.1016/j.jmb.2004.08.100. [DOI] [PubMed] [Google Scholar]
- 20.Collaborative Computational Project 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 21.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucl Acids Res. 2006;34:W116–W118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang J, Edelsbrunner H, Woodward C. Anatomy of protein pockets and cavities: measurement of binding site geometry and implications for ligand design. Protein Sci. 1998;7:1884–1897. doi: 10.1002/pro.5560070905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Güntert P, Mumenthaler C, Wüthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]