

Abstract
In this work, we describe two novel approaches to utilize the dynamic structure information implicitly contained in large crystal structure data sets. The first approach visualizes both consistent as well as variable ligand-induced changes in ligand-bound compared with apo protein crystal structures. For this purpose, information was mined from B-factors and ligand-induced residue displacements in multiple crystal structures, minimizing experimental error and noise. With this approach, the mechanism of action of non-nucleoside reverse transcriptase inhibitors (NNRTIs) as an inseparable combination of distortion of protein dynamics and conformational changes of HIV-1 reverse transcriptase was corroborated (a combination of the previously proposed “molecular arthritis” and “distorted site” mechanisms). The second approach presented here uses “consensus structures” to map common binding features that are present in a set of structures of NNRTI-bound HIV-1 reverse transcriptase. Consensus structures are based on different levels of structural overlap of multiple crystal structures and are used to analyze protein–ligand interactions. The structures are shown to yield information about conserved hydrogen bonding interactions as well as binding-pocket flexibility, shape, and volume. From the consensus structures, a common wild type NNRTI binding pocket emerges. Furthermore, we were able to identify a conserved backbone hydrogen bond acceptor at P236 and a novel hydrophobic subpocket, which are not yet utilized by current drugs. Our methods introduced here reinterpret the atom information and make use of the data variability by using multiple structures, complementing classical 3D structural information of single structures.
Keywords: ligand-induced conformational changes, pocket characterization, flexibility, B-factors, working mechanism
Introduction
The availability of crystal structures in both public archives [such as the Protein Data Bank (PDB)1] as well as proprietary repositories (such as within pharmaceutical companies) is growing at a phenomenal speed. Crystal structures can provide a wealth of experimental data to the scientist, but the information obtained is static and cannot accurately depict the actual dynamic properties of the protein and its ligand.2 Additional information that can provide insights into the dynamics is implicitly contained within a larger group of crystal structures of the same protein, as this set of structures captures (part of) the dynamically accessible conformation space of the protein. The challenge resides in how to mine this wealth of data. In the work presented here, we will introduce two different methods for mining large sets of ligand–protein crystal structures.
Our first approach mines data from B-factor values and ligand-induced residue displacements. In a single structure, information concerning the dynamics is provided by B-factors, which reflect the fluctuations of atoms around their average position in the crystal.3 However, B-factors are also influenced by experimental error, temperature and crystal quality. Therefore, it is per se difficult to distinguish this dynamic information from measurement errors and artifacts in situations where only a single structure is studied. The utilization of multiple structures can alleviate this problem,4 provided that the B-factor values are normalized before comparing different structures.5
A closely related second approach is mapping ligand-induced changes in residue orientation by comparing apo structures with ligand bound structures.6 Similarly, when only one pair of structures, that is, one apo structure and one ligand-bound structure are compared, it is difficult to distinguish useful information from experimental artifacts. Our hypothesis governing the current work was that the simultaneous analysis of several apo and ligand-bound structures will lead to a better understanding of information common to all structures, highlighting trends and distinguishing them from artifacts or noise.
The third approach is to analyze the common spatial and pharmacophoric interaction properties of the available crystal structures, which we named “consensus structures.” Existing approaches to derive a consensus structure have been aimed at mapping common features of a group of known ligands and creating a consensus pharmacophore.7–9 However, this ligand-based approach does not take any protein information into account, resulting in the inability of such approaches to extract protein-related information. Furthermore, it has already been shown that consensus information derived from several protein crystal structures can indeed extrapolate beyond the original data.10,11 Consensus structures are based on the aligned ligand binding pockets of multiple ligand-bound crystal structures and allow analysis of the shape and pharmacophoric patterns present in all of the structures, as well as the differences between them. Consensus structures combine information about different binding site geometries to identify key features responsible for ligand binding. Isocontour consensus surfaces that visualize features common to a minimum percentage of the total structures used are obtained from these consensus structures and allow visualization of the degree of conservation of the protein or protein features.
In a case study, we applied the above methods to a data set consisting of human immunodeficiency virus Type 1 reverse transcriptase (HIV-1 RT) structures in complex with non-nucleoside reverse transcriptase inhibitors (NNRTIs). HIV-1 RT is one of the most studied drug targets known today and was the first target identified in the treatment of infection with HIV-1.12 As a result, a large number of crystal structures are available in the PDB, rendering this target suitable for our first case study.1 NNRTIs are noncompetitive inhibitors of HIV-1 RT acting on an allosteric binding pocket with high specificity. However, the nature of HIV-1 replication leads to a quick onset of resistance of HIV-1 toward NNRTIs.13,14 This resistance forms an increasing problem in the treatment of HIV-1 infection and is mainly caused by point mutations in the protein.13–16 HIV-1 RT is a heterodimer, consisting of a large 560-residue subunit (p66) and a smaller 440-residue subunit (p51). The catalytic site on the p66 unit consists of a conserved YMDD motif and a third aspartic acid (residues D110, Y183, M184, D185, and D186). The rather flexible pocket is not present in the apo form of the enzyme and is only created upon binding of an NNRTI to HIV-1 RT, thus reducing enzyme flexibility.17 Furthermore, it has been shown that the flexibility of HIV-1 RT depends on its ligation state, and it is increased upon DNA binding.18
The information mined from the B-factors and ligand-induced changes of the HIV-RT crystal structures enabled us to explore the mechanism of NNRTI inhibition in more detail. The consensus structure analysis resulted in the identification of conserved hydrophobic and hydrogen bonding features that provided new insights and design options for HIV-1 RT inhibition by NNRTIs.
Results and Discussion
B-factor analysis
Firstly, normalized B-factors of NNRTI-bound enzymes were compared with the corresponding values in apo enzymes. Our results show a significant decrease in B-factors of the entire pocket upon NNRTI binding, indicating smaller fluctuations and a stiffer protein backbone in this region. This reduction in flexibility is in agreement with earlier MD simulations.17,19 Although B-factors of most residues respond variably upon ligand binding (see Fig. 1), the loop containing two of the catalytic site residues (D185 and D186) and the neighboring residues, that is, residues 181 to 188, show a significant decrease in flexibility (see also Supporting Information Fig. S1). The restriction of conformational change of this loop by NNRTIs, which was proposed to be the mechanism of action by Das et al.,20 is fully supported by our results on the significantly decreased B-factors of this region. In contrast, the region between residues L228 and L234 undergoes a consistent average increase in flexibility. This region contains the “primer grip” residue M230 and the increase would be unfavorable for its function retaining the growing DNA strand. A principal component analysis (PCA) of the normalized B-factor distributions over the residues shows that all structures form a cluster, which shows significant diversity (but no outliers) and that the three apo structures are nearest neighbors (Supporting Information Fig. S2). A heat map of the normalized B-factor profiles of all structures can be found in Supporting Information Figure S3. To compare our findings to DNA-bound protein, all crystal structures were compared with three different DNA-bound structures. However, contrary to the consistent profile observed in the ligand bound or apo structures the overall B-factor profile differs between these DNA-bound structures. Therefore, we do not have enough data to draw firm conclusions on flexibility changes upon DNA binding. When we separated all NNRTI-bound structures into wild type (wt) and mutated structures a trend was observed that resistance-conferring point mutations lead to a partial restoration of flexibility of the catalytic site region (Supporting Information Fig. S1). This trend is consistent with the findings of Zhou et al.17
Figure 1.
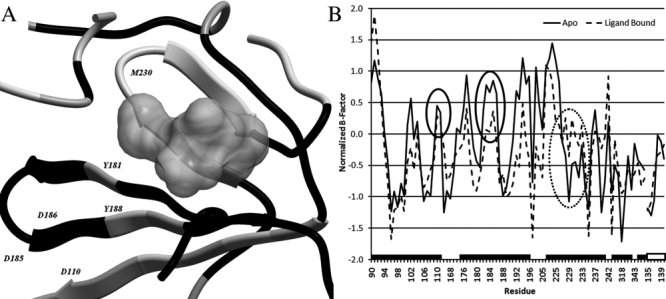
Changes in average B-factor as a result of ligand binding projected on the backbone ribbon. White residues indicate an increase (≥0.2), gray indicates no change (between 0.2 and −0.2), and black indicates a decrease (≤−0.2) (PDB:1FK9). (A) The gray volume represents the NNRTI Efavirenz. The entire pocket region has a lower average normalized B-factor in the ligand-bound compared to the apo form while the profile remains comparable (B). The catalytic residues undergo a decrease in B-factor (black circles), while the primer grip region containing residue W229 and M230 undergoes an increase upon ligand binding (dashed black circle). The black bars on the horizontal axis indicate continuous residues, the white filled bar indicates the residues on the p51 subunit. Each tick marks a separate residue the precise residue numbers can be found in the materials and methods, p66 and p51 have been separated by a gap.
This type of flexibility information is useful when implementing protein flexibility in, for example, docking calculations.21,22 Awareness about the flexible residues enables focusing computational expense on only these parts of the protein, as still allowing induced fit to take place to the required degree.
Ligand induced displacement analysis
We next analyzed the residue displacements resulting from ligand binding. This analysis confirms that all NNRTIs induce a similar binding pocket into the protein when compared to the apo structure. This holds true for both the backbone carbon alpha atoms as well as the movement of the center of mass of the residues (Supporting Information Figs. S4–S11). The only exception is observed in the Capravirine structure; however, as we only had one structure available, we cannot confirm this to be an effect characteristic for the ligand or for this particular crystal structure. Hence, this structure was removed from further analysis. Next, the residue center of mass absolute displacement distances were examined. Figure 2(A) depicts the pocket backbone colored according to the values obtained from the calculation of the displacement upon ligand binding. A PCA analysis of the displacement vectors shows that the ligand-bound structures cluster together, even more than do the apo structures (Supporting Information Fig. S4), confirming the formation of a common pocket upon ligand binding.
Figure 2.
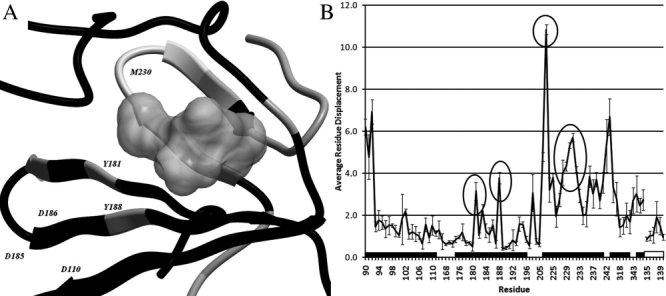
The backbone of the NNRTI pocket, colored according to the average residue displacement (PDB:1FK9). Black indicates a small average displacement, ≤2 Å, gray indicates a medium displacement, between 2 and 4 Å, and white indicates a large displacement ≥4 Å (A). The gray volume represents the NNRTI Efavirenz. Residues Y181, Y188, K223, and M230 all undergo a relatively large movement upon ligand binding (B, black circles). The residues between L228 and L234 all undergo a large displacement upon ligand binding, this correlates with the results from the B-factor analysis. The residue axis is labeled identically to Figure 1.
Summarizing our findings for NNRTI binding, firstly the shift of the catalytic acid residues in Figure 2(B) is conserved. Secondly, the known shifts as a result of the flip of residues Y181, Y188 and the known upward movement of W229 and M230 are confirmed for all NNRTIs.23 The entire β12 sheet (P225–P236) known to be in contact with the growing DNA template via residue M230,20,24–26 is displaced upward into the DNA binding groove, adapting to the ligand upon binding. However, the displacement of the sheet is a rotation pivoting around residues P226 and L234/H235, which remain relatively in place. This suggests how the sheet adapts itself to the size of the bound NNRTI as moving the primer grip away from the nucleotide binding region. The mutations present in the mutated RT structures do not appear to have a significant influence on residue movement. (For detailed heat maps indicating the shift per residue see Supporting Information Figs. S6–S10) Interestingly, the distance measured does not necessarily correlate with these residues being involved in resistance mutations. However, it is striking that some of the residues that are detected to move relatively large distances in all structures are in known locations for resistance conferring mutations. Because these residues undergo the largest changes resulting from NNRTI binding, changes occurring in these residues are most likely to perturb NNRTI binding.
Upon DNA binding, the position of the catalytic site loop carrying residues Y181 through Y188 is consistently shifted (Fig. 3). The loop maintains the overall conformation suggesting a hinge-like movement during catalysis. Furthermore, the known shift of the catalytic loop induced by NNRTI binding is in fact opposite to the path of the downward movement induced by DNA binding. (Supporting Information Figs. S6–S11) We compared the shift on each cartesian axis as a result of either DNA binding or NNRTI binding. The figures clearly show that the catalytic loop and especially residues 184–186 move in an opposite direction when compared to the apo structures in both the case of Cα and centroid movement on both the x- and z-axes. In the case of the y-axis, the result is less pronounced.
Figure 3.
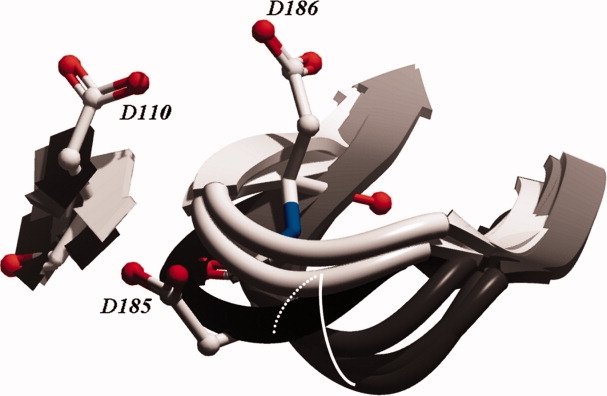
Overview of the changes occurring at the catalytic site as a result of DNA binding (three gray ribbons), NNRTI binding (two black ribbons) compared with the apo position (two white ribbons). The three aspartic acids that are visible are part of the DNA bound conformation (1RTD). In the apo structures, these residue side chains are placed similarly, however in the NNRTI bound structures D186 points toward D110, D110 points outward, and D185 points downward. Upon DNA binding, the loop containing the residues moves along the path indicated by the white curve, in the presence of an NNRTI the loop moves along the path indicated by the dashed white curve.
NNRTI working mechanism
Combining the above findings on the movement and flexibility of HIV-RT residues upon binding of an NNRTI allows us to elaborate on previously proposed working mechanisms of NNRTIs. The working mechanism has been proposed to be either the result of a catalytic site distortion26–30 or a more rigid protein.19,29,30 From our flexibility analysis, we observe that the binding of an NNRTI to HIV-1 RT leads to a shift in flexibility of residues around the binding site, with mobile residues become more rigid, whereas rigid residues becoming more mobile. In addition, upon NNRTI binding the catalytic triad residues and their neighboring residues, D110-V111 and Y181-Y188, but also the primer grip region, M230 and surrounding residues, undergo a consistent displacement opposite to the displacement induced by DNA binding. As a result the conformation of the catalytic loop is distorted, enlarging the distance between primer grip and the nucleotide binding site. These changes were found to be consistent among all crystal structures studied. At the catalytic site, the 4 Å average movement of the aspartic acids is a large distance as the three-dimensional orientation of the catalytic motif and the nucleotide is crucial in catalyzing the reaction. Mendieta et al.31 have found that the Mg2+ ions stabilize the catalytic complex and lower the catalytic attack distance to 3 Å. These essential Mg2+ ions were missing in all crystal structures containing an NNRTI, indicating that the changed orientation of the aspartic acids might not be able to contain these ions.
The large movement of the primer grip away from the catalytic site upon NNRTI binding and the increase in flexibility are consistent among all structures. Both of these changes are likely to lead to a decreased reaction rate as they inhibit the function of retaining the DNA strand. Therefore, we conclude that the NNRTIs disrupt both protein conformation and dynamics and that it is this combination that inhibits the function of HIV-RT. Thus, we propose a working mechanism for NNRTIs that is a combination of both the rigid protein, distorted-catalytic-site and distorted primer grip region theories. The bound NNRTI stabilizes the region surrounding the catalytic site in a conformation not able to perform catalysis. The effects on protein conformation and dynamics cannot be seen independently as one directly influences the other. Therefore, it could be speculated that an increase in flexibility as a result of point mutations allows the primer grip and catalytic site to move closer together restoring catalytic activity. As a result this could lead to resistance of the particular HIV-RT mutant form. Our results support this theory.
When this manuscript was revised, Paris et al.32 published a large scale comparison of NNRTI crystal structures. Although their superposition is based on only a subset of residues and we left out the structures where an NNRTI was soaked out of the pocket, their main conclusions are generally in line with ours. Moreover, our results indicate that NNRTI binding influences not only primer grip movement but also distortion of the catalytic site and changes in protein dynamics and that these are all in fact required for inhibitory activity.
Consensus structures
In the second part of our work on mining information from multiple crystal structures, we present the results from the consensus structures creation. The difference between two extreme situations of conservation values is visualized in Figure 4. The surface visualizing low conservation is shown as a green wire grid. This surface shows all parts of the three-dimensional space covered by at least 10% of the protein structures, including different side-chain orientations. This surface features a rather large protein volume, and consequently the empty pocket volume in this map is smaller than in the high-conservation map. The low-conservation surface thus describes the unity of conformational space accessible to the protein in any one of the dataset structures. The surface visualizing high conservation is shown as a solid green mesh. This surface contains the volume covered by at least 50% of the structures. It suggests the volume to which the protein binding pocket can be extended, describing the most conserved side-chain-occupied space. This volume does not occur in any of the individual structures, and this high-conservation surface represents the largest possible binding pocket a hypothetical ligand could occupy. In addition, combining high and low conservation surfaces in one view can be used to locate regions of flexibility by visually comparing highly conserved side-chain orientations, where there is little difference between high- and low-conservation surfaces, to side-chains that can move more freely, showing a large difference between high- and low-conservation surfaces.
Figure 4.

Difference between surfaces that represent low conservation and high conservation. All the grid points that are occupied in at least 10% of the structures are shown as a green wire grid (low conservation points). All the grid points that represent at least 50% of the structures are shown as a green solid mesh (high conservation points). Both represent an extreme value and are overlaid on the PDB structure 1FK9, depicted with its ligand Efavirenz. The wire grid surface shows all possible side chain locations (low conservation), whereas the solid mesh surface only shows the most conserved side chain location.
When the consensus creation procedure is repeated on our set of NNRTIs, the surfaces visualize the space taken up by the different NNRTIs within HIV-1 RT. Here the low-conservation surface visualizes the unity of conformational space accessible to the NNRTIs in any one of the dataset structures, and a larger volume than present in any of the individual structures. The high-conservation surface visualizes the common volume that is used by all NNRTIs.
Consensus pocket
Van der Waals (VdW) consensus structures can visualize common features present in all structures, including crystal structures and optionally even homology models carrying point mutations.33 The resulting consensus pocket shape can be regarded as a target shape for high affinity ligands because it represents the maximal theoretically accessible volume. As the highly flexible pocket adapts to each different NNRTI and there is no standardized wild-type pocket, consensus structures can provide this standardized pocket. Based on the degree of conservation of the surfaces, an estimate can be made of the space available for NNRTI binding and this can be related to the actual volume of NNRTIs. From Table I, it can be concluded that second-generation NNRTIs, Rilpivirine, Capravirine, and Delavirdine make better use of the maximal theoretical available space (66%, 69%, and 75%, respectively). Given our consensus structures, even larger NNRTIs than Delavirdine are theoretically possible. Coincidentally, as finalizing this manuscript, this has been experimentally confirmed by Sweeney et al.34
Table I.
Volumetric Information Relating the Size of the Consensus Structures to the Size of NNRTIs
| Volumetric object | Volume (Å3) |
|---|---|
| NNRTI consensus (50% occupancy) | 103 |
| NNRTI consensus (30% occupancy) | 296 |
| NNRTI consensus (10% occupancy) | 578 |
| 739W94 | 267 |
| Nevirapine | 274 |
| 1051U91 | 275 |
| Efavirenz | 305 |
| HEPT | 311 |
| Alpha-Apa | 315 |
| PETT2 | 315 |
| PETT1 | 333 |
| HBY097 | 334 |
| MKC442 | 336 |
| UC781 | 341 |
| 9-Chloro-TIBO | 344 |
| 8-Chloro-TIBO | 344 |
| TNK6123 | 364 |
| Rilpivirine | 379 |
| TNK651 | 391 |
| Capravirine | 400 |
| Delavirdine | 432 |
Overview of the volumes contained within the different NNRTI consensus structures compared with the volume of NNRTIs in their bound confirmation as it was obtained from the crystal structure.
New pocket features
The combination of consensus structures of the pocket with consensus structures of the combined NNRTIs can pinpoint locations within the pocket that are not or inefficiently used by current drugs, shown in Figure 5. The figure was created combining a VdW surface of the binding pocket and a VdW NNRTI surface, both visualizing low conservation. A small conserved sub-pocket is visible in the low conservation protein surface, which means that it is present in all crystal structures. Located between residues P95, P97, L100, and W229 on the “A” chain it might be the appropriate place to add an additional methyl group to a ligand to fill this lipophilic pocket. None of the NNRTIs from the studied crystal structures contacts the residues surrounding the sub-pocket, as illustrated for Efavirenz. Analogously, VdW consensus structures can also be applied to other drug targets in the rational design of new ligands, leading to derivatives that occupy nonused pocket space, thus maximizing the contact surface and interaction potential.
Figure 5.
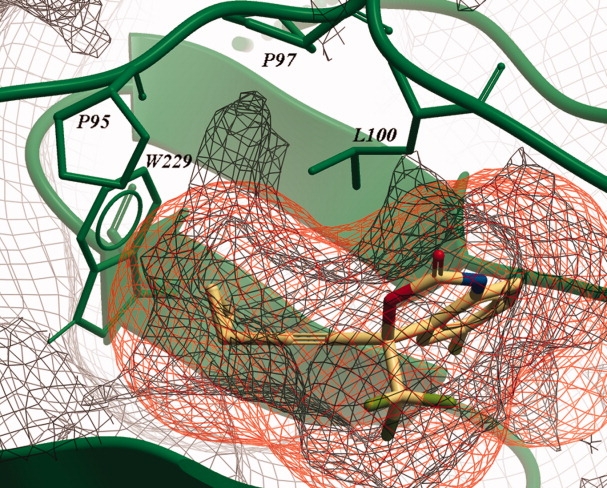
The protein VdW consensus surface is shown at 10% occupancy (green wire grid) and is superimposed on the 10% occupancy NNRTI VdW consensus surface (yellow wire grid). The PDB structure 1FK9 is shown in green and with yellow carbon atoms its ligand Efavirenz. Located between the residues P95, P97, L100, and W229 is a sub-pocket unused by current NNRTIs. This sub-pocket is in fact an extension of the known cavity located between Y181, Y188, and W229.
Hydrogen bonding information
The surfaces created based on hydrogen bonding (HB) occupancy visualize the conserved HB potential within the pocket or located on the NNRTIs, giving information about HB potential that can be exploited in drug discovery projects. Similar to VdW surfaces, variation of the conservation level identifies conserved HB locations. On our HIV-RT dataset for example, known backbone-NH HB interactions at K101 and K103 [Fig. 6(A)] combined with the known backbone carbonyl HB at K101 and electrostatic interactions at H235 [Fig. 6(B)] were confirmed using the consensus structures. Furthermore, a conserved carbonyl HB acceptor not used by NNRTIs can be identified at K102, although this acceptor is accessible in only a few of the ligand bound structures. Most interestingly, we found that large NNRTIs that lead to a large shift of the β12-sheet (P225–P236) break the HB between backbone-NH at K103 and backbone carbonyl at P236. Although the backbone-NH at K103 has been described to participate in HB interactions to NNRTIs,35 the concurrently available carbonyl at P236 has not. Use of this HB acceptor could provide an additional backbone interaction to an NNRTI, which might lead to an improved resistance profile. As some viral strains carry a K101P mutation, which does not allow the backbone-NH HB to be formed,36 it is of high importance to identify additional common interaction sites in the NNRTI pocket that can be exploited. Although a P236L mutant has been identified, this will not change the potential for a backbone HB interaction. Additionally, this mutant has been described as having low replication fitness.37
Figure 6.
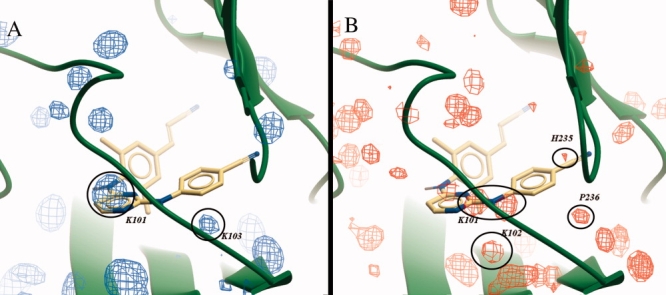
Consensus surfaces visualizing all conserved HB donors surrounding the pocket (A) and all conserved HB acceptors (B) among the selection of crystal structures. For reasons of clarity, only surfaces visualizing 20% occupancy are shown. The three dimensional surfaces are superimposed on the PDB structure 2ZD1, the green ribbon indicating the backbone, and Rilpivirine depicted by yellow carbons.
Materials and methods
Dataset
A total of 47 crystal structures were used in our analysis (Table II). Crystal structures were obtained from the PDB and grouped according to bound NNRTI. The selection included several structures of the NNRTIs approved for clinical use, namely, Nevirapine, Efavirenz, and Delavirdine. Several structures of apo HIV-1 RT have been used (Group a) three of which contain a bound DNA fragment (Group b). Apo structures 1JLE and 1RTJ were obtained by soaking out the NNRTI and are, therefore, not native apo structures. Accordingly, in an analysis of the structures, the orientation of the backbone was found to show more similarities with ligand-bound forms of HIV-RT than with the apo form. For this reason, structures 1JLE and 1RTJ were omitted from our dataset. Out of the selected structures several contained a point mutation in the NNRTI pocket. From Groups a and c in Table II B-factors were extracted in Molsoft ICM.38 The consensus structures were created from a sub-selection of all 40 NNRTI-bound crystal structures, including 23 different NNRTIs (Group c, Table II). For all NNRTIs, the charge at pH 7, was calculated using the Marvin Beans pKa prediction tool by ChemAxon,39 and all were found to be uncharged.
Table II.
Summary of the PDB Structures that were Used in all Analyses
| PDB Code | Group | Mutation | Drug | Class | Resolution(Å) |
|---|---|---|---|---|---|
| 1DLO | a | A172K | n/l | Apo | 2.70 |
| 1HMV | a | n/p | n/l | Apo | 3.20 |
| 1HQE | a | K103N | n/l | Apo | 2.70 |
| 1QE1 | a | M184I | n/l | Apo | 2.85 |
| 1N6Q | b | n/p | n/l | DNA Bound | 3.00 |
| 1RTD | b | n/p | n/l | DNA Bound | 3.20 |
| 2HMI | b | n/p | n/l | DNA Bound | 2.80 |
| 1RTH | c | n/p | 1051U91 | 1051U91 | 2.20 |
| 1JLQ | c | n/p | 739W94 | 739W94 | 3.00 |
| 1VRU | c | n/p | Alpha-Apa | Alpha-Apa | 2.40 |
| 1EP4 | c | n/p | Capravirine | Capravirine | 2.50 |
| 2ZD1 | c | n/p | Rilpivirine | DAPY | 1.80 |
| 2ZE2 | c | L100I/K103N | Rilpivirine | DAPY | 2.90 |
| 3BGR | c | K103N/Y181C | Rilpivirine | DAPY | 2.10 |
| 1KLM | c | n/p | Delavirdine | Delavirdine | 2.65 |
| 1FK9 | c | n/p | Efavirenz | Efavirenz | 2.50 |
| 1FKO | c | K103N | Efavirenz | Efavirenz | 2.90 |
| 1IKW | c | n/p | Efavirenz | Efavirenz | 3.00 |
| 1JKH | c | Y181C | Efavirenz | Efavirenz | 2.50 |
| 1BQM | c | n/p | HBY097 | HBY097 | 3.10 |
| 1C1C | c | n/p | TNK6123 | HEPT | 2.50 |
| 1JLA | c | Y181C | TNK651 | HEPT | 2.50 |
| 1RT1 | c | n/p | MKC442 | HEPT | 2.55 |
| 1RT2 | c | n/p | TNK651 | HEPT | 2.55 |
| 1RTI | c | n/p | HEPT | HEPT | 3.00 |
| 1FKP | c | K103N | Nevirapine | Nevirapine | 2.90 |
| 1JLB | c | Y181C | Nevirapine | Nevirapine | 3.00 |
| 1JLF | c | Y188C | Nevirapine | Nevirapine | 2.60 |
| 1S1U | c | L100I | Nevirapine | Nevirapine | 3.00 |
| 1S1X | c | V108I | Nevirapine | Nevirapine | 2.80 |
| 1VRT | c | n/p | Nevirapine | Nevirapine | 2.20 |
| 2HND | c | K101E | Nevirapine | Nevirapine | 2.50 |
| 2HNY | c | E138K | Nevirapine | Nevirapine | 2.50 |
| 1DTQ | c | n/p | PETT1 | PETT | 2.80 |
| 1DTT | c | n/p | PETT2 | PETT | 3.00 |
| 1JLC | c | Y181C | PETT2 | PETT | 3.00 |
| 1HNV | c | n/p | 8TIBO | TIBO | 3.00 |
| 1REV | c | n/p | 9TIBO | TIBO | 2.60 |
| 1TVR | c | n/p | 9TIBO | TIBO | 3.00 |
| 1UWB | c | Y181C | 8TIBO | TIBO | 3.20 |
| 1JLG | c | Y188C | UC781 | UC | 2.60 |
| 1RT4 | c | n/p | UC781 | UC | 2.90 |
| 1RT5 | c | n/p | UC10 | UC | 2.90 |
| 1RT6 | c | n/p | UC38 | UC | 2.80 |
| 1RT7 | c | n/p | UC84 | UC | 3.00 |
| 1S1T | c | L100I | UC781 | UC | 2.40 |
| 1S1W | c | V106A | UC781 | UC | 2.70 |
The table shows a summary of the PDB structures that were used in the analyses. All structures were subdivided into three groups: a, which contained apo-enzymes, b, which contained apo enzymes bound to a DNA fragment, and c, which contained NNRTI bound structures. Furthermore, mutations present in the NNRTI binding pocket were identified, if none were present “n/p” was used. Structures marked with “n/l” did not contain an NNRTI. Finally, all NNRTIs were subdivided into 13 classes as shown in the table.
Computational details
All structural experiments and visualizations were performed using ICM. In all structures the NNRTIs were checked for inconsistencies and the bond orders were confirmed (A workflow of the performed experiments is given in Supporting Information Fig. S12). A multiple sequence alignment was created within ICM using default options, verifying that no mutations were present around the NNRTI pocket other than the known single mutations within the PDB structures. Hydrogens were added to the PDB by ICM object conversion, which contains hydrogen bond optimization, protonation state optimization of His residues, and rotamer optimization of Asn, Gln, and His residues. Subsequently, all RT crystal structures were superimposed based on alignment of the backbone atoms of selected residues. This selection was made using a 12 Å sphere around the largest NNRTI, Dlv, in PDB structure 1KLM. This included the following residues on chain A: 90–111, 161, 164, 168, 171, 172, 175–196, 198, 199, 201, 205, 222–240, 242, 315–321, 343, 348–350 and on chain B: 135–140. Visualizations and calculations were performed using the superimposed structures.
B-factor analysis
The influence of experimental error and conditions was minimized by normalizing the B-factors of every residue in the crystal structures similar to Yuan et al.,5 where we used full residue B-factors instead of Cα-only B-factors. This normalization uses the standard deviation over all B-factors per structure to allow a comparison of the values of different crystal structures.5 After normalization, the B-factors from all ligand bound structures were combined and their mean value per residue was calculated, the same was done for the apo structures. Because of the very high temperature factors, structures 1HMV and 2ZE2 were omitted from these experiments. The average difference values between apo and ligand bound normalized B-factors per residue were binned in three classes: lower (≤−0.2), virtually unchanged (between −0.2 and 0.2), and higher (≥0.2).
Ligand induced displacement analysis
Ligand-induced conformational changes were characterized in three ways. Firstly, for each of the superimposed structures a scalar representing the mean distance for each pocket residue between the centroid in the ligand-bound conformation and the centroid of the two apo structures was determined. The obtained average centroid displacement distances were binned into three classes: small (≤2 Å), medium (between 2 Å and 4 Å), and large (≥4 Å). Secondly, the three-dimensional displacement vector between residue Cα positions in the ligand-bound conformation and the apo structures was split up over all three Cartesian axes to determine the backbone movement on each specific axis. Thirdly, the procedure was repeated between residue centroid positions in the ligand-bound conformation and the apo structures. Thus, the movement of the backbone and the side chains were both taken into account as well as changes in orientation of side chains (which are known to be relatively independent from backbone movements).40
Conversion of crystal structures to three-dimensional occupancy maps
The crystal structures were converted to three-dimensional occupancy maps by placing the structures in a grid box. A VdW occupancy value was assigned to each grid point and was normalized to a value between 0 and 1 using the internal auto trim function of ICM. This quasi-binary scaling enabled a comparison between occupancy maps obtained from different crystal structures.38 For HB maps, the values were scaled between 0 and 1 for acceptors and between 0 and −1 for donors, similar to the VdW scaling.
Creation of consensus potentials and structures
Consensus structures were created from the initial superpositions by adding up the individual occupancy maps of the set of crystal structures. This step was performed separately for the RT binding pocket residues and the bound NNRTI structures. When several structures containing the same NNRTI were present, the average value of all structures containing that specific NNRTI was used. Thereby each NNRTI class contributed equally to the final occupancy map and the domination of a single class of NNRTIs over the others was avoided. Occupancy maps for the set of crystal structures were created for VdW, HB donors and HB acceptors, from the occupancy maps of each individual structure and named consensus structures. Isocontour surfaces were created at different levels, corresponding to visualization of different degrees of conservation.
Conclusion
In conclusion, both methods presented in this work, the analysis of B-factors and ligand induced residue displacement as well as the analysis of steric and pharmacophoric properties from multiple crystal structures show how the growing number of crystal structures available can be mined efficiently to generate novel hypotheses for lead optimization. Although the amount of information available in these analyses increases with the number of structures included, the use of a handful of structures can already provide insights. Our findings were also supported by novel, more sterically demanding NNRTIs that were published by Sweeney et al. just as this manuscript had been finalized.
Our methods facilitated the identification of the working mechanism of NNRTIs as a combination of two mechanisms that were previously suggested (the “molecular arthritis” and “distorted catalytic site” hypotheses). In addition, our consensus structures were able to extract conserved locations of interest from the crystal structures without the need for molecular dynamics. The different types of consensus structures can complement each other and provide a useful overview of the interaction between a class of compounds and its target protein. Using this method, we identified a novel backbone hydrogen bond acceptor at P236 and a novel hydrophobic subpocket.
Acknowledgments
We thank Andrew Orry (Molsoft L.L.C), Anik Peeters, Luc Geeraert, Ann Vos, and Carlo Boutton (Tibotec-Virco BVBA) for their helpful discussions. A.B. was funded by the Dutch Top Institute Pharma, project number: 01-105.
References
- 1.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knegtel RM, Kuntz ID, Oshiro CM. Molecular docking to ensembles of protein structures. J Mol Biol. 1997;266:424–440. doi: 10.1006/jmbi.1996.0776. [DOI] [PubMed] [Google Scholar]
- 3.Yuan Z, Bailey TL, Teasdale RD. Prediction of protein B-factor profiles. Proteins. 2005;58:905–912. doi: 10.1002/prot.20375. [DOI] [PubMed] [Google Scholar]
- 4.Wampler JE. Distribution analysis of the variation of B-factors of X-ray crystal structures; temperature and structural variations in lysozyme. J Chem Inf Comput Sci. 1997;37:1171–1180. doi: 10.1021/ci9702252. [DOI] [PubMed] [Google Scholar]
- 5.Yuan Z, Zhao J, Wang Z-X. Flexibility analysis of enzyme active sites by crystallographic temperature factors. Protein Eng. 2003;16:109–114. doi: 10.1093/proeng/gzg014. [DOI] [PubMed] [Google Scholar]
- 6.Keller PA, Leach SP, Luu TTT, Titmuss SJ, Griffith R. Development of computational and graphical tools for analysis of movement and flexibility in large molecules. J Mol Graph Model. 2000;18:235–241. doi: 10.1016/s1093-3263(00)00028-0. 299. [DOI] [PubMed] [Google Scholar]
- 7.Richmond N, Abrams C, Wolohan P, Abrahamian E, Willett P, Clark R. GALAHAD: 1. Pharmacophore identification by hypermolecular alignment of ligands in 3D. J Comput-Aided Mol Des. 2006;20:567–587. doi: 10.1007/s10822-006-9082-y. [DOI] [PubMed] [Google Scholar]
- 8.Totrov M. Atomic property fields: generalized 3D pharmacophoric potential for automated ligand superposition, pharmacophore elucidation and 3D QSAR. Chem Biol Drug Des. 2008;71:15–27. doi: 10.1111/j.1747-0285.2007.00605.x. [DOI] [PubMed] [Google Scholar]
- 9.O'Brien SE, Brown DG, Mills JE, Phillips C, Morris G. Computational tools for the analysis and visualization of multiple protein-ligand complexes. J Mol Graph Model. 2005;24:186–194. doi: 10.1016/j.jmgm.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 10.Powers RA, Shoichet BK. Structure-based approach for binding site identification on AmpC beta-lactamase. J Med Chem. 2002;45:3222–3234. doi: 10.1021/jm020002p. [DOI] [PubMed] [Google Scholar]
- 11.Nichols SE, Domaoal RA, Thakur VV, Tirado-Rives J, Anderson KS, Jorgensen WL. Discovery of wild-type and Y181C mutant non-nucleoside HIV-1 reverse transcriptase inhibitors using virtual screening with multiple protein structures. J Chem Inf Model. 2009;49:1272–1279. doi: 10.1021/ci900068k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitsuya H, Weinhold KJ, Furman PA, St Clair MH, Lehrman SN, Gallo RC, Bolognesi D, Barry DW, Broder S. 3′-Azido-3′-deoxythymidine (BW A509U): an antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc Natl Acad Sci USA. 1985;82:7096–7100. doi: 10.1073/pnas.82.20.7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Richman DD, Havlir D, Corbeil J, Looney D, Ignacio C, Spector SA, Sullivan J, Cheeseman S, Barringer K, Pauletti D. Nevirapine resistance mutations of human immunodeficiency virus type 1 selected during therapy. J Virol. 1994;68:1660–1666. doi: 10.1128/jvi.68.3.1660-1666.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Havlir DV, Eastman S, Gamst A, Richman DD. Nevirapine-resistant human immunodeficiency virus: kinetics of replication and estimated prevalence in untreated patients. J Virol. 1996;70:7894–7899. doi: 10.1128/jvi.70.11.7894-7899.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rhee S-Y, Fessel WJ, Zolopa AR, Hurley L, Liu T, Taylor J, Nguyen DP, Slome S, Klein D, Horberg M, Flamm J, Follansbee S, Schapiro JM, Shafer RW. HIV-1 protease and reverse-transcriptase mutations: correlations with antiretroviral therapy in subtype B isolates and implications for drug-resistance surveillance. J Infect Dis. 2005;192:456–465. doi: 10.1086/431601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson V, Brun-Vezinet F, Clotet B, Gunthard H, Kuritzkes D, Pillay D, Schapiro J, Richman D. Update of the drug resistance mutations in HIV-1: December 2008. Top HIV Med. 2008;16:138–145. [PubMed] [Google Scholar]
- 17.Zhou Z, Madrid M, Evanseck JD, Madura JD. Effect of a bound non-nucleoside RT inhibitor on the dynamics of wild-type and mutant HIV-1 reverse transcriptase. J Am Chem Soc. 2005;127:17253–17260. doi: 10.1021/ja053973d. [DOI] [PubMed] [Google Scholar]
- 18.Madrid M, Lukin JA, Madura JD, Ding J, Arnold E. Molecular dynamics of HIV-1 reverse transcriptase indicates increased flexibility upon DNA binding. Proteins. 2001;45:176–182. doi: 10.1002/prot.1137. [DOI] [PubMed] [Google Scholar]
- 19.Bahar I, Erman B, Jernigan RL, Atilgan AR, Covell DG. Collective motions in HIV-1 reverse transcriptase: examination of flexibility and enzyme function. J Mol Biol. 1999;285:1023–1037. doi: 10.1006/jmbi.1998.2371. [DOI] [PubMed] [Google Scholar]
- 20.Das K, Sarafianos SG, Clark AD, Jr, Boyer PL, Hughes SH, Arnold E. Crystal structures of clinically relevant Lys103Asn/Tyr181Cys double mutant HIV-1 reverse transcriptase in complexes with ATP and non-nucleoside Inhibitor HBY 097. J Mol Biol. 2007;365:77–89. doi: 10.1016/j.jmb.2006.08.097. [DOI] [PubMed] [Google Scholar]
- 21.Carlson HA. Protein flexibility and drug design: how to hit a moving target. Curr Opin Chem Biol. 2002;6:447–452. doi: 10.1016/s1367-5931(02)00341-1. [DOI] [PubMed] [Google Scholar]
- 22.Carlson HA, McCammon JA. Accommodating protein flexibility in computational drug design. Mol Pharmacol. 2000;57:213–218. [PubMed] [Google Scholar]
- 23.Hsiou Y, Ding J, Das K, Clark AD, Jr, Hughes SH, Arnold E. Structure of unliganded HIV-1 reverse transcriptase at 2.7 A resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure. 1996;4:853–860. doi: 10.1016/s0969-2126(96)00091-3. [DOI] [PubMed] [Google Scholar]
- 24.Das K, Bauman JD, Clark AD, Jr, Frenkel YV, Lewi PJ, Shatkin AJ, Hughes SH, Arnold E. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: strategic flexibility explains potency against resistance mutations. Proc Natl Acad Sci USA. 2008;105:1466–1471. doi: 10.1073/pnas.0711209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rodriguez-Barrios F, Balzarini J, Gago F. The molecular basis of resilience to the effect of the Lys103Asn mutation in non-nucleoside HIV-1 reverse transcriptase inhibitors studied by targeted molecular dynamics simulations. J Am Chem Soc. 2005;127:7570–7578. doi: 10.1021/ja042289g. [DOI] [PubMed] [Google Scholar]
- 26.Maga G, Radi M, Zanoli S, Manetti F, Canciano R, Hubscher U, Spadari S, Falciani C, Terrazas M, Vilarrasa J, Botta M. Discovery of non-nucleoside inhibitors of HIV-1 reverse transcriptase competing with the nucleoside substrate. Angew Chem Int Ed Engl. 2007;46:1810–1813. doi: 10.1002/anie.200604165. [DOI] [PubMed] [Google Scholar]
- 27.Esnouf R, Ren J, Ross C, Jones Y, Stammers D, Stuart D. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat Struct Biol. 1995;2:303–308. doi: 10.1038/nsb0495-303. [DOI] [PubMed] [Google Scholar]
- 28.Balzarini J. Current status of the non-nucleoside reverse transcriptase inhibitors of human immunodeficiency virus type 1. Curr Top Med Chem. 2004;4:921–944. doi: 10.2174/1568026043388420. [DOI] [PubMed] [Google Scholar]
- 29.Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 30.Sarafianos SG, Marchand B, Das K, Himmel DM, Parniak MA, Hughes SH, Arnold E. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J Mol Biol. 2009;385:693–713. doi: 10.1016/j.jmb.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mendieta J, Cases-Gonzalez CE, Matamoros T, Ramirez G, Menendez-Arias L. A Mg2+-induced conformational switch rendering a competent DNA polymerase catalytic complex. Proteins. 2008;71:565–574. doi: 10.1002/prot.21711. [DOI] [PubMed] [Google Scholar]
- 32.Paris KA, Haq O, Felts AK, Das K, Arnold E, Levy RM. Conformational landscape of the human immunodeficiency virus type 1 reverse transcriptase non-nucleoside inhibitor binding pocket: lessons for inhibitor design from a cluster analysis of many crystal structures. J Med Chem. 2009;52:6413–6420. doi: 10.1021/jm900854h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wegner JK, Vlijmen HV, Boutton CWM. 2008. Phenotype prediction method. Patent WO2008065180A1; EP2007063047.
- 34.Sweeney ZK, Harris SF, Arora N, Javanbakht H, Li Y, Fretland J, Davidson JP, Billedeau JR, Gleason SK, Hirschfeld D, Kennedy-Smith JJ, Mirzadegan T, Roetz R, Smith M, Sperry S, Suh JM, Wu J, Tsing S, Villaseñor AG, Paul A, Su G, Heilek G, Hang JQ, Zhou AS, Jernelius JA, Zhang F-J, Klumpp K. Design of annulated pyrazoles as inhibitors of HIV-1 reverse transcriptase. J Med Chem. 2008;51:7449–7458. doi: 10.1021/jm800527x. [DOI] [PubMed] [Google Scholar]
- 35.Wang Z, Wu B, Kuhen KL, Bursulaya B, Nguyen TN, Nguyen DG, He Y. Synthesis and biological evaluations of sulfanyltriazoles as novel HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg Med Chem Lett. 2006;16:4174–4177. doi: 10.1016/j.bmcl.2006.05.096. [DOI] [PubMed] [Google Scholar]
- 36.Parkin NT, Gupta S, Chappey C, Petropoulos CJ. The K101P and K103R/V179D mutations in human immunodeficiency virus type 1 reverse transcriptase confer resistance to nonnucleoside reverse transcriptase inhibitors. Antimicrob Agents Chemother. 2006;50:351–354. doi: 10.1128/AAC.50.1.351-354.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gerondelis P, Archer RH, Palaniappan C, Reichman RC, Fay PJ, Bambara RA, Demeter LM. The P236L delavirdine-resistant human immunodeficiency virus type 1 mutant is replication defective and demonstrates alterations in both RNA 5′-end- and DNA 3′-end-directed RNase H activities. J Virol. 1999;73:5803–5813. doi: 10.1128/jvi.73.7.5803-5813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.ICM. Version 3.6d. La Jolla: Molsoft L.L.C.; 2009. [Google Scholar]
- 39.Marvin Beans pKa Prediction tool. Version 4.1. Budapest: ChemAxon; 2006. [Google Scholar]
- 40.Najmanovich R. Side-chain flexibility in proteins upon ligand binding. Proteins. 2000;39:261–268. doi: 10.1002/(sici)1097-0134(20000515)39:3<261::aid-prot90>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]