

Abstract
The development of a kinase structural database, the kinase knowledge base (KKB), is described. It covers all human kinase domain structures that have been deposited in the Protein Data Bank. All structures are renumbered using a common scheme, which enables efficient cross-comparisons and multiple queries of interest to the kinase field. The common numbering scheme is also used to automatically annotate conserved residues and motifs, and conformationally classify the structures based on the DFG-loop and Helix C. Analyses of residue conservation in the ATP binding site using the full human-kinome–sequence alignment lead to the identification of a conserved hydrogen bond between the hinge region backbone and a glycine in the specificity surface. Furthermore, 90% of kinases are found to have at least one stabilizing interaction for the hinge region, which has not been described before.
Keywords: protein kinases, structure-based drug design, crystallography: X-ray, structural informatics, binding sites, databases: protein, protein conformation, catalytic domain, sequence analysis: protein, protein binding
Introduction
The availability of and public access to X-ray and NMR structures of protein and nucleic acid drug targets through the Protein Data Bank (PDB, http://www.rcsb.org)1 has enabled the development of the field of structure-based drug discovery (SBDD). Many successes of receptor-based lead discovery2 and lead optimization3,4 have been described in the literature, and more recently SBDD has played a pivotal role in the development of fragment-based drug discovery5.
The number of structures available through the PDB currently approaches 60,000 (August, 2009) and the PDB website has undergone significant enhancements in recent years, such as addition of ligand-based (sub)structure searches and sequence-based (BLAST/FASTA) searches to the already available PDB identifier, keyword, author, reference, and other text searches. Despite these enhancements, retrieval of all structures of a particular protein target is not always straightforward and requires a significant amount of manual analysis of retrieved structures. Moreover, in the realm of drug discovery, specifically in the quest of highly specific inhibitors targeting a single-protein target, there is often an interest to analyze and compare many targets and off-targets of a protein family. Within the PDB framework, this is virtually impossible to accomplish.
With these limitations to the PDB structural database, a number of protein family targeted structural databases have been developed to augment the information from the PDB with additional annotations specific to the family of interest. For example, a peptidase database, MEROPS6, and an antibody-focused structural database, SACS7 have been described. The Vertex group has published on a kinase database that covers only a subset of available kinase X-ray structures. In this database, 426 crystal structures are available with kinase-specific annotations like DFG-in/out and Helix C in/out.8 In addition to these protein-family focused databases, protein–ligand interaction focused databases have been developed that allow querying on specific atomic interactions present in protein–ligand complexes,9–11 a feature not available through the PDB. Other databases have integrated known binding affinities of ligands with the relevant complex structures.12–14
We have recently developed both a general, ligand-focused protein relational database (PRDB) and protein family focused knowledge bases, which in essence are subsets of PRDB. Currently knowledge bases have been developed for the matrix metalloproteins (MMP's) and kinases (Mobilio et al., Submitted).15 This article outlines in details the derivation of the kinase knowledge base (KKB) and the methods developed to annotate individual kinase structures. The KKB contains all crystallized catalytic domain structures that have appeared in the public domain.
Because of their involvement in critical signaling pathways, kinases have become popular drug targets since the 1980's,16 resulting in an explosion of available crystal structures in recent years (Supporting Information Fig. 1). A number of marketed small-molecule drugs target the ATP binding site of kinases.17 A high-level overview of the development of the KKB is shown in Figure 1, describing the use of BLAST searches to identify kinase catalytic domain crystal structures and the development and use of a kinome-wide multiple sequence alignment to derive a common numbering scheme. The latter is subsequently used to renumber all kinase crystal structures and to annotate conserved motifs and the different structural states observed for the kinase catalytic domain (Fig. 2). The common numbering scheme enables a number of queries specific to kinases, as shown in Figure 3.
Figure 1.
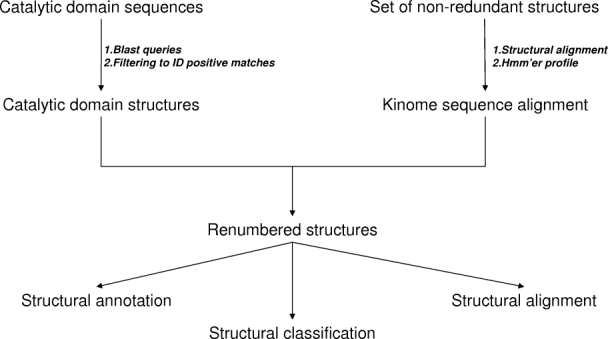
Overview of derivation of KKB schematic of the development of the KKB, starting from the catalytic domain sequences to identify available structures in the PDB and the structural alignment of a limited set of kinases to allow for a kinome-wide sequence alignment. Subsequently, all available structures were renumbered and annotated.
Figure 2.
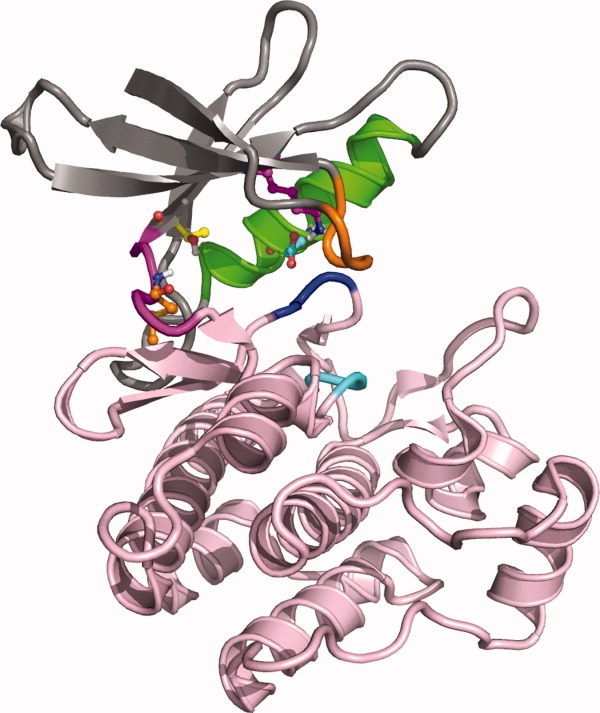
Overview of kinase catalytic domain structure of Abl kinase (2GQG) with the N-terminal lobe shown on top (mostly gray ribbon) and C-terminal lobe at the bottom (pink ribbon). The hinge region, which connects the two lobes, is shown in magenta on the left-hand side. Helix C is shown in green, the glycine-rich loop in orange. The conserved DFG (dark blue ribbon) and HRD motifs (cyan ribbon) are highlighted as well. Residues explicitly shown are the hinge region donor residue (orange carbons), the gatekeeper residue (yellow carbons), the catalytic lysine .(magenta carbons), and the Helix C acid (cyan carbons)
Figure 3.
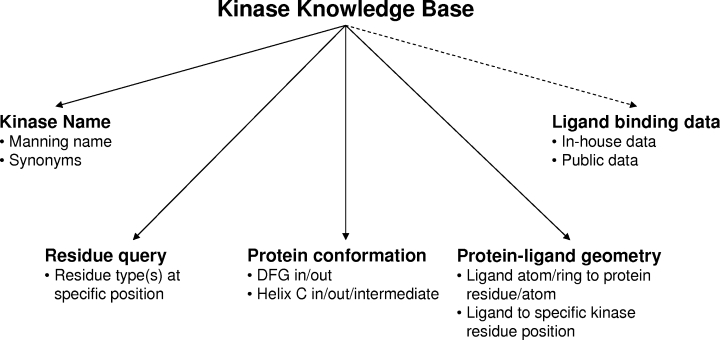
Overview of kinase-specific queries enabled in KKB. The various queries enabled by the KKB are highlighted. Kinases can be retrieved by either the Manning name or through searching the synonyms list. The common numbering scheme enables residue-based queries, which retrieve all kinases with a particular residue at a particular position in the structure. Kinases with particular conformations for the DFG motif or Helix C can also be retrieved. Retrieval of kinases that form specific interactions with a ligand have been enabled through integration with PRDB and the common numbering scheme further enhances this capability. Finally, ligands retrieved from the KKB can be linked to available in-house or public data through another in-house application (signified by dashed arrow) using the ligand hashcodes.
Results
Using the human kinase catalytic domain sequences from Manning et al.18 and BLAST searches (Fig. 1), 1150 X-ray structures were identified containing 1648 chains (August, 2009). These 1150 structures cover 148 different kinases. Because the BLAST matches between the human sequences and the sequence from the protein structure are not required to be a 100% match, mutant enzymes and nonhuman structures are retrieved as well. The database currently consists of 843 human catalytic domains. The remaining ∼300 structures are mostly from bovine, mouse, rat, and chicken sources. Despite the fuzzy matching of sequence to structure, the criteria are strict enough to correctly distinguish closely related kinase structures such as the different p38 kinases and CDK's.
The multiple sequence alignment generated using the HMMER profile results in a sequence length of 2030 amino acid positions, but this is a sparsely occupied sequence as the average number of occupied positions is 259 across the 1150 crystal structures.
The renumbered and original PDB files, both on a per-chain basis, are structurally aligned to a reference structure, in this case 1ATP, with the alignment algorithm TM-Align.19 The TM-align algorithm focuses on parts of the structures that are closer in space, making it less sensitive to structural changes. The correlation between backbone RMSD and the percentage of the residues that are used in the alignment is shown in Supporting Information Figure 2. This data was extracted from the TM-align output and is not stored in the database. The TM-align RMSD is calculated over the backbone atoms of the residues used in the structural alignment. As to be expected, other PKA structures have low RMSD's when aligned to 1ATP, generally less than 1 Å, and high sequence similarity. However, several PKA structures show significant structural changes based on the RMSD (1.5–2.0 Å). These turn out to be PKA structures of the APO form (e.g., 1J3H and 1SYK), which undergoes a significant conformational change compared with the ligand-bound form.20,21 Not surprisingly, structures with RMSD's between 1 Å and 2 Å (23% of KKB structures) have the highest sequence similarity to PKA, between 25% and 50%. The majority of the kinases have RMSD's in the 2.0–2.5 Å (43%) and 2.5–3.0 Å (26%) range. The average sequence similarity is 30% across all the structures (median 26%).
One of the mechanisms that regulate kinase catalytic activity is the conformational change that occurs in the DFG motif, which can flip from an “in” conformation in which residue 1111, usually a phenylalanine, is buried in the kinase structure, to the “out” conformation in which the phenylalanine is exposed in the ATP binding pocket (Fig. 4). The aspartic acid of the DFG motif moves in the opposite direction, becoming buried in the DFG-out conformation. In the DFG-out conformation, the phenylalanine blocks binding of ATP. This results in inactivity of the kinase. This movement can be described using the distance between the Cα atom of the hinge region donor residue (residue 486) and the Cα atom of the phenylalanine. A distance ≥15 Å is observed in kinases in the DFG-in conformation, whereas a distance of ≤13.5 Å is observed in the DFG-out conformation (Supporting Information Fig. 3). At intermediate distances, classification is achieved by analyzing the conformation of the backbone and side chain of the aspartic acid, through measurement of the phi, psi, and chi angles. The automatic classification scheme classifies 109 kinases (74% of 148 kinases) in the DFG-in conformation only, 15 (10.1%) in DFG-out only, and 24 kinases (16.2%) are found in both the DFG-in and out conformations (Supporting Information Table 2).
Figure 4.
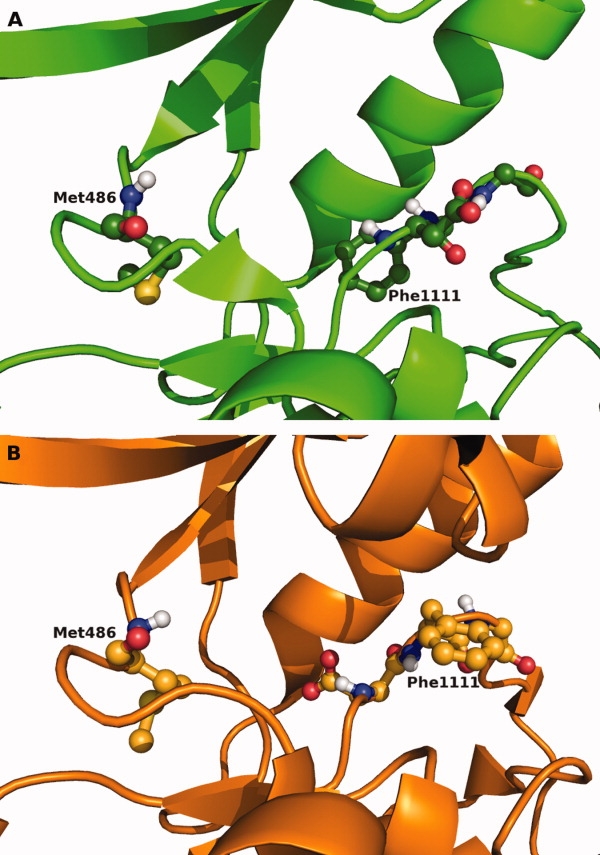
Comparison of DFG-in and DFG-out structures. Side-by-side comparison of the DFG-in (a) (2G2I) and DFG-out (b) (2FO0) conformations of Abl kinase. In the DFG-in conformation, Phe1111 is buried deep in the protein and pointing away from the ATP binding site. In the DFG-out conformation, Phe1111 occupies the ATP binding site, preventing ATP binding.
A trademark of active kinase structures is the existence of a salt bridge between the catalytic lysine (residue 189) and the acid from Helix C (residue 265). In the so-called “Helix C in” conformation [Supporting Information Figs. 4 and 5(A)], it is this salt bridge that stabilizes the conformation of the catalytic lysine. This in turn helps the lysine to stabilize the α and β phosphates of ATP, which is necessary to present the γ phosphate for catalysis. Movement of Helix C, which results in a disruption of the salt bridge, is another mechanism to regulate the catalytic activity of kinases. The conformation in which the Helix C is rotated away from the binding site is the so-called “Helix C out” conformation [Fig. 5(C)]. However, in many kinase structures, the Helix C has not moved away from the position observed in active kinases, yet the hallmark salt bridge is not present. This is the state which we call a “Helix C intermediate” conformation, which is the result of only movement of the side chain of the glutamic acid [Fig. 5(B)]. This movement results in the acid moiety pointing away from the catalytic lysine and the ATP binding site.
Figure 5.

Close-up of the different Helix C conformations comparison of Helix C position and glutamic acid position in the Helix C in (a) (3F6X), Helix C intermediate (b) (3D7U), and Helix C out conformations (c) (1FMK) for Src kinase. The in form clearly shows the presence of the salt bridge between the catalytic lysine and the glutamic acid. In the intermediate form, Helix C is in the same position as in the “in” form but the glutamic acid side chain is pointing toward the solvent rather than the ATP binding site, resulting in disruption of the salt bridge. In the Helix C out position, both the helix and the glutamic acid have moved away from the ATP binding pocket.
Based on these observations, parameters were derived to automatically classify Helix C in these three conformations (Supporting Information Fig. 5). For a number of kinases classification of the Helix C conformation is not possible, due to (partial) absence of the catalytic lysine or the Helix C acid. At very short and long distances between the quaternary nitrogen of the lysine side chain and the acid oxygen's, structures are classified as Helix C in or out, respectively. At intermediate distances, the angle of the acid side chain is determined to assess whether the acid is pointing toward or away from the binding site. If the acid is pointing away from the binding site, the kinase is considered to be in an intermediate Helix C conformation. Of the 148 kinases with available crystal structures, 105 are found that only occupy a single Helix C conformation. Of these, 88 are found in the Helix C in conformation (59.0 %), and the remaining 17 in the Helix C out conformation (11.5%). Twenty-four kinases are found to occupy two Helix C conformations, with 14 occupying the in and intermediate conformations, eight occupying the in and out conformations and the remaining two in the intermediate and out conformations. Seven kinases are found that occupy all three conformations, including Src kinase (Supporting Information Figs. 4 and 5). Twelve kinases are not classified because the catalytic lysine or Helix C acid is (partially) undefined in the X-ray structure (Supporting Information Table 3).
Available structures for kinases with annotated active site cysteines, as identified by Gray and coworkers22 (Supporting Information Table 3 in Gray and coworkers22) were visually analyzed and the common residue number for each of the active site cysteines was identified. The common numbering scheme and the structural overlays highlighted that a number of groups correspond to the same residue position in the kinome-wide sequence alignment, as indicated in Table I. A new group was also identified, which contains FMS and FLT3. They both contain two hinge region cysteines, one at the hinge-region hydrogen-bond donor position (Groups 3B–E) and the other at the adjacent residue (new Group 3G). FMS contains a third active-site cysteine next to the DFG motif (Group 4). Group 3F, which contains EGFR, is associated with two common residue numbers that are structurally equivalent when overlaid. All members of this group identified by Gray and coworkers are associated with common residue 497. However, the cysteine in MAP2K7 (2DYL), which was originally classified as belonging to Group 3E, overlays perfectly with the cysteine found in EGFR and other members of Group 3F (Supporting Information Fig. 6). This observation results in reclassification of MAP2K7 into Group 3F and the addition of the second common residue number that identifies members of this group.
Table I.
Reactive Cysteines Found in the Active Site and the Corresponding Common Residue Number for Each
| Reactive cysteine group | Common residue number | Active site location |
|---|---|---|
| 1A | 71 or 72 | G-rich loop |
| 1B | 76 | G-rich loop |
| 1C | 77 | G-rich loop |
| 2A | 79 | β-strand after G-rich loop |
| 2B | 81 | β-strand after G-rich loop |
| 2C | 84 | β-strand after G-rich loop |
| 3A | 482 | Hinge region |
| 3B,3C, 3D,3E | 486 | Hinge region/HB donor |
| 3F | 497 or 491 | Extension of hinge region |
| 3Ga | 487 | Hinge region |
| 4 | 1105 | Directly precedes DFG motif |
The common residue number of the reactive cysteine for each group and an approximate location in the active site are shown.
Newly identified group, not identified by Gray and coworkers.22
Discussion
Over the past two decades kinases have become important drug targets and the availability of X-ray structures has increased exponentially. The availability of structural information makes kinases ideal targets for SBDD techniques, however, within the PDB framework retrieval of relevant structures is not always straightforward. In addition, a number of queries useful for structure-based design are not available. To further aid SBDD, we have developed a kinase knowledge base, which combines structural information from the public domain with kinase-specific annotations.
Because of its integration with the in-house developed PRDB, ligand-based or protein–ligand distance queries are available in the KKB (Mobilio et al., Submitted).15 The schemas describing both PRDB and the protein-family focused subsets of PRDB are presented in detail elsewhere (Mobilio et al., Submitted). The development of a kinome-wide common numbering scheme enhances these queries made accessible through integration with PRDB further, for example, all kinase structures that have an interaction between a ligand and a particular residue position can be retrieved. The results from such a query can be used to analyze how different chemotypes interact with, for example, the catalytic lysine or the gatekeeper residue in different kinases. Visual analysis of the retrieved structures has been streamlined by pre-aligning all structures onto a common reference frame (1ATP) using the TM-align algorithm.19 In addition, ligands, waters, and other (solvent) molecules that are far from the active site have been removed. Just as in PRDB, retrieved structures can be visualized directly with Benchware 3D Explorer23 or downloaded and visualized with any other molecular viewer. Other queries enabled by the common numbering scheme are identification of all kinases with a particular residue(s) at a particular position (Fig. 3), which allows an assessment of conservation and could give insight into inhibitor specificity, and identification of cysteine residues in the active site. Reactive cysteines are potential targets for covalent inhibitor design.
The common numbering scheme also enabled the automatic classification of the different conformations observed for the DFG motif (Fig. 4, Supporting Information Fig. 3) and Helix C (Fig. 5, Supporting Information Figs. 4 and 5). We describe three different conformations for Helix C based on visual analysis of kinase-catalytic–domain structures. In the “Helix C in” conformation, the salt bridge is present between the catalytic lysine and the conserved acid of Helix C. In the out conformation, Helix C has moved away significantly from the ATP binding site and it is defined based on the large separation between the catalytic lysine (common residue 189) and the Helix C acid (common residue 265). The Helix C intermediate conformation retains the same position for the helix, but the glutamic acid is pointing away from the binding site and thus the salt bridge has been disrupted. These observations have been translated in an automatic classification scheme for the three Helix C conformations based on the distance between the Helix C acid and the catalytic lysine and the conformation of the acid side chain. Because the Helix C intermediate conformation overlaps with the Helix C in conformation, conformational classification cannot be achieved by structural clustering or RMSD assessments. Not surprisingly, the vast majority of kinases, 109 out of 148, have been found in the active or DFG-in/Helix C in conformation.
The link between the Manning name, an abbreviation of the full kinase name, and the list of synonyms facilitates retrieval of all structures of a kinase and does not require knowledge of all the different possible nomenclatures for that kinase. For example, searching for “Abl,” for Abelson kinase, against the “synonyms” field gives exactly the same number of hits in KKB as the query with “Abelson” as the criterion, namely, 27 PDB structures. Through the PDB website, the Abl and Abelson keyword searches result in different numbers of hits (61 and 36, respectively). Because the kinase genes contain other domains in addition to the kinase domain, many of the hits retrieved from the PDB do not necessarily contain a catalytic domain and additional analysis of the results is required, which can be time consuming.
Ligand hashkeys24 have been calculated for all heteroaromatic residues in the PDB and these can be used to quickly retrieve available binding data from the in-house database and the public domain through the GVK database.25 This enables linking of structural information to structure-activity relationships in a straightforward manner.15
Visual analysis of the kinases identified as having one or more cysteines in the active site based on work by Gray and coworkers22 led to the merging of four groups into a single one (Table I). Furthermore, for Group 3F, which contains EGFR and BTK that are being targeted with covalent inhibitors,26,27 we identified another kinase that based on structural overlays should be a member of this group. To the best of our knowledge, MAP2K7 has not been identified as being a member of this group until now. MAP2K7 contains a cysteine at position 491 in the common numbering scheme, but structurally this position overlay perfectly with position 497, which is the cysteine in EGFR and other members of this group (Supporting Information Fig. 6). Thus, MAP2K7 is a potential off-target for irreversible inhibitors of the kinases in Group 3F. Based on analysis of the kinome-wide sequence alignment only MAP2K7 has this cysteine and the structural overlap appears to be the result of a 2-residue insertion in MAP2K7 (Supporting Information Fig. 7), which is not present in any other kinase, including other MAP kinases (Supporting Information Fig. 8).
Residue conservation for active site residues was analyzed based on the full kinome-sequence alignment (Table II), which is not stored in the database. However, the same analysis can be done using the stored active site residues in the database, although the analysis would be less complete due to missing residues. Average sequence conservation in the binding site is ∼50% based on the geometric mean. For each position, a physical–chemical character was assigned based on the highest percentage or percentages of the residue types that most frequently are observed at each position. Figure 6 shows a close-up of the ATP binding site and each active site residue is colored according to the assigned physical–chemical character observed across the human kinome [Fig. 6(A)]. Figure 6(B) shows only the most (single residue ≥70% occurrence) and least conserved positions (≤30%). Eleven sites out of 24 are strongly conserved (≥70%) and five sites are highly variable (≤30%). The region around the hinge region is mostly hydrophobic and a strong preference for an aromatic residue is observed for the residue that precedes the hinge-region hydrogen-bond donor residue. This so-called hydrophobic enclosure of the adenine binding site enhances the hydrogen bonding interactions to the hinge region.28 Residues 181 and 974, which form the top and bottom of the adenine binding site, are highly conserved. The conservation is probably a result of steric constraints: Larger hydrophobic residues would prevent ATP from binding. The ribose pocket and the surrounding region have significant polar character to it. The conserved positive and negative character for the catalytic lysine and Helix C acid and DFG motif are as expected.
Table II.
Analysis of Sequence Conservation for Kinase Active Site Residues
| Common residue | Number of different residues found | Highest % of occurrence | Residue with highest occurrence | Site character | Location in active site |
|---|---|---|---|---|---|
| 70 | 7 | 48% | Leucine | Hydrophobic | β-strand before G-rich |
| 71 | 6 | 88% | Glycine | Flexible | G of G-rich loop |
| 72 | 20 | 18% | Lysine | Polar | X of G-rich loop |
| 73 | 8 | 93% | Glycine | Flexible | G of G-rich loop |
| 77 | 15 | 62% | Phenylalanine | Aromatic | X of G-rich loop |
| 81 | 8 | 87% | Valine | Hydrophobic | β-strand after G-rich |
| 181 | 10 | 87% | Alanine | Hydrophobic | β-strand after G-rich |
| 189 | 6 | 95% | Lysine | Positive | Catalytic lysine |
| 265 | 9 | 91% | Glutamic acid | Negative | Helix C acid |
| 352 | 14 | 52% | Valine | Hydrophobic | b-strand below hinge |
| 461 | 15 | 36% | Methionine | Mixed | Gatekeeper |
| 479 | 14 | 76% | Glutamic acid | Negative | Hinge/Gatekeeper +1 |
| 482 | 18 | 41% | Tyrosine | Aromatic | Hinge/Gatekeeper +2 |
| 486 | 15 | 26% | Methionine | Hydrophobic | Hinge h-bond donor |
| 487 | 16 | 18% | Glutamic acid | Mixed | Hinge/gatekeeper +4 |
| 492 | 18 | 38% | Glycine | Flexible | Specificity surface |
| 494 | 18 | 72% | Glycine | Flexible | Specificity surface |
| 497 | 14 | 32% | Aspartic acid | Polar | Ribose pocket |
| 505 | 20 | 30% | Aspartic acid | Negative | Specificity surface |
| 966 | 15 | 28% | Glutamic acid | Polar | Loop bottom binding site |
| 967 | 7 | 95% | Asparagine | Polar | Loop bottom binding site |
| 974 | 7 | 77% | Leucine | Hydrophobic | Bottom hinge region |
| 1105 | 12 | 29% | Alanine | Mixed | DFG -1 |
| 1108 | 10 | 93% | Aspartic acid | Negative | D of DFG |
| Average | 12.7 | 59% | |||
| Mean | 11.7 | 51.4% |
Summary of analysis of residue conservation in the active site. Residue conservation is calculated across the full-kinome based on the kinome-sequence alignment. For each active site position, the total number of residues found at that position is shown and the residue with the highest occurrence and its occurrence percentage. The mean was calculated as the geometric mean. Based on the occurrence rate, number, and types of residues found at each position, the site is assigned an overall physical–chemical character, or if glycine is the dominant residue, the site is marked as “flexible.” The location in the binding site for each position is described.
Figure 6.
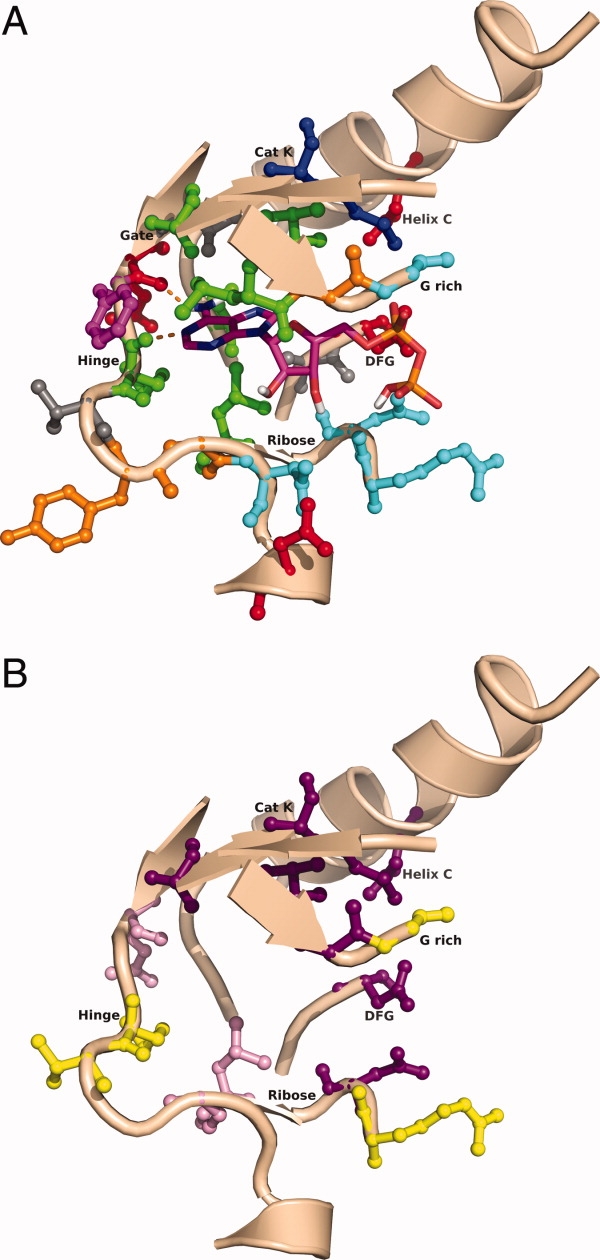
Analysis of residue conservation in the ATP binding site close-up of Abl binding site in complex with ADP (2G2I). All active site residues are shown and are color-coded based on the observed physical–chemical character (a). Hydrophobic sites are shown in green, aromatic sites in magenta, positive sites in dark blue, negative sites in red, polar sites in cyan, mixed sites in gray, and flexible sites in orange. In (b) only highly conserved (≥70%) and highly variable (conservation ≤ 30%) sites are shown. Residues conserved between 70% and 80% are shown in pink, residues conserved between 80% and 100% are shown in purple, and residues conserved ≤30% are shown in yellow.
The specificity surface, directly after the hinge region, has significant inherent flexibility due to the preference of glycine at this position. The strong preference for glycine at position 494 was investigated further by analyzing the phi, psi angles occupied by this glycine using the renumbered structures from the database. The database structures were further processed to calculate distances and dihedral angles with an OEChem script. As is shown in the Ramachandran plot [Supporting Information Fig. 9(A)], the dihedral angle space sampled covers only two regions, which only glycine can occupy. Comparison of the Ramachandran plot of Gly494 to that of all other glycines in kinase crystal structures [Supporting Information Fig. 9(B)] confirms that Gly494 only samples a small portion of the conformational space usually available to glycines. Further investigation of the distance between the backbone nitrogen of residue 494 and the backbone carbonyl oxygen of hinge region residue 486 and the angle between the backbone-NH and the carbonyl of residues 494 and 486 shows that the backbone conformation of Gly494 allows for the formation of a hydrogen bond in 64% of the structures (N—O distance ≤3.5 Å) (Fig. 7 and 8). Even at longer distances, when a hydrogen bond cannot be formed, there still is a strong conservation of the directionality with the backbone-NH of glycine 494 pointing toward the backbone carbonyl of residue 486 of the hinge region. Thus, when a hydrogen bond cannot be formed, a short-range electrostatic interaction between glycine 494 and the hinge region backbone carbonyl helps stabilize the conformation of the hinge region. The distance between the backbone nitrogen of glycine 494 is found to be within 4.0 Å of the backbone carbonyl oxygen in 82% and within 5.0 Å in 94% of the structures. This is too small for a water molecule to help mediate the interaction between glycine 494 and the hinge region. Ligands can form a hydrogen bonding interaction to the carbonyl of residue 486, but this would not disrupt the interaction between glycine 494 and the carbonyl of hinge region residue 486 (e.g., 2OFV or 2QUV).
Figure 7.
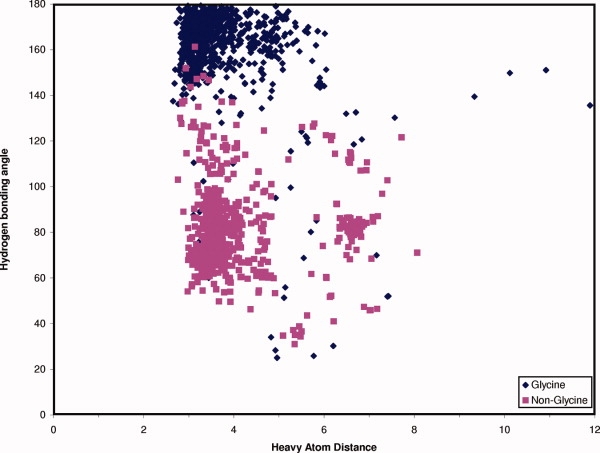
Analysis of hydrogen bonding potential between the hinge region and the specificity surface plot of the heavy atom distance between the backbone carbonyl of residue 486 of the hinge region and the backbone-NH of residue 494 and the angle formed between the backbone-NH of residue 494 and the carbonyl of residue 486. The results for glycine at residue 494 are shown in blue and the results for other residues than glycine at position 494 are shown in magenta. Hydrogen's were added with the reduce_build script from Molprobity.29
Figure 8.
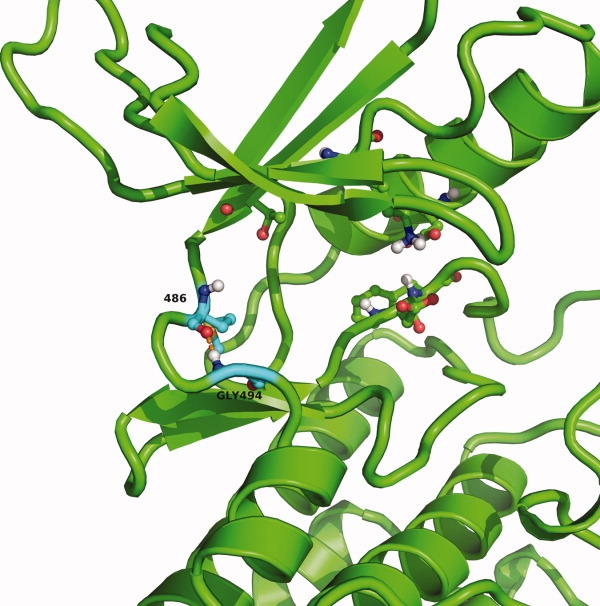
Conserved hydrogen bond of the hinge region and the specificity surface close-up of binding site of EGFR (2ITY) showing a number of conserved residues, like the catalytic lysine, the DFG region and the gatekeeper. Shown with cyan carbons are the hinge region donor residue 486 and the specificity surface residue glycine 494. The conserved hydrogen bond between residues 486 and 494 is shown in orange.
If residue 494 is not a glycine, the backbone-NH of residue 494 is pointing away from the carbonyl, as shown by the angle in Figure 7 and the Ramachandran plot [Supporting Information Fig. 9(C)], and thus a hydrogen bond cannot be formed. The conserved hydrogen bond or electrostatic interaction possible with glycine at position 494 is likely the driving force behind its conservation as the interaction will help stabilize the conformation of the hinge region and the turn into the specificity surface.
The other residues found at position 494 also sample a narrow amount of conformational space [Supporting Information Figs. 9(C) and 7] with the backbone-NH pointing away from the backbone carbonyl of residue 486, making a hydrogen bonding interaction impossible. The backbone conformation of non-glycine residues at position 494 forces the side chain to point away from the ATP binding site. Other backbone conformations that would present the side chain to the binding site would not be compatible with the tight turn made by the hinge region/specificity surface.
The strong preference for glutamic acid for the hinge region residue adjacent to the gatekeeper residue is the result of the formation of a conserved salt bridge with a lysine [common residue 1098 (Fig. 9)].30 This lysine is present in 76% of kinases and arginine, which can also form a salt bridge to glutamic acid, is found in 7% of kinases. This salt bridge probably aids in presenting the backbone carbonyl to the ATP binding site, which allows the carbonyl to form a hydrogen bond to the adenine of ATP.
Figure 9.
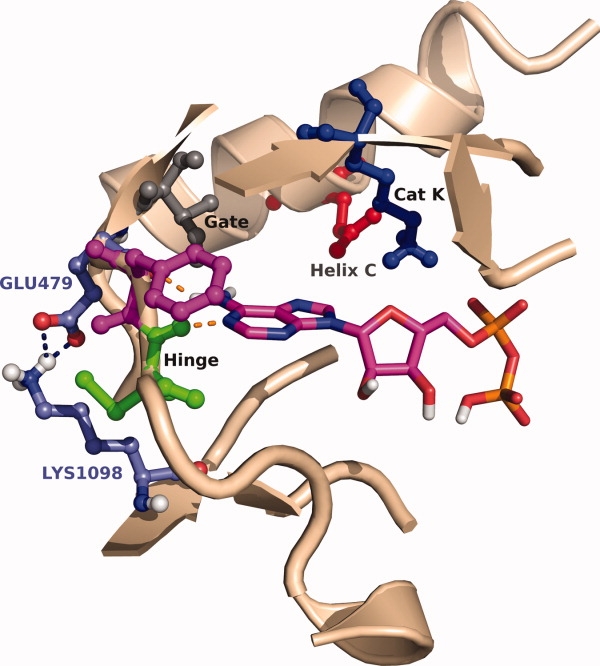
Conserved salt bridge of hinge region residue 479 close-up of Abl binding site (2G2I), which illustrates the conserved salt bridge across the kinome between glutamic acid with common residue 479 (316 in Abl) and lysine with common residue 1098 (378 in Abl).
The hinge region conformation can thus be stabilized by two interactions, either the salt bridge at the beginning of the hinge region between glutamic acid (or aspartic acid) 479 and lysine (or arginine) 1098, or by a hydrogen bonding or short-range electrostatic interaction between the backbone-NH of glycine 494 in the specificity surface and the backbone carbonyl of hinge region residue 486. A kinome-wide sequence analysis shows that 57% of kinases have both stabilizing interactions, 19% of kinases have only the salt bridge stabilization, and 14% of kinases have only glycine 494 to help stabilize the hinge region. Thus, only 10% of kinases do not have either of these conserved interactions, so stabilization of the hinge region appears to be important in helping to form the hinge region to aid in the formation of the critical hydrogen bonding interactions with the adenine ring of ATP (Supporting Information Table 4).
The KKB has been used in drug discovery projects to investigate a number of different hypotheses. An in-house X-ray structure of a high-throughput screening hit revealed the hinge-interaction binding motif. Initially, it was thought this binding motif was very common within kinases. A substructure search against the KKB identified 51 X-ray structures with the same hinge-interaction motif. However, analysis of the overlaid structures revealed that in 50/51 structures the core was flipped compared with the in-house compound. This led the team to further explore this potentially novel hinge-interaction motif.
In a number of projects the common numbering scheme and kinome-wide sequence alignment have been used to assess the potential effects of introduction of interactions with certain residues on selectivity over other kinases in the human kinome. Additional use cases have been described elsewhere.15
Methods
Sequence-based entry retrieval
The starting point for identification of kinase catalytic domain crystal structures are the human eukaryotic protein kinase catalytic domain sequences from http://www.kinase.com,18 where atypical protein kinases have been excluded. BLAST searches were performed for each of the 594 catalytic domain sequences using an in-house mirror of the PDB. Positive identification of correct matches between kinase sequence and crystal structure were done using the E-Values, Scores (in bits) and the percentage identities and percentage positives in the BLAST output. Two different criteria were used to identify crystal structures of kinase catalytic domains. Structures are identified as a match (1) if the E-Value ≤ 1e-145 and Score > 450 or (2) if the E-Value > 1e-145 but E-Value ≤ 1e-100 and % identity and % positive ≥ 90%. Finally, because a number of PDB codes are associated with multiple kinases, these redundant matches are eliminated based on a comparison of the BLAST E-Value of all PDB code matches to distinct kinase sequences and the match with the most significant E-Value is retained. No resolution or other quality cut-offs are applied for inclusion in the database,
Kinome-wide sequence alignment
To obtain a kinome-wide sequence alignment of the 594 catalytic domain sequences, a HMM-profile was built using hmmbuild in HMMER.31 A total of 18 nonredundant kinase structures were used (Supporting Information Table 1)32. The 18 X-ray structures were aligned using the Protein Structure Alignment tool in Maestro 8.033 and the resulting alignment was exported for use by hmmbuild. After the profile was built, hmmalign31 was used to create the kinome-wide multiple sequence alignment of all 594 kinase catalytic domain sequences.
Chain-based processing
All PDB files are processed on a chain-by-chain basis, so a single RCSB entry can result in multiple entries in the KKB. This allows alignment of each crystallized catalytic domain onto a common reference structure. cAMP-dependent kinase (Manning name PKACa) was used as the reference frame for the structural alignments (1ATP). The TM-Align algorithm was used to create the structural alignments.19
As there is no geometric relationship between the heteroatoms and the various protein chains, an algorithm was developed to remove solvent molecules and ligands that are far away from each chain's active site. The algorithm uses the coordinates of the backbone nitrogen of the hinge-region donor residue (common number 486) to calculate the distance of nonprotein atoms and molecules (identified through the HETATM record identifier) to the active site. Water molecules, which have “HOH” as the residue name, are retained if the distance between backbone nitrogen and the water oxygen is ≤10 Å. For nonwater heteroatoms, other solvent molecules and ligands, including the co-crystallized ligand, the center-of-mass is calculated. Subsequently the distance between the backbone nitrogen donor atom and the center-of-mass of the heteroaromatic residue is calculated. If the distance is ≤20 Å the HETATM is kept. The larger cut-off for HETATM records ensures retention of allosteric inhibitors as, for example, found in MEK1 and MEK2 (Manning name MAP2K1 and MAP2K2, respectively) kinases (e.g., 1S9I and 1S9J).
Naming of entries in KKB
Each entry in the KKB is associated with a PDB/chain identifier combination. Each unique PDB/chain identifier is linked to the Manning name as the standard name for all kinase structures in the database. The Manning name has been linked to the standard name in the GVK Bio database,25 which gives access to a number of synonyms for each kinase. This enables a large number of ways to query for kinases through full and partial synonyms or the Manning name.
Development of common numbering scheme
The common numbering scheme is derived from the kinome-wide HMMER alignment. For each PDB file, the renumbering consists of two main steps. In the first step, the residue numbers and residue names for the corresponding kinase is taken from the kinome-wide sequence alignment. We will call this the Manning sequence. The Manning residue numbers for each position is the common numbering scheme for that kinase, which will subsequently be used to renumber the PDB file in the second step. In the second step, the Manning sequence needs to be matched to the sequence of the residues present in the PDB file. To do this a 3-residue query sequence from the Manning sequence is used to find the corresponding sequence in the PDB file. When a match is found, the residues in the PDB file can be renumbered according to the common numbering scheme. If no match is found, the second triplet from the Manning sequence is used as the query, and so forth. After an initial 3-residue match has been found, subsequent residues will be renumbered after matching residue names in the PDB file to those in the Manning sequence to account for missing residues in the X-ray structure.
Structural annotations
Kinase catalytic domains contain a number of conserved residues, residue motifs, and structural features. Using the common numbering scheme, these can be easily annotated. Conserved residues that have been annotated are the catalytic lysine (common residue 189) and the Helix C acid (usually glutamic acid; common residue 265), the gatekeeper residue (common residue 461), and the hinge-region hydrogen-bond donating residue (common residue 486). In addition, active site residues are defined based on the distance to staurosporine in a CDK2 crystal structure (1AQ1). Residues that are within 4 Å of staurosporine in ≥150 structures (out of 900 analyzed unique chains) are considered active site residues. A total of 24 residues make up the active site, with common numbers 70, 71, 72, 73, 77, 81, 181, 189, 265, 352, 461, 479, 482, 486, 487, 492, 494, 497, 505, 966, 967, 974, 1105, and 1108.
Conserved annotated motifs include the glycine-rich loop, with the sequence of GXGXXG (glycines at positions 71, 73, and 78), the hinge region (common residues 479, 482, 486, 487, and 492), the HRD motif (common residues 929, 930, and 931), and the DFG motif (common residues 1108, 1111, and 1114).
Residues commonly found to be part of the Helix C have been annotated as well (common residues 248–300). The maximum number of residues in Helix C is 22.
Different active site positions, where reactive cysteines have been observed, have been identified by Gray and coworkers.22 The common residue number(s) corresponding to each of the groups is identified based on crystallized structures for each group.
Structural classification schemes
To classify the conformation of the DFG motif into DFG-in and DFG-out, OEChem Python34 scripts were developed to extract the phi, psi, and chi angles for the aspartic acid (common residue 1108) and the distance between the hinge region donor residue (common residue 486) backbone nitrogen and the Ca atom of the DFG motif phenylalanine (common residue 1111). When the distance between the hinge region backbone and the phenylalanine is short (≤13.5 Å), the DFG motif is in the “out” conformation. If the separation is long (≥15.0 Å) the DFG motif is in the “in” conformation. At intermediate distances, the phi and psi angles of the phenylalanine determine whether the loop is in the in or out conformation.
To classify the conformation of Helix C, the distance between the terminal nitrogen of the catalytic lysine (common residue 189) and the Helix C acid (common residue 265) is determined in addition to the chi angle of the Helix C acid. At short (≤4 Å) and long (≥8.5 Å) distances the Helix C is considered to be in the “in” and “out” conformation, respectively. At intermediate distances, the chi angle of the Helix C acid is determined to assess whether the side chain is pointing toward the ATP binding pocket or away from it. When the acid is pointing toward the binding site, the Helix C is considered to be in the “in” conformation, if it is pointing away the Helix C is considered to be in the “intermediate” conformation.
Conclusions
The availability of crystallographic data of protein targets can significantly enhance the drug discovery process. The development of the KKB as part of the in-house PRDB further enhances structure-based drug design and the analysis of structure-activity relationships through kinase-specific annotations of the structures and by making retrieval and comparison of a large number of kinase structures trivial. The developed common numbering scheme allows many kinome-wide structural queries and enabled the automatic classification of the different conformational states of kinase catalytic domains.
The availability of a sequence alignment of the full human kinome has enabled an analysis of the conservation of active site residues, which led to the identification of a conserved hydrogen bond or short-range electrostatic interaction between the hinge region and the specificity surface present in kinases with a glycine at position 494. Furthermore, it was shown that 90% of kinases have one or two stabilizing interactions to help form the hinge region. To the best of our knowledge, the conservation, and thus importance, of stabilizing interactions around the hinge region has not been described before.
As we evolve knowledge and understanding of inhibitor binding through the availability of additional ligand-bound crystal structures, we can readily evolve the KKB to capture new molecular features.
Acknowledgments
N. Brooijmans thanks Eric Bennett for pointing out the TM-align algorithm and Dennis Powell for pointing out the annotation of reactive cysteines done by Gray and coworkers. The authors thank Gary Walker and our GVK Bio collaborators Rupesh Kowticwar, Jyothi Mankala, Satish Palli, Sairam Punyamantula, Maneesh Thatipally, and Reji John for their contributions to the development of PRDB and KKB. They also thank Eric Feyfant and Jack Bikker for helpful discussions.
References
- 1.Deshpande N, Addess KJ, Bluhm WF, Merino-Ott JC, Townsend-Merino W, Zhang Q, Knezevich C, Xie L, Chen L, Feng Z, Green RK, Flippen-Anderson JL, Westbrook J, Berman HM, Bourne PE. The RCSB protein data bank: a redesigned query system and relational database based on the mmCIF schema. Nucleic Acids Res. 2005;33:D233–D237. doi: 10.1093/nar/gki057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alvarez JC. High-throughput docking as a source of novel drug leads. Curr Opin Chem Biol. 2004;8:365–370. doi: 10.1016/j.cbpa.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Bohacek RS, McMartin C, Guida WC. The art and practice of structure-based drug design: a molecular modeling perspective. Med Res Rev. 1996;16:3–50. doi: 10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 4.Hardy LW, Malikayil A. The impact of structure-guided drug design on clinical agents. Curr Drug Discov. 2003:15–20. December. [Google Scholar]
- 5.Hajduk PJ. Fragment-based drug design: how big is too big? J Med Chem. 2006;49:6972–6697. doi: 10.1021/jm060511h. [DOI] [PubMed] [Google Scholar]
- 6.Rawlings ND, Morton FR, Kok CY, Kong J, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2008;36:D320–D325. doi: 10.1093/nar/gkm954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allcorn LC, Martin AC. SACS–self-maintaining database of antibody crystal structure information. Bioinformatics. 2002;18:175–181. doi: 10.1093/bioinformatics/18.1.175. [DOI] [PubMed] [Google Scholar]
- 8.Jacobs MD, Caron PR, Hare BJ. Classifying protein kinase structures guides use of ligand-selectivity profiles to predict inactive conformations: structure of lck/imatinib complex. Proteins. 2008;70:1451–1460. doi: 10.1002/prot.21633. [DOI] [PubMed] [Google Scholar]
- 9.Hendlich M, Bergner A, Gunther J, Klebe G. Relibase: design and development of a database for comprehensive analysis of protein-ligand interactions. J Mol Biol. 2003;326:607–620. doi: 10.1016/s0022-2836(02)01408-0. [DOI] [PubMed] [Google Scholar]
- 10.Bergner A, Gunther J, Hendlich M, Klebe G, Verdonk M. Use of Relibase for retrieving complex three-dimensional interaction patterns including crystallographic packing effects. Biopolymers. 2001;61:99–110. doi: 10.1002/1097-0282(2001/2002)61:2<99::AID-BIP10075>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 11.Schreyer A, Blundell T. CREDO: a protein-ligand interaction database for drug discovery. Chem Biol Drug Des. 2009;73:157–167. doi: 10.1111/j.1747-0285.2008.00762.x. [DOI] [PubMed] [Google Scholar]
- 12.Hu L, Benson ML, Smith RD, Lerner MG, Carlson HA. Binding MOAD (Mother of all databases) Proteins. 2005;60:333–340. doi: 10.1002/prot.20512. [DOI] [PubMed] [Google Scholar]
- 13.Liu T, Lin Y, Wen X, Jorissen RN, Gilson MK. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic Acids Res. 2007;35:D198–D201. doi: 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X, Liu M, Gilson MK. BindingDB: a web-accessible molecular recognition database. Comb Chem High Throughput Screen. 2001;4:719–725. doi: 10.2174/1386207013330670. [DOI] [PubMed] [Google Scholar]
- 15.Brooijmans N, Mobilio D, Walker G, Nilakantan R, Denny RA, Feyfant E, Diller D, Bikker JA, Humblet C. Structural informatics approaches to mine kinase knowledge-bases. Drug Discov Today. doi: 10.1016/j.drudis.2009.11.005. (in press). doi: 10.1016/j.drudis.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 16.Cohen P. Protein kinases–the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 17.Bikker JA, Brooijmans N, Wissner A, Mansour TS. Kinase domain mutations in cancer: implications for small molecule drug design strategies. J Med Chem. 2009;52:1493–1509. doi: 10.1021/jm8010542. [DOI] [PubMed] [Google Scholar]
- 18.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akamine P, Madhusudan J, Xuong NH, Ten Eyck LF, Taylor SS. Dynamic features of cAMP-dependent protein kinase revealed by apoenzyme crystal structure. J Mol Biol. 2003;327:159–171. doi: 10.1016/s0022-2836(02)01446-8. [DOI] [PubMed] [Google Scholar]
- 21.Wu J, Yang J, Kannan N, Madhusudan NH, Ten Eyck LF, Taylor SS. Crystal structure of the E230Q mutant of cAMP-dependent protein kinase reveals an unexpected apoenzyme conformation and an extended N-terminal A helix. Protein Sci. 2005;14:2871–2879. doi: 10.1110/ps.051715205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tripos, L.P. Benchware 3D Explorer 2.5. Saint Louis: Tripos L.P.; 2008. [Google Scholar]
- 24.Nilakantan R, Bauman N, Haraki KS. Database diversity assessment: new ideas, concepts, and tools. J Comput-Aided Mol Des. 1997;11:447–452. doi: 10.1023/a:1007937308615. [DOI] [PubMed] [Google Scholar]
- 25. GVK. GVK Bio Databases. Available at: http://www.gvkbio.com/informatics.html August 2009.
- 26.Wissner A, Mansour TS. The development of HKI-272 and related compounds for the treatment of cancer. Arch Pharm. 2008;341:465–477. doi: 10.1002/ardp.200800009. [DOI] [PubMed] [Google Scholar]
- 27.Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, Mendonca RV, Sweeney MD, Scott KC, Grothaus PG, Jeffery DA, Spoerke JM, Honigberg LA, Young PR, Dalrymple SA, Palmer JT. Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. Chem MedChem. 2007;2:58–61. doi: 10.1002/cmdc.200600221. [DOI] [PubMed] [Google Scholar]
- 28.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 29.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, III, Snoeyink J, Richardson JS, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kannan N, Neuwald AF. Did protein kinase regulatory mechanisms evolve through elaboration of a simple structural component? J Mol Biol. 2005;351:956–972. doi: 10.1016/j.jmb.2005.06.057. [DOI] [PubMed] [Google Scholar]
- 31.Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 32.Sheinerman FB, Giraud E, Laoui A. High affinity targets of protein kinase inhibitors have similar residues at the positions energetically important for binding. J Mol Biol. 2005;352:1134–1156. doi: 10.1016/j.jmb.2005.07.074. [DOI] [PubMed] [Google Scholar]
- 33. Schrodinger (2007) Maestro 8.0. Manual. New York, NY: Schrodinger, p294.
- 34. OpenEye Scientific Software(2008) OEChemTK 1.6.0. Santa Fe, NM: OpenEye Scientific Software, Inc.