

Abstract
Hepatitis B virus (HBV) DNA polymerase (HDP) is a pharmacological target of intense interest. Of the seven agents approved in USA for the treatment of HBV infections, five are HDP inhibitors. However, resistance development against HDP inhibitors, such as lamivudine and adefovir, has severely hurt their efficacy to treat HBV. As a step toward understanding the mechanism of resistance development and for gaining detailed insights about the active site of the enzyme, we have built a homology model of HDP which is an advance over previously reported ones. Validation using various techniques, including PROSTAT, PROCHECK, and Verify-3D profile, proved the model to be stereochemically significant. The stability of the model was studied using a 5 ns molecular dynamics simulation. The model was found to be sufficiently stable after the initial 2.5 ns with overall root mean squared deviation (RMSD) of 4.13 Å. The homology model matched the results of experimental mutation studies of HDP reported in the literature, including those of antiviral-resistant mutations. Our model suggests the significant role of conserved residues, such as rtLys32, in binding of the inhibitors, contrary to previous studies. The model provides an explanation for the inactivity of some anti-HIV molecules which are inactive against HDP. Conformational changes which occurred in certain binding pocket amino acids helped to explain the better binding of some of the inhibitors in comparison to the substrates.
Keywords: comparative modeling, sequence, enzyme, molecular dynamics, triphosphate, infectious disease, resistance
Introduction
Hepatitis B is among the top 10 infectious diseases in the world. An estimated 350 million people worldwide are chronically infected with the hepatitis B virus (HBV).1 As patients age, hepatitis B can cause serious infection, cirrhosis of the liver, liver cancer, and liver failure, leading to death. Although HBV infection can be largely prevented by use of a safe and effective vaccine, not everyone is vaccinated and there is no vaccine for the people who are already chronically infected. Therefore, there is continuous need for antiviral drugs to suppress viral replication or eliminate infection.
HBV DNA polymerase (HDP) has been of considerable interest as a target for treatment of HBV infections in the last few years. Currently, seven agents are approved for the treatment of chronic HBV. Interferon alfa-2b (IFN) and long-acting pegylated interferon (peginterferon) alfa-2a (PEG-IFN) are two biological products which have shown very good anti-HBV activity. Efficacy of IFN is improved by conjugation with polyethylene glycol. However, issues of practicality, high cost, effectiveness, and severe adverse reactions have limited the use of these agents for the treatment of chronic HBV. Apart from the two biologics, five nucleoside/nucleotide analogs have been approved for the treatment of HBV infections,2 and they are all HDP inhibitors. Lamivudine, a small molecule inhibitor initially developed for treatment of human immunodeficiency virus Type 1 (HIV-1), showed very good inhibitory activity against HDP. In addition to lamivudine, adefovir dipivoxil, entecavir, telbivudine, and tenofovir (Fig. 1) are the small molecules available in the market in USA,2,3 and several others are in clinical studies. Subsequent to entry into the cell, these inhibitors undergo phosphorylation by cellular kinases to their bioactive form. Severini et al. determined that lamivudine inhibits viral replication by acting as a chain terminator of both the reverse transcriptase (RT) and DNA polymerase activities of Duck HDP.4 Subsequently, all the nucleotide/nucleoside analogs that have been approved for the treatment of human HBV infections were demonstrated to inhibit HDP.
Figure 1.
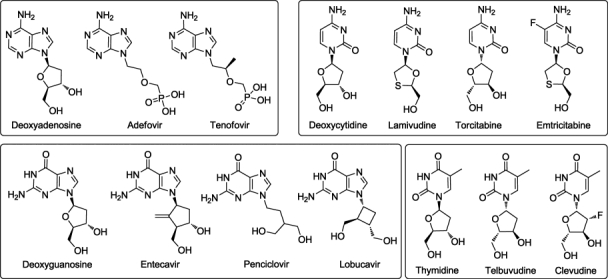
HDP-inhibitors used for the docking studies. The inhibitors are classified based on the nucleoside present. The natural substrates are shown on the left of each box and the corresponding competitive inhibitors are shown to the right of the most closely corresponding natural substrate.
Accurate knowledge of the 3D structure of HDP would be valuable for understanding the molecular basis of many of its properties, including mechanisms of polymerization, inhibition, and drug resistance, and for interpretation of clinical and biochemical data. However, attempts to determine the X-ray structure of HDP have not yet been successful.
Mutation studies reported in the literature provide valuable information about the structure and function of proteins and provide particularly useful clues about the homology of a protein when its 3D structure is not available. In HIV-1 RT, several key point mutations, including K65R, K70R, L74V, Q151M, M184I/V, and T215Y/F, are reported to be associated with resistance to nucleoside (nucleotide) analogs. These mutations (termed TAMs for Thymidine-Associated Mutations) are located in the active site where the incoming nucleotide interacts with the enzyme.5 Several mutations in corresponding positions have been reported in HDP. For example, the most common lamivudine resistance mutation rtM204I/V in HDP, which occurs in the important YMDD motif, corresponds to the M184I/V lamivudine resistant mutation in HIV-1 RT. This mutation is mainly accompanied in HDP by rtL180M. Substitution of a β-branched amino acid (I, V, or T) for methionine at position 204 along with the rtL180M mutation confers a high degree of resistance to lamivudine and other L-nucleoside analogs. Many of the residues involved in key protein-ligand interactions in HIV-1 RT are conserved between the HIV-1 RT structure and HDP. These include the YMDD motif, the catalytic triad of aspartic acids residues, rtM250 and rtG251. All these data suggest that HIV-1 RT shares sufficient structural and functional similarity with HDP to serve as an excellent template to build a 3D structure for HDP.
Homology modeling is a very good tool for prediction of the 3D structure of a protein for which an experimental structure is not available.6 Good sequence homology between HDP and HIV-1 RT has enabled several researchers to build 3D homology models of the structure of the HDP catalytic domain,7–9 which were used to provide insights about the binding mode of various inhibitors and developing resistance. Das et al. aligned the sequence of HDP to HIV-1 RT and used an HIV-1 RT X-ray structure 1RTD.pdb for building a tertiary structure of HDP, which was used to reveal the mechanism of HDP resistance to lamivudine and emtricitabine.7 Chu and coworkers further utilized the same model for MD studies to explain the mechanism of action of clevudine and adefovir sensitivity and resistance.10–12 Bartholomeusz et al. developed another alignment of the sequence of HDP to HIV-1 RT and then also used 1RTD.pdb for building their model for HDP, and used it for mapping out the effects on the HDP model of various mutations reported in the literature.8 Langley et al. reported docking studies of entecavir using a homology model of HDP and its various mutants. Those authors used the same sequence alignment as Das et al. and prepared their 3D model using 1RTD as a template.9
In this article, we report a different homology model of HDP based on a better alignment and a more recent HIV-1 RT X-ray structure. Our model can be used to explain all mutations in HDP reported to date as well as the inactivity of some known HIV-1 RT inhibitors against HDP. The model was validated using docking and various protein-validation tools. The stability of the model was assessed using molecular dynamics simulation with a 5 nanosecond production run. The model was stable and can serve as a good starting point for further investigations of mechanistic insights of the enzyme.
Results and Discussion
Homology model of HDP
Alignment
The sequence alignment of HDP with HIV-1 RT is shown in Figure 2. Previous studies have reported that the K65R mutation in HIV-1 RT confers reduced susceptibility to a number of NRTIs including dideoxynucleotides (ddN), lamivudine,13 and tenofovir.14,15 In the reported crystal structures of HIV-1 RT, the side chain of K65 is involved in ionic interaction with the γ-phosphate of the incoming nucleotide.16 The K65R mutation causes changes in the electrostatic interactions and destabilizes the positioning and alignment of the phosphate groups in the active site, slowing the catalytic step.17 This mutation confers intermediate resistance against ddC, tenofovir, adefovir, and lamivudine, when associated with other mutations. Although no mutational data has been reported to date for the lysine corresponding to K65R in HDP, rtK32, it could be expected to play a similar role to that played by K65 in HIV-1 RT for binding of similar analogs and the development of resistance to inhibitors. While carrying out the sequence alignment, special attention was paid to this aspect. As shown in the alignment (Fig. 2), rtK32 of HDP is aligned with K65 of HIV-1 RT. In the final model, as in the template, rtK32 was involved in ionic interactions with the incoming nucleotide, vide infra. In the previous homology models built by Das et al.7 and Bartholomeusz et al.8 K66 instead of K65 of HIV-1 RT was considered to be a conserved residue. Alignment of K65 with rtK32 led to insertion of a dipeptide, instead of the tripeptide insertion predicted by Das et al., after S69. Most experimental studies have reported a dipeptide, and not a tripeptide, insertion between residues 69 and 70, conferring resistance to many NRTIs.18,19 Considering all aforementioned observations, we believe that rtK32 is an important residue playing a significant role in binding of nucleotides and known HDP inhibitors. This hypothesis could be proved by mutation studies of this residue.
Figure 2.

The sequence alignment of HBV polymerase and HIV-1 RT used in construction of the model. Conserved residues are highlighted with gray background.
De Clercq has reviewed anti-HBV activity of many HIV-1 NRTIs. According to his findings, many NRTIs including ddN's, AZT, and d4T did not show any activity against HBV.20 For HIV-1 RT, the L74V mutation is responsible for development of resistance to ddN's. In agreement with previous results, our alignment also explains the inactivity of ddN's and thymidine analogs. This resistance is conferred mainly by L74V and Q151M mutations in HIV-1 RT; the corresponding amino acids in HDP are rtV43 and rtM171. The previously published alignments considered Y115F to be a conserved substitution.7,8 Although there is considerable similarity between the two aromatic amino acids, Tyr and Phe, the substitution could still be significant in this case, since studies have shown that this substitution in combination with L74V mutation confers HIV-1 RT resistance against abacavir.21 Our final model included loops of length 26, 11, and 9 residues, whereas the model of Das et al.7 had corresponding loops of 26 and 23 residues, while Bartholomeuz et al.8 had one long loop of 46 residues.
Salient features of homology model
The final model of HDP is shown as a ribbon diagram (in two different orientations) in Figure 3. Like HIV-1 RT and other (RNA- as well as DNA-dependent) polymerases, the modeled HDP folds in a classic “right hand” shape with fingers, palm, and thumb subdomains. Very good similarity was found in the “palm” subdomain of HIV-1 RT and HDP. Various motifs such as YMDD and MGY are conserved. Many key protein–NTP and protein–DNA interactions are conserved in the resultant HDP model. The overall subdomain structure of the model is consistent with the findings reported by O'Reilly and Kao.22 The finger subdomain is composed of two α-helices containing residues rt1–49 and rt90–rt172. The thumb subdomain contains residues rt268–rt351 and is mostly α-helical. The palm subdomain contains residues from rt50 to rt89 and rt173 to rt267 and is dominated by β-sheets with very few α-helices.
Figure 3.
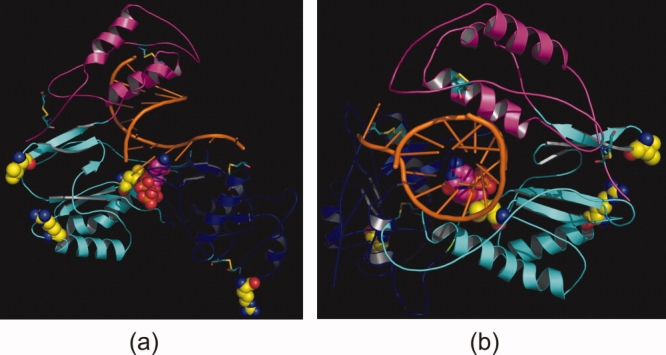
The HBV polymerase is shown as a ribbon diagram (a and b show two different orientations) with the fingers (rt1–49 and rt90–rt172), palm (rt50–rt89 and rt173–rt267), and thumb (rt268–rt351) subdomains in pink, cyan, and blue, respectively. The bound double-stranded DNA template primer is shown as a stick model in orange ribbon mode, and dCTP is in magenta (with P, N and O atoms in orange, blue and red, respectively). The four proposed disulfide links are represented by stick models. The four residues present in the disallowed region in the Ramachandran plot are shown in yellow space-filling model. [Color figure can be viewed in the online issue, which is available at http://www.interscience.wiley.com.]
All of the NRTIs are prodrugs and must undergo intracellular conversion to mono-, di-, and, finally, active triphosphates through the action of cellular kinases. The nucleotide binding site is buried in the center of the protein. The site is formed from all three domains together, along with the DNA template. The three aspartate residues of the catalytic triad are rtD83, rtD205, and rtD206, corresponding in HIV-1 RT to D110, D185, and D186, respectively. These aspartate residues are responsible for the coordination of the metal ion which participates in the nucleotidyl transfer reaction. One of the Mg+2 ions is coordinated by aspartate residues, the 3′OH of the last nucleotide of the DNA primer, and the α-phosphate of the incoming nucleotide. The other Mg+2 ion is coordinated by the aspartate residues, the backbone carbonyl of rtV84, and the phosphate of the incoming nucleotide. The two Mg+2 ions are very important for the activity of the enzyme.
One of the most studied mutations in HIV-1 RT is a mutation in the YMDD motif, M182V/I. The mutation of the corresponding Met residue in HDP has also been reported. These mutations are responsible for resistance to lamivudine which both HIV-1 RT and HDP can develop. Various crystallographic analyses for HIV-1 RT23 and molecular modeling studies on homology-modeled HDP7,24 have shown that Cβ-side chain amino acids, such as Val, Ile, or Thr, can cause steric hindrance to the incoming lamivudine nucleotide. As seen from the model, rtM204 is present in the active site in close proximity to the ligand, next to two aspartate residues of the catalytic triad.
Model assessment
The final homology model of HDP was evaluated with various tools including PROCHECK and PROFILE3D analysis. The Ramachandran plot of the model is shown in supporting information (Fig. S1). A total of 70.7% of the residues were found in the most favored regions (compared with 72.3% in the HIV-1 RT template), 25.6% in additionally allowed regions (26.2%), and 2.4% in the generously allowed regions (1.2%). Only four residues were found in the disallowed regions. Analysis of the model revealed that three of those four outlying residues were located far from the putative active site of the protein. rtM204 of the active site YMDD motif was also found in the disallowed region. Detailed analysis of this residue revealed that the corresponding Met residue in many HIV-1 RT protein complexes was also found in the disallowed region, suggesting that this is not an error but rather a valid conserved feature of the protein structure. The overall PROCHECK score of the model was −0.34, indicating that the molecular geometry is stereochemically acceptable. Figure S2 (supporting information) shows a ribbon diagram of the model colored by Verify score.25 The total Verify score of the modeled structure was 100.5 (Expected High Score = 171.4; Expected Low Score = 77.1). The results showed that our modeled structure was of comparable quality to standard PDB structures. Overall root mean squared deviation (RMSD) of the modeled structure was 1.02 Å (0.98 Å without including the three long loops).
Docking and binding pose validation
Once the model was prepared and the quality of the structure validated, we carried out docking of known substrates/inhibitors of HDP into the active site. As aforementioned, the nucleosides used in the docking studies were modified into their bioactive form (nucleotides). Four different models were used as input for docking, depending upon the nucleotides present in the ligand. Table I shows docking scores of all ligands used in the study. The docking studies revealed that the incoming nucleotides (substrate or inhibitor) docked well into the active site. The binding pose of CTP is shown in Figure 4. The incoming nucleotide fit well in the complex, making π-stacking interactions with the primer DNA base and hydrogen bonds with the complementary DNA base in the template. A number of hydrogen bonds stabilized the complex. The nitrogen base of the incoming nucleotide (CTP in this case) exhibited three hydrogen bonds with the complementary base pair in the active site. The phosphate binding region of the pocket was lined by backbone NH's of various amino acids. The γ-phosphate formed a hydrogen bond with the backbone amide NH of rtA86, and made ionic interactions with rtK32. The β-phosphate hydrogen bonded with the backbone amide NH group of rtA86, and the α-phosphate of the ligand was involved in ionic interactions with rtR41 in the active site. The phosphate backbone was involved in metal chelating interactions with two Mg+2 ions present in the active site. Other ligands used in the docking studies were found to have similar interactions and binding pose to CTP.
Table I.
Docking Scores of the Molecules Used in Docking Studies Using Four Different HDP Models
| Predynamics score |
Postdynamics score |
||||||
|---|---|---|---|---|---|---|---|
| HDP model | Ligand | Gold score | Chem score | ΔG | Gold score | Chem score | ΔG |
| HDP_A | Clevudine | 127.17 | 17.90 | −37.59 | 115.81 | 11.73 | −41.71 |
| Telbivudine | 130.83 | 17.99 | −36.90 | 126.94 | 13.05 | −37.98 | |
| TTP | 115.80 | 16.50 | −26.25 | 131.34 | 24.85 | −40.25 | |
| HDP_C | PCVTP | 136.54 | 25.33 | −40.06 | 143.06 | 17.15 | −41.48 |
| ETV | 126.45 | 13.78 | −32.79 | 155.20 | 25.95 | −56.67 | |
| LBVTP | 135.43 | 23.41 | −44.55 | 163.77 | 21.66 | −41.42 | |
| GTP | 149.22 | 15.72 | −30.78 | 168.77 | 20.96 | −38.44 | |
| HDP_T | Tenofovir | 114.17 | 10.85 | −35.81 | 146.43 | 24.33 | −39.76 |
| Adefovir | 116.76 | 11.70 | −34.98 | 138.63 | 21.98 | −33.53 | |
| ATP | 121.86 | 6.97 | −32.60 | 145.68 | 19.22 | −33.17 | |
| HDP_G | Emtricitabine | 131.90 | 24.98 | −45.45 | 138.58 | 27.20 | −43.20 |
| Lamivudine | 130.32 | 19.06 | −30.35 | 139.30 | 32.78 | −52.89 | |
| Torcitabine | 125.51 | 27.63 | −39.35 | 136.58 | 28.16 | −42.16 | |
| CTP | 131.33 | 26.26 | −42.55 | 158.55 | 26.94 | −36.96 | |
Figure 4.
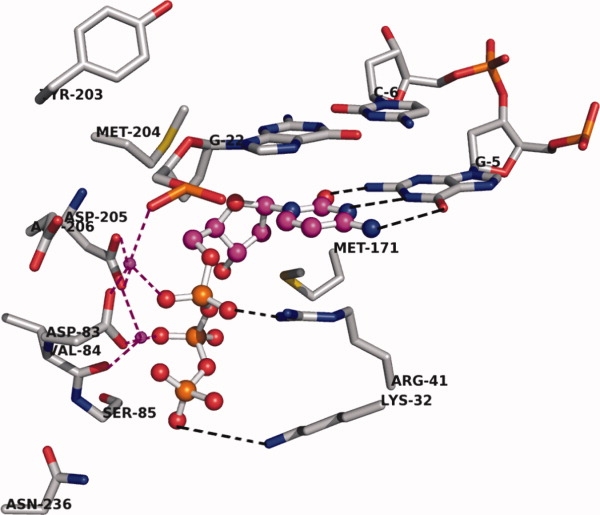
Close-up view of the active site of HDP before MD simulation. Two Mg+2 ions are shown in magenta colored small spheres. The ligand, CTP, is shown with magenta carbon in ball-and-stick mode. Mg coordination is shown with magenta dotted lines. Hydrogen bonding interactions are shown with black dotted lines. Only the important amino acids in the active site are shown; rtR41 and rtK32 are involved in ionic interactions with the phosphate group. Backbone and hydrogen atoms are removed for clarity. [Color figure can be viewed in the online issue, which is available at http://www.interscience.wiley.com.]
The docking studies provided insights into the binding of ligands to the active site and subsequent conformational changes in the active site. Lamivudine is a nucleoside, which contains an L-configuration oxathiolane ring. Docking studies revealed the specific orientation of the heterocyclic ring which is in close proximity to the side-chain of rtM204 (Fig. 5). As discussed earlier, mutation of this residue leads to resistance development. As shown in Figure 5(b), replacement of rtM204 with a β-branched amino acid (Val, Thr, or Ile) would make unfavorable contacts (steric hindrance) of the heterocycle with the amino acid, thus inducing a repositioning of the inhibitor in the active site. The oxathiolane ring of lamivudine was observed to occupy a small hydrophobic pocket formed at the rear of the dNTP binding site in the model. This pocket is lined with hydrophobic residues like rtA87, rtF88, rtP177, rtL180, and rtM204, along with the last nucleotide of the primer DNA strand. These findings are consistent with those reported by Langley et al.9
Figure 5.

(a) Binding of lamivudine in the active site of HDP before MD simulation. All the interactions present in the natural nucleotide are shown by lamivudine. Its oxathiolane ring is in close proximity to rtM204. The surface of the ligand is shown in wire mesh, colored with the electrostatic potential. (b) Replacement of rtM204 with Ile causes unfavorable contacts of the oxathiolane ring with Ile, which is a β-branched amino acid. [Color figure can be viewed in the online issue, which is available at http://www.interscience.wiley.com.]
Molecular dynamics simulations
The relative stability of the whole model can be measured as the root mean square deviation (RMSD) of backbone atoms from the initial structure as a function of time. The model built with Swiss-Pdb Viewer was taken as the initial structure for comparison. The RMSD in Figure 6 was calculated for each MD snapshot (one every 4 ps) after the coordinates of the model were superimposed on the coordinates of the initial structure. The overall RMSD of the homology model after simulation for 5.0 ns was ∼3.8 Å. As seen from the figure, the modeled structure rapidly deviated from the initial structure during the first nanosecond of the simulation run. This increase in the RMSD was because of the optimization of interactions within the protein structure, as well as with the water solvent. After ∼2.5 ns the total RMSD had stabilized at 3.5–4.0 Å. The average RMSD of the backbone in the model during 2.5–5 ns of the production run was 3.69 Å. Hence, this is a satisfactory structural model.
Figure 6.
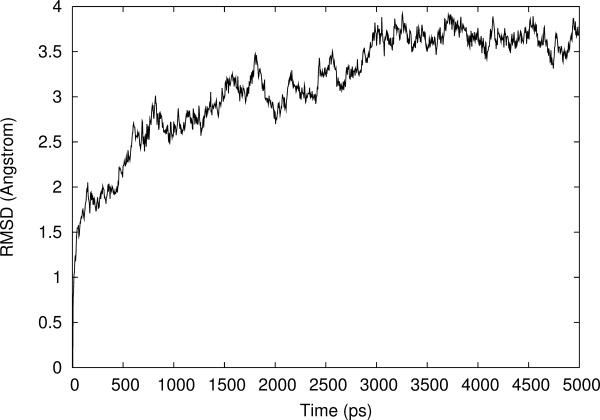
Root Mean Squared deviation (RMSD) of Cα backbone of HDP homology model during the molecular dynamics simulation relative to the initial homology model.
Figure 7 shows fluctuations in hydrogen bonding distances in H-bonding partners (distances between heavy atoms). Hydrogen bonding interactions found in the initial model were retained during the MD simulation. Analysis of the trajectories revealed that the hydrogen bonding interactions shown by CTP with the initial model were maintained throughout the simulation. Hydrogen bonds between the C:G pair were preserved during the whole simulation, showing 100% occupancy [Fig. 7(a,b,c)]. A hydrogen bond between rtR41 and the α phosphate O was also retained throughout the simulation, showing an overall occupancy of ∼82% [Fig. 7(d)]. The side-chain amide of rtN236 was located ∼7 Å away from the γ phosphate of CTP in the initial model. However, during the first 500 ps of the MD simulation production run, the loop containing rtN236 moved toward the active site and after that the side-chain amide group of rtN236 was seen to be involved in hydrogen bonding with the γ-phosphate of the binding nucleotide with more than 76% occupancy [Fig. 7(e)]. The rtN236T mutation has been reported to be responsible for resistance development against adefovir.26 The rtK32 terminal NH2 showed ionic interactions with the terminal phosphate group of the incoming nucleotide. This ionic interaction (measured in terms of H-bond distance) showed only ∼47% occupancy but nevertheless this suggests that rtK32 is also important in ligand recognition by HDP.
Figure 7.
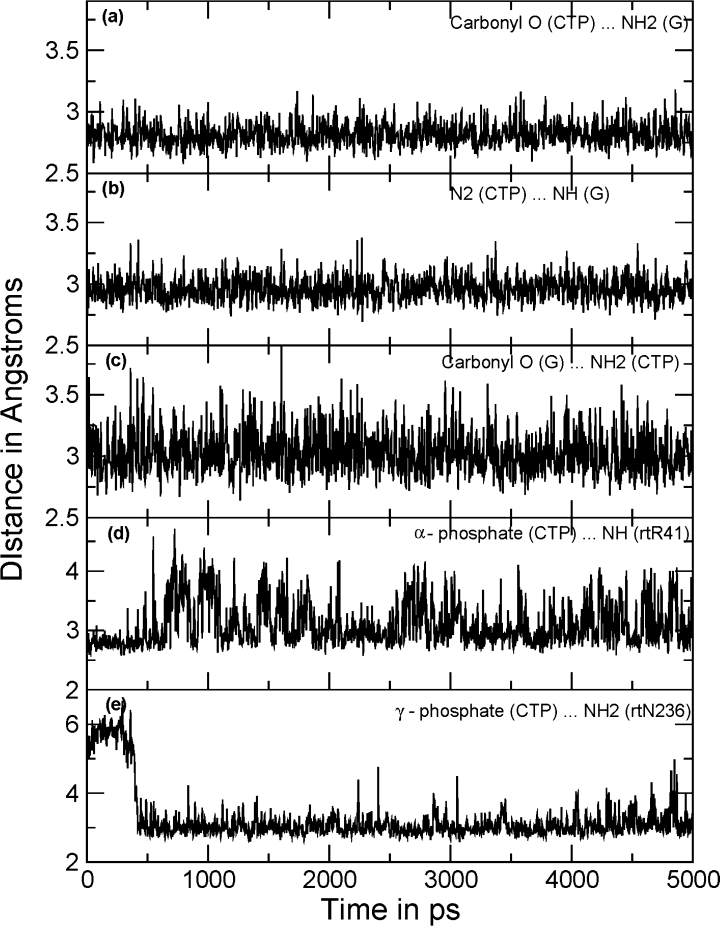
Distance plots between the hydrogen bonding atoms. All the distances shown in the figure are between heavy atoms (O…N or N…N). Three hydrogen bonds between the C:G pair were retained throughout the simulations showing 100% occupancy: (a) between carbonyl O (CTP) and NH2 of G; (b) between N2 of CTP and NH of G; (c) and between NH2 of CTP and carbonyl O of G. Panel (d) shows variations in distance between the α-phosphate of CTP and the terminal guanidine group of rtR41. (e) The residue rtN236 was distant in the beginning of the MD simulation; loop movement pushed the side-chain amide toward the γ-phosphate of CTP as depicted.
The structures from before and after MD simulation are shown in Figure 8. Overall, considering the secondary structure of the protein throughout the simulation, the protein fold was not disturbed to a large extent. Only the secondary structure of the longest loop changed significantly: it lost its α-helix and hence showed structure after equilibration which was quite different from the initial structure. Das et al.7 proposed the presence of an α-helix in this loop, similar to that predicted from the algorithms used in our work. However, Das et al. restricted their studies only to model building and subsequent biochemical characterization and did not carry out MD simulations with their HDP homology model. Hence, it is difficult to compare their reported model with our model after MD simulation. The overall energetic analysis, RMSD, and RMS Cα fluctuations analysis (not shown) suggest that our HDP homology model has acceptable stability.
Figure 8.
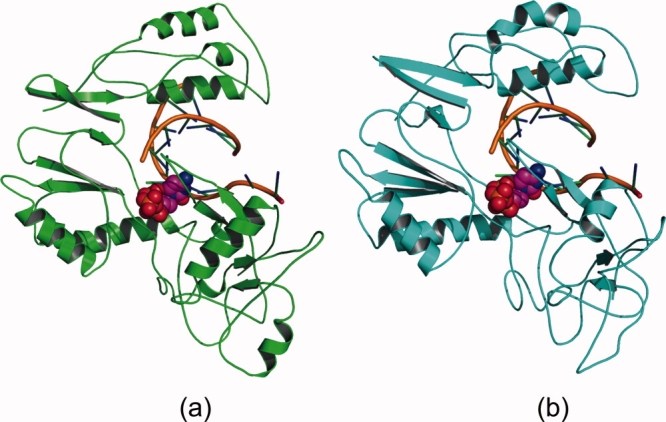
Cartoon picture of overall fold of the homology model (a) before (green) and (b) after ∼4.5 ns (cyan) of the 5 ns MD simulation. Ligand is shown in space-filled mode in the active site. [Color figure can be viewed in the online issue, which is available at http://www.interscience.wiley.com.]
Docking after MD simulation
Docking into the protein structure obtained after 5 ns of MD simulation (described above) produced very similar binding poses to those before the MD simulation for most of the ligands. However, some changes were noticeable, such as for entecavir. Docking before MD simulation revealed that the exocyclic double bond of entecavir occupied a similar pocket to that of the oxathiolane ring in lamivudine (mentioned above), forming hydrophobic interactions with the side-chains of hydrophobic amino acids [Fig. 9(a)]. Conformational changes in amino acid side-chains during the MD simulation generated an additional small cavity lined by hydrophobic residues. Before dynamics, the pocket was not obvious and not available for the docking. Molecular docking after MD simulation gave a more precise picture of the binding of entecavir into the active site. As seen from Figure 9(b), the exocyclic double bond of entecavir is buried in the pocket, thus increasing van der Waals interactions between ligand and enzyme. This resulted in an increase in the predicted binding affinity (increase in GoldScore by 28 and decrease in ΔG by −24). Langley and coworkers have reported a similar binding pose for entecavir, and studied in detail the mechanism of action of entecavir in chain termination.9
Figure 9.
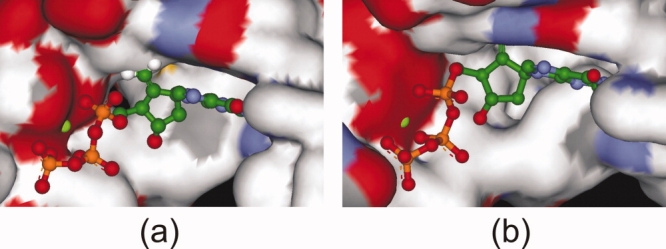
GOLD predicted binding pose of entecavir in the active site of HDP: (a) homology model before MD simulation and (b) after MD simulation of the model. Entecavir is shown in ball-and-stick mode. The surface of the active site is colored by atom type. The five-membered ring exocyclic double bond occupies a small hydrophobic pocket which is generated only after MD simulation (see text for details). [Color figure can be viewed in the online issue, which is available at http://www.interscience.wiley.com.]
The sulfur atom of the oxathiolane ring in lamivudine occupied the same pocket. The increased hydrophobic interactions in the pocket caused a higher docking score and decreased ΔG for lamivudine for postdynamics docking. The increased binding affinity of TTP to the enzyme after MD simulations was because of fewer steric clashes of TTP with the active site and more hydrophobic interactions.
Few experimental binding studies have been reported for the inhibitors we treat in this work and the reported binding data are not consistent. For example, the Ki's for HDP inhibition from Das et al.7 and Langley et al.9 disagree for lamivudine, 250 nM versus 4.4 nM, and for adefovir, 100 nM versus 22.2 nM, so it is not possible even to state which inhibitor binds better. There is no single report which studies binding of all the inhibitors, which would be the most useful for comparison with our calculated results.
Materials and Methods
Homology modeling
Building a 3D model for a protein includes a number of steps: (1) sequence alignment of target with template; (2) generation of initial homology model; (3) model refinement; and (4) model evaluation.
The entire amino acid sequence database available in the Protein Data Bank (PDB) was used in the search. Secondary structure prediction was performed using the PSIPRED server27 and various algorithms available in SYBYL7.2.28 The results were used to assist analysis for the sequence alignment. Sequence alignment was then carried out with CLUSTALW1.8.29 The obtained alignment was then refined manually.
Template selection
Template selection was a very crucial step. Numerous HIV-1 RT crystal structures are available in the PDB database. From the structures available, 1RTD30 (3.2 Å), 1T0516 (2.8 Å), and 2HMI31 (2.8 Å) were selected. Although other structures with higher resolution are available, we chose to use these structures because each contains a nucleoside triphosphate (NTP) as a bound ligand in the active site, whereas most of the other available structures either are complexes with non-nucleoside ligands or are holoenzymes. Out of the three structures, 1T05 was chosen as the final template for homology modeling since it contains a bound inhibitor. The remaining two structures were used in sequence alignment.
Generation of initial model
Following the alignment, the backbone coordinates of the residues in HDP were generated with the stand-alone version of Swiss-Pdb Viewer 3.7.32 Regions in the HIV-1 RT enzyme that aligned with the structurally conserved regions (SCRs) among the three templates were generated by copying the backbone coordinates from 1T05. A loop search was carried out using all the structures in the PDB. For the loop with 25 amino acids, secondary structure prediction using the PHD server27 (ProteinPredict) suggested the presence of an alpha helix in the loop. Following construction of the initial backbone model, initial side-chain conformers were generated. Prediction of disulfide linkages in the model was carried out using DISULFIND.33 Based on the distances between cysteine residues and prediction by DISULFIND, 4 disulfide bonds were assigned, the same as those assigned by Das et al.7
Model refinement
Once the initial model was ready, it was then transferred to SYBYL7.2. The coordinates of the DNA model and Mg+2 ions were transferred from the 1RTD.pdb structure. The resultant model consisted of the quaternary complex of HDP-dTTP with DNA and two Mg+2 ions. The complex was energy minimized with the Amber02 force field in SYBYL7.2. The minimization was carried out in a step-wise manner first with hydrogen minimization, next side-chain minimization and then backbone minimization. Active site minimization was carried out with constraints on Mg+2 coordinating amino acids and ligands to maintain octahedral geometry around the ions. The whole protein structure, without any constraints, was further minimized for 500 steps using the steepest-descent method, followed by 1000 steps using the conjugate-gradient method.
Model assessment
The model quality was assessed using a variety of validation tools, including PROSTAT and PROFILE3D modules of InsightII.25 The reliability of the model was further evaluated using docking simulations and binding analysis.
Molecular docking
The minimized protein–ligand–DNA complex was used for docking studies with the GOLD program.34 The active site was defined as a sphere of radius 15 Å surrounding the terminal carbon of rtM204. The ligands used in docking are shown in Figure 1. All of these NRTIs must undergo intracellular conversion to the mono-, di-, and finally, active triphosphates through the action of cellular kinases. Hence, the ligands were modified into their corresponding bioactive form (triphosphate) before docking. All the ligands were sketched and minimized using the MMFF94 force field and charges in SYBYL7.2.28 The docking was nucleotide analog-specific, using the four models described above with appropriate nitrogen base present. For the docking of CTP, lamivudine, and other cytosine analogs, the nucleotide present in the active site was GTP, and so on. Two different scoring functions were used for docking and ranking of the ligands, GoldScore and ChemScore.
Molecular dynamics
Minimization and molecular dynamics (MD) of docked conformation–HDP complexes were carried out using the sander module and amber03 force field in AMBER8.0.35
The potential energy of the whole system (the model in explicit water solvent) during the equilibration phase of the MD simulation was constant which demonstrated the overall stability of the model. The 5 ns MD simulation brought the potential energy to a relatively stable value. The trajectories from the MD simulation were analyzed using the ptraj module of AMBER8.0. The model stability was checked after each step, monitoring the root mean square deviation (RMSD) of the backbone as well as the temperature, density and energy (total, kinetic and potential) of the system. The overall atom fluctuation of the system was calculated by determining RMS fluctuations.
Post-dynamics docking
The final conformation obtained from MD (5 ns) was used for the next stage of docking. The protein for each of the four models was minimized using the Amber02 force-field and Amber charges in SYBYL7.2 for 100 iterations to clean up the geometry. The cleaned structures were then used for docking using GOLD with the same procedures as described above.
Electronic supporting information
The supporting information includes detailed procedures for model building, including model assessment and refinement, as well as the Ramachandran plot and PROFILE3D validation of the newly generated model.
Conclusions
Hepatitis B is a growing problem worldwide and available treatments are limited by ineffectiveness, side effects, and development of resistance. Hence, there is continuous need for new drugs, especially for ones which are not prone to resistance development. Hepatitis B virus DNA polymerase has been shown to be a valid target for treatment of this disease. We have built a stereochemically significant homology model of HDP. There were two main differences from previously reported homology models of HDP: our sequence alignment made sure to match properly a conserved K residue found in HIV-1 RT, which shows important salt bridge interactions with the γ-phosphate of an incoming nucleotide. We believe that, analogous to the role of K65 in HIV-1 RT, rtK32 plays a considerable role in the binding of nucleotides and known HDP inhibitors. Mutation studies of this residue will help to demonstrate its role in binding of inhibitors and its possible role in resistance development. Second, we used 1T05 as a 3D-structure template; this is a higher resolution structure compared with the previously used template (1RTD) and is appropriate since it contains a co-crystallized inhibitor, tenofovir. The homology model was validated using various techniques, including PROSTAT, PROCHECK, and Verify-3D profile. The model matched available experimental evidence from mutation studies of HDP, including for resistant mutations. Our model was helpful for explaining the inactivity of certain anti-retroviral molecules against HDP. The stability of the model was studied using a 5 ns molecular dynamics simulation with one of the substrates (CTP). The model was sufficiently stable after the initial 2.5 ns with an overall RMSD of 4.13 Å. We carried out two different docking studies, before and after the MD simulation; both docking runs produced very similar docking poses for 4 substrates and 10 inhibitors. Conformational changes during the MD simulations led to formation of a small additional hydrophobic pocket near the nucleotide binding site. The exocyclic alkene moiety of entecavir and the sulfur atom of lamivudine occupied this newly formed hydrophobic pocket, explaining the better binding affinity of these inhibitors in comparison to the natural substrates.
Although our modeling results provided explanation for the various resistant mutants of HDP and successfully predicted binding conformations of known inhibitors of HDP, further work is required to fully understand the mechanisms of resistance. The putative 3D structure of HDP and binding poses of the inhibitors should provide a basis for further investigation and understanding of the reaction mechanism of the nucleotidyl transfer to DNA primer as well as of the development of resistance to various existing inhibitors, for example using precise binding energy calculations and QM/MM molecular dynamics simulations.
References
- 1.Lok ASF, Mcmahon BJ. Chronic Hepatitis B. Hepatology. 2007;45:507–539. doi: 10.1002/hep.21513. [DOI] [PubMed] [Google Scholar]
- 2.Borgia G, Gentile I. Treating chronic Hepatitis B: today and tomorrow. Curr Med Chem. 2006;13:2839–2855. doi: 10.2174/092986706778521995. [DOI] [PubMed] [Google Scholar]
- 3.Dienstag J, Easley C, Kirkpatrick P. Telbivudine. Nat Rev Drug Discov. 2007;6:267–268. doi: 10.1038/nrd2295. [DOI] [PubMed] [Google Scholar]
- 4.Severini A, Liu XY, Wilson JS, Tyrrell DL. Mechanism of inhibition of duck hepatitis B virus polymerase by (-)- β-L-2′, 3′-dideoxy-3′-thiacytidine. Antimicrob Agents Chemother. 1995;39:1430–1435. doi: 10.1128/aac.39.7.1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deval J, Courcambeck J, Selmi B, Boretto J, Canard B. Structural determinants and molecular mechanisms for the resistance of HIV-1 RT to nucleoside analogues. Curr Durg Metabol. 2004;5:305–316. doi: 10.2174/1389200043335478. [DOI] [PubMed] [Google Scholar]
- 6.Daga PR, Patel RY, Doerksen RJ. Template-based protein modeling: Recent methodological advances. Curr Top Med Chem. 2010;10:84–94. doi: 10.2174/156802610790232314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Das K, Xiong X, Yang H, Westland CE, Gibbs CS, Sarafianos SG, Arnold E. Molecular modeling and biochemical characterization reveal the mechanism of Hepatitis B virus polymerase resistance to Lamivudine (3TC) and Emtricitabine (FTC) J Virol. 2001;75:4771–4779. doi: 10.1128/JVI.75.10.4771-4779.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartholomeusz A, Tehan BG, Chalmers DK. Comparison of the HBV and HIV polymerase, and antiviral resistance mutations. Antiviral Therapy. 2004;9:149–160. [PubMed] [Google Scholar]
- 9.Langley DR, Walsh AW, Baldick CJ, Eggers BJ, Rose RE, Levine SM, Kapur AJ, Colonno RJ, Tenney DJ. Inhibition of Hepatitis B virus polymerase by Entecavir. J Virol. 2007;81:3992–4001. doi: 10.1128/JVI.02395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chong Y, Chu CK. Understanding the unique mechanism of L-FMAU (Clevudine) against Hepatitis B virus: molecular dynamics studies. Bioorg Med Chem Lett. 2002;12:3459–3462. doi: 10.1016/s0960-894x(02)00747-3. [DOI] [PubMed] [Google Scholar]
- 11.Yadav V, Chu CK. Molecular mechanisms of adefovir sensitivity and resistance in HBV polymerase mutants: a molecular dynamics study. Bioorg Med Chem Lett. 2004;14:4313–4317. doi: 10.1016/j.bmcl.2004.05.075. [DOI] [PubMed] [Google Scholar]
- 12.Sharon A, Chu CK. Understanding the molecular basis of HBV drug resistance by molecular modeling. Antiviral Res. 2008;80:339–353. doi: 10.1016/j.antiviral.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bazmi HZ, Hammond JL, Cavalcanti SCH, Chu CK, Schinazi RF, Mellors JW. In vitro selection of mutations in the human immunodeficiency virus Type 1 reverse transcriptase that decrease susceptibility to (-)-β -D-dioxolane-guanosine and suppress resistance to 3′-azido-3′-deoxythymidine. Antimicrob Agents Chemother. 2000;44:1783–1788. doi: 10.1128/aac.44.7.1783-1788.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gu Z, Gao Q, Fang H, Salomon H, Parniak MA, Goldberg E, Cameron J, Wainberg MA. Identification of a mutation at codon 65 in the IKKK motif of reverse transcriptase that encodes human immunodeficiency virus resistance to 2′, 3′-dideoxycytidine and 2′, 3′-dideoxy-3′-thiacytidine. Antimicrob Agents Chemother. 1994;38:275–281. doi: 10.1128/aac.38.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu Z, Salomon H, Cherrington JM, Mulato AS, Chen MS, Yarchoan R, Foli A, Sogocio KM, Wainberg MA. K65R mutation of human immunodeficiency virus type 1 reverse transcriptase encodes cross-resistance to 9-(2- phosphonylmethoxyethyl)adenine. Antimicrob Agents Chemother. 1995;39:1888–1891. doi: 10.1128/aac.39.8.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tuske S, Sarafianos SG, Clark AD, Ding J, Naeger LK, White KL, Miller MD, Gibbs CS, Boyer PL, Clark P, Wang G, Gaffney BL, Jones RA, Jerina DM, Hughes SH, Arnold E. Structures of HIV-1 RT-DNA complexes before and after incorporation of the anti-AIDS drug tenofovir. Nat Struct Mol Biol. 2004;11:469–474. doi: 10.1038/nsmb760. [DOI] [PubMed] [Google Scholar]
- 17.Selmi B, Boretto J, Sarfati SR, Guerreiro C, Canard B. Mechanism-based suppression of dideoxynucleotide resistance by K65R human immunodeficiency virus reverse transcriptase using an alpha-boranophosphate nucleoside analogue. J Biol Chem. 2001;276:48466–48472. doi: 10.1074/jbc.M107003200. [DOI] [PubMed] [Google Scholar]
- 18.Larder BA, Bloor S, Kemp SD, Hertogs K, Desmet RL, Miller V, Sturmer M, Staszewski S, Ren J, Stammers DK, Pauwels R. A family of insertion mutations between codons 67 and 70 of human immunodeficiency virus type 1 reverse transcriptase confer multinucleoside analog resistance. Antimicrob Agents Chemother. 1999;43:1961–1967. doi: 10.1128/aac.43.8.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menendez-Arias L, Matamoros T, Cases-Gonzalez CE. Insertions and deletions in HIV-1 reverse transcriptase: consequences for drug resistance and viral fitness. Curr Pharm Des. 2006;12:1811–1825. doi: 10.2174/138161206776873608. [DOI] [PubMed] [Google Scholar]
- 20.De Clercq E. Perspectives for the treatment of Hepatitis B virus infections. Int J Antimicrob Agents. 1999;12:81–95. doi: 10.1016/s0924-8579(99)00060-6. [DOI] [PubMed] [Google Scholar]
- 21.Ray AS, Basavapathruni A, Anderson KS. Mechanistic studies to understand the progressive development of resistance in human immunodeficiency virus Type 1 reverse transcriptase to Abacavir. J Biol Chem. 2002;277:40479–40490. doi: 10.1074/jbc.M205303200. [DOI] [PubMed] [Google Scholar]
- 22.O'Reilly EK, Kao CC. Analysis of RNA-dependent RNA polymerase structure and function as guided by known polymerase structures and computer predictions of secondary structure. Virology. 1998;252:287–303. doi: 10.1006/viro.1998.9463. [DOI] [PubMed] [Google Scholar]
- 23.Sarafianos SG, Das K, Clark AD, Ding J, Boyer PL, Hughes SH, Arnold E. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with beta-branched amino acids. Proc Natl Acad Sci USA. 1999;96:10027–10032. doi: 10.1073/pnas.96.18.10027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chong Y, Stuyver L, Otto MJ, Schinazi RF, Chu CK. Mechanism of antiviral activities of 3′-substituted L-nucleosides against 3TC-resistant HBV polymerase: a molecular modelling approach. Antiviral Chem Chemother. 2003;14:309–319. doi: 10.1177/095632020301400603. [DOI] [PubMed] [Google Scholar]
- 25.Bowie JU, Luthy R, Eisenberg D. A method to identify protein sequences that fold into a known three-dimensional structure. Science. 1991;253:164–170. doi: 10.1126/science.1853201. [DOI] [PubMed] [Google Scholar]
- 26.Dando T, Plosker G. Adefovir dipivoxil: a review of its use in chronic Hepatitis B. Drugs. 2003;63:2215–2234. doi: 10.2165/00003495-200363200-00007. [DOI] [PubMed] [Google Scholar]
- 27.Bryson K, McGuffin LJ, Marsden RL, Ward JJ, Sodhi JS, Jones DT. Protein structure prediction servers at University College London. Nucl Acids Res. 2005;33:W36–W38. doi: 10.1093/nar/gki410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tripos Associates. SYBYL7.2. St. Louis, MO: Tripos Associates Inc; 2006. [Google Scholar]
- 29.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL-W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang H, Chopra R, Verdine GL, Harrison SC. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: Implications for drug resistance. Science. 1998;282:1669–1675. doi: 10.1126/science.282.5394.1669. [DOI] [PubMed] [Google Scholar]
- 31.Ding J, Das K, Hsiou Y, Sarafianos SG, Clark AD, Jacobo-Molina A, Tantillo C, Hughes SH, Arnold E. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody fab fragment at 2.8 A resolution. J Mol Biol. 1998;284:1095–1111. doi: 10.1006/jmbi.1998.2208. [DOI] [PubMed] [Google Scholar]
- 32.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 33.Vullo A, Frasconi P. Disulfide connectivity prediction using recursive neural networks and evolutionary information. Bioinformatics. 2004;20:653–659. doi: 10.1093/bioinformatics/btg463. [DOI] [PubMed] [Google Scholar]
- 34.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 35.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The AMBER biomolecular simulation programs. J Comput Chem. 2005;26:1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]