

Abstract
Low expression and instability during isolation are major obstacles preventing adequate structure-function characterization of membrane proteins (MPs). To increase the likelihood of generating large quantities of protein, C-terminally fused green fluorescent protein (GFP) is commonly used as a reporter for monitoring expression and evaluating purification. This technique has mainly been restricted to MPs with intracellular C-termini (Cin) due to GFP's inability to fluoresce in the Escherichia coli periplasm. With the aid of Glycophorin A, a single transmembrane spanning protein, we developed a method to convert MPs with extracellular C-termini (Cout) to Cin ones providing a conduit for implementing GFP reporting. We tested this method on eleven MPs with predicted Cout topology resulting in high level expression. For nine of the eleven MPs, a stable, monodisperse protein-detergent complex was identified using an extended fluorescence-detection size exclusion chromatography procedure that monitors protein stability over time, a critical parameter affecting the success of structure-function studies. Five MPs were successfully cleaved from the GFP tag by site-specific proteolysis and purified to homogeneity. To address the challenge of inefficient proteolysis, we explored expression and purification conditions in the absence of the fusion tag. Contrary to previous studies, optimal expression conditions established with the fusion were not directly transferable for overexpression in the absence of the GFP tag. These studies establish a broadly applicable method for GFP screening of MPs with Cout topology, yielding sufficient protein suitable for structure-function studies and are superior to expression and purification in the absence GFP fusion tagging.
Keywords: membrane protein expression, green fluorescent protein, fluorescence-detection size exclusion chromatography FSEC, pWarf, glycophorin A
Introduction
Membrane proteins perform a host of cellular processes ranging from energy transduction to the transport of otherwise impermeable molecules across the membrane bilayer. Alterations in membrane protein function are the underlying cause for many human diseases and membrane proteins make up 40% of all pharmaceutical drugs targets currently under investigation.1 Thus, a clear public health need exists to understand their structure and function. Currently, detailed characterization of membrane proteins is more arduous than for soluble proteins due to their hydrophobic nature, which makes them difficult to express, purify and stabilize in detergent micelles. In recent years, a number of advances have been made to overcome these challenges, in particular, the implementation of green fluorescent protein (GFP) as a C-terminal fusion reporter for monitoring protein expression, purification and stability.2–4
In 2001, Drew et al. used the GFP fusion tag to monitor expression of bacterial inner membrane proteins via whole cell fluorescence, eliminating the need for time consuming purifications and immunoblots to decipher optimal expression conditions.3 More recently, Gouaux and colleagues have expanded the membrane protein-GFP fusion methodology to identify ideal protein-detergent complexes through fluorescence-detection size exclusion chromatography (FSEC),4 laying the foundation for four recent membrane protein crystal structures.5–8 Despite these advances, use of the GFP fusion technology in Escherichia coli (E. coli) has been mostly limited to membrane proteins with an intracellular C-terminus (Cin)3,9,10 stemming from GFP's inability to mature and fluoresce when positioned in the periplasmic space of E. coli.11 Topology prediction data for 29 genomes analyzed using the TransMembrane Hidden Markov Model (TMHMM) methodology shows 35% of all multispanning membrane proteins have Cout topology12—a percentage much too large to ignore. Thus, developing a GFP fusion technology applicable to the complete membrane proteome, regardless of C-termini topology, would be advantageous.
Wright and colleagues used the GFP technology on sodium sugar transporters (SGLT), which have predicted Cout topologies, by repositioning the extracellular C-termini using a Glycophorin A (GpA, a single transmembrane spanning protein) fusion.2,13 Expanding upon these early results, we engineered a set of vectors for monitoring expression, detergent screening and stability testing of both Cin and Cout proteins with GFP fluorescence. We have termed these vectors pWarf(−) and pWarf(+) [Fig. 1 and Supporting Information Figs 1(A,B)].
Figure 1.
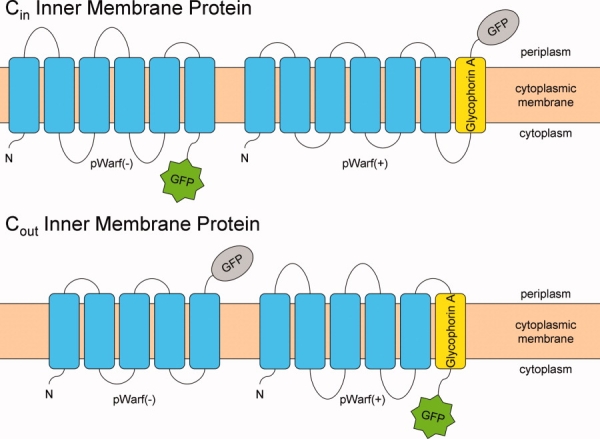
Schematic of membrane proteins expressed with pWarf vector system. The pWarf(−) fuses GFP to the C-terminus of the inner membrane protein, while the pWarf(+) fuses GpA and subsequent GFP to the C-terminus of the inner membrane protein. The GFP is fluorescent in the cytoplasm and nonfluorescent in the periplasm. [Color figure can be viewed in the online issue, which is available at http://www.interscience.wiley.com.]
In this article, we present a streamlined methodology for monitoring expression, purification and detergent stabilization using GFP for all membrane proteins, regardless of C-terminal topology. In total, we have been able to isolate pure and homogeneous samples from 54% of the target proteins with Cout topology (in excess of milligram quantities) that will permit structure-function characterization.
Results
Expanding upon previous reports using GFP methodology, we present a streamlined GFP methodology that addresses expression, purification and stability of membrane proteins with both Cin and Cout topology using the pWarf vector system. The primary achievement is the ability to monitor expression of Cout topology membrane proteins by inserting GpA, which repositions the C-terminus to the intracellular space, permitting GFP fluorescence.
Expression screening and optimization of membrane proteins using the pWarf vector system
Building upon previously described reports using GFP as a reporter for monitoring expression and purification,3–10,14–16 we established a comprehensive, streamlined approach for confirming topology, optimizing expression, and screening stable protein-detergent complexes for both Cin and Cout membrane proteins. The pWarf vector system consists of two vectors, pWarf(−) and pWarf(+), that were constructed by modifying the pET28 vector described in Drew et al.9 (Supporting Information Fig. 1). To validate the system, twelve inner membrane transport proteins (eleven Cout and one Cin) from the E. coli genome were selected using topology prediction data from Daley et al. (Table I).17 To test the robustness of the system, the target proteins were selected from four distinct families (IT, HAAAP, CaCA, and MFS) with a diverse range of substrates and transport mechanisms (Table I).18 All genes were successfully cloned into both vectors and subjected to expression screening and optimization. Whole-cell GFP fluorescence was monitored to quantify expression levels based on a method previously reported,9 where the whole-cell GFP fluorescence was directly proportional to membrane protein yield.
Table I.
E. coli Proteins Used in This Study
| Target | Accession # | Functional category | MW (kDa) | # TM predicted | C-term localization | No. of amino acids | Family | TC # |
|---|---|---|---|---|---|---|---|---|
| ArsB | P0AB93 | Arsenic efflux pump | 45 | 11 | out | 429 | ITa | 2.A.45 |
| DsdX | P08555 | D-serine transporter | 47 | 12 | out | 445 | ITa | 2.A.8.1.5 |
| GntP | P0AC94 | High-affinity gluconate transporter | 47 | 10 | out | 447 | ITa | 2.A.8 |
| GntU | P0AC96 | Low-affinity gluconate transporter | 46 | 11 | out | 446 | ITa | 2.A.8 |
| LldP | P33231 | L-lactate permease | 59 | 12 | out | 551 | ITa | 2.A.14.1.1 |
| Mtr | P0AAD2 | Tryptophan permease | 44 | 11 | out | 414 | HAAAPb | 2.A.42.1.2 |
| SdaC | P0AAD6 | Serine transporter | 47 | 11 | out | 429 | HAAAPb | 2.A.42.2.1 |
| TdcC | P0AAD8 | Threonine/serine transporter | 49 | 11 | out | 443 | HAAAPb | 2.A.42.2.2 |
| TyrP | P0AAD4 | Tyrosine permease | 43 | 11 | out | 403 | HAAAPb | 2.A.42.1.1 |
| ChaA | P31801 | Calcium/proton antiporter | 39 | 11 | out | 366 | CaCAc | 2.A.19.1.1 |
| YrbG | P45394 | Putative calcium/proton antiporter | 35 | 9 | out | 325 | CaCAc | 2.A.19.5.1 |
| LacY | P02920 | Lactose permease | 47 | 10 | in | 417 | MFSd | 2.A.1.5.1 |
Ion Transporter Superfamily.
Hydroxy/Aromatic Amino Acid Permease Family.
Ca2+:Cation Antiporter Family.
Major Facilitator Superfamily.
Initial expression screening was performed in triplicate on three expression cell strains [BL21(DE3), C41(DE3), and C43(DE3)] to confirm the preferred topology and to quantify the yield for each test protein using standard expression conditions (Supporting Information Table I). Expression of all test proteins yielded high level fluorescence in a single vector [pWarf(−) for Cin and pWarf(+) for Cout] with negligible fluorescence in the alternate vector [Fig. 2(A)]. These results show that Glycophorin A successfully repositions the GFP to the cytoplasmic environment for proper maturation, providing a quick, convenient method to identify C-terminal topology as an alternative to the troublesome alkaline phosphatase method currently used for topological determination.17 This initial expression trial identified plausible targets for the isolation of milligram quantities of protein—a prerequisite for structure-function studies.
Figure 2.
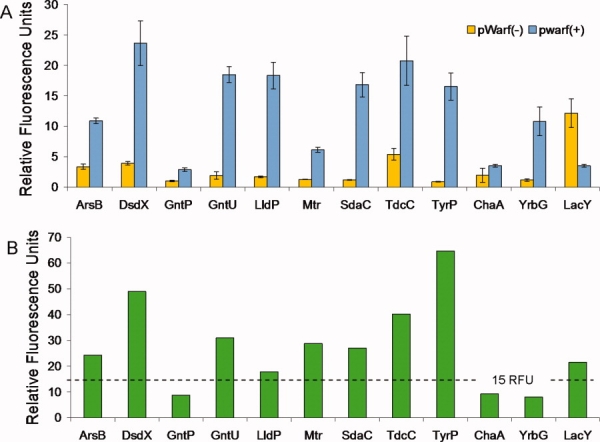
Whole cell fluorescence measurements from expression in pWarf vector system. A: Each test protein was expressed in the pWarf(−) and pWarf(+) vectors using standard conditions (LB, 0.5 mM IPTG, 4 h induction) and the fluorescence values relating to expression are shown in yellow and blue, respectively. The standard deviation was calculated from the triplicate measurements and shown as error bars. B: For each test protein, the appropriate expression vector was selected and subjected to an optimization screen to increase expression. The fluorescence values relating to optimized expression are shown in green. From left to right, the first eleven proteins have a predicted Cout topology, and the rightmost protein, LacY, has Cin topology. The dashed line indicates the 15 RFUs criterion for protein purification. [Color figure can be viewed in the online issue, which is available at http://www.interscience.wiley.com.]
To explore the possibility of increasing the expression threshold, a comprehensive optimization screen was developed. Specifically, each protein was subjected to 72 expression variations examining optimal cell strain, media, inducer concentration, and time of induction (Supporting Information Table II). By expanding upon the previously reported optimization procedures,10 we aimed to: (A) identify the best conditions for overexpression of each target protein; (B) determine the extent to which optimization increases yields over standard expression conditions; and (C) explore general trends for increasing expression yields to be applied to other membrane proteins. In most cases, the expression levels doubled after optimization with the highest expressing protein, TyrP, experiencing a nearly a fourfold increase (from 17 to 65 RFU's) in fluorescence after optimization [Fig. 2(B)]. Additionally, the relationship between yield and expression conditions was explored for each protein using a four-way Analysis of Variance (ANOVA) test.19
The ANOVA analysis is a common statistical test used to determine the significance of each variable alone (a main effect) as well as the interactions between variables. The ANOVA was calculated for each protein and the P-values (P ≤ 0.05 indicates statistical significance) were reported in Table IV to highlight which main effects and higher order interactions are significant. It is important to note that the variables identified empirically to give the highest level of expression for a protein correspond to the significant variables identified via the ANOVA and these interactions are detailed in the Supporting Information. However, because a variety of membrane proteins have been used in this study, the ANOVA results reported here will examine overall trends in expression rather than specific interactions for a single protein.
Table IV.
Compilation of ANOVA Findings for Expression of Each GpA-GFP-Fusion Protein
| Interactions | ArsB | DsdX | GntP | GntU | LldP | Mtr | SdaC | TdcC | TyrP | ChaA | YrbG | LacY | Total |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| x1: Cell strain | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 11 | |
| x2: Growth medium | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 | 0.03 | 0.04 | 0.00 | 0.00 | 11 | |
| x3: Inducer concentration | 0.03 | 0.00 | 0.00 | 0.00 | 0.01 | 5 | |||||||
| x4: Time of induction | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 | 0.00 | 11 | |
| x1x2 | 0.00 | 0.00 | 0.00 | 0.00 | 0.05 | 0.02 | 0.01 | 0.00 | 8a | ||||
| x1x3 | 0.05 | 0.00 | 0.00 | 3 | |||||||||
| x1x4 | 0.03 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.01 | 0.00 | 10a | ||
| x2x3 | 0.00 | 0.00 | 0.00 | 3 | |||||||||
| x2x4 | 0.02 | 0.00 | 0.00 | 0.00 | 0.01 | 0.03 | 0.00 | 7a | |||||
| x3x4 | 0 | ||||||||||||
| x1x2x3 | 0.01 | 0.00 | 0.00 | 3 | |||||||||
| x1x2x4 | 0.00 | 0.02 | 0.00 | 0.00 | 0.00 | 0.03 | 6a | ||||||
| x1x3x4 | 0 | ||||||||||||
| x2x3x4 | 0.00 | 0.01 | 2 | ||||||||||
| x1x2x3x4 | 0.00 | 1 |
The values reported are the Probability > F or P-values. A value P ≤ 0.05 is statistically significant.
Indicates interactions that were statistically significant for at least half of the test proteins.
The highest order interaction, observed for half of the test proteins (six proteins), is a three-way interaction between cell strain, growth medium and time of induction. There are also numerous two-way interactions (up to 10 proteins) involving cell strain, growth medium and/or time of induction as highlighted in Table IV. These findings suggest, protein overexpression requires higher order interactions where two or more variables in combination act together to achieve higher levels of expression. Notably, inducer concentration was rarely found as a significant variable in higher order interactions (≤3 proteins) and is likely not as an important variable for the screening process. Overall, the analysis identified cell strain, growth medium and time of induction as significant variables for increasing expression and therefore should be preferentially screened for optimizing protein overexpression.
Detergent screening and stability analysis using fluorescence-detection size exclusion chromatography
A prerequisite for structure-function characterization is a large quantity of protein isolated in a detergent that renders the protein stable and monodisperse in an aqueous solution. Often the best detergent for a particular protein is one that mimics its native membrane environment. Identifying such a detergent, however, is still done empirically. Traditional detergent selection methods are both time and resource intensive, requiring milligrams of pure protein. However, the fluorescence-detection size-exclusion chromatography (FSEC) strategy has recently emerged as an efficient method for identifying suitable detergents, bypassing a number of obstacles that previously confronted this characterization procedure.4 The FSEC strategy involves solubilizing the crude membrane preparation with a panel of detergents and applying the samples to a gel-filtration column, where the GFP-fusion elution profile is monitored using fluorescence spectroscopy. Analysis of the elution profile indicates the dispersity and stability of the protein, where a stable, monodisperse protein normally yields a single, Gaussian peak, and a polydisperse protein generally yields multiple, asymmetrical peaks that are detrimental to structure-function studies.4–6,14
Using the FSEC strategy, the eleven Cout proteins were subjected to a panel of four detergents most commonly used for structure-function studies [n-dodecyl-N,N-dimethylamine-N-oxide (LDAO), n-octyl-β-d-glucopyranoside (OG), n-decyl-β-d-maltoside (DM), and n-dodecyl-β-d-maltoside (DDM)].20,21 To streamline the process, the crude membrane fraction was divided, solubilized in each of the four detergents, and independently loaded on a size-exclusion column equilibrated with buffer containing the respective detergent. Using this procedure we were able to screen all four detergents in half a day. The FSEC profiles were routinely analyzed by Gaussian peak fitting,22 expanding on earlier work by Kawate et al.,4 as a means to standardize the procedure for determining the dispersity of the solubilized GFP-fusion proteins. Ten of the eleven test proteins were monodisperse in at least one detergent (Table II). The sole exception, GntP, had no detectable fluorescence, possibly due to the detergent's inability to extract the sample from its native membrane environment. Notably, 9 of the 10 remaining proteins were soluble and monodisperse in DM and DDM, and half were well maintained in OG. ChaA was the only protein to have a good elution profile in all four detergents. This data is summarized in Table II and a representative FSEC trace for DsdX stabilized in DM is shown in Figure 3(A).
Table II.
Detergent Screening Analysis via Gaussian Peak Fitting
| Initial solubilization |
48-h post solubilization |
||||
|---|---|---|---|---|---|
| Protein | Detergent | Number of Gaussian functions required | R2 | Number of Gaussian functions required | R2 |
| ArsB | LDAO | 5 | 1.00 | — | — |
| OG | 1 | 0.98 | 2 | 0.96 | |
| DM | 1 | 0.99 | 1 | 0.99 | |
| DDMa | 1 | 1.00 | 1 | 0.98 | |
| DsdX | LDAO | 3 | 1.00 | — | — |
| OG | 5 | 0.97 | — | — | |
| DMa | 1 | 1.00 | 1 | 0.99 | |
| DDM | 1 | 1.00 | 3 | 0.98 | |
| GntPb | — | — | — | — | |
| GntU | LDAO | 3 | 1.00 | — | — |
| OG | 6 | 0.99 | — | — | |
| DMa | 1 | 1.00 | 1 | 1.00 | |
| DDM | 1 | 0.97 | 1 | 0.99 | |
| LldP | LDAO | 4 | 1.00 | — | — |
| OG | 2 | 0.95 | — | — | |
| DMa | 1 | 1.00 | 1 | 0.99 | |
| DDM | 1 | 1.00 | 3 | 0.99 | |
| Mtr | LDAO | 4 | 0.99 | — | — |
| OG | 1 | 0.99 | 3 | 0.96 | |
| DM | 1 | 0.99 | 1 | 0.99 | |
| DDMa | 1 | 1.00 | 1 | 0.99 | |
| SdaC | LDAO | 2 | 0.99 | — | — |
| OG | 1 | 0.99 | 2 | 0.98 | |
| DM | 1 | 0.99 | 1 | 0.99 | |
| DDMa | 1 | 1.00 | 1 | 0.98 | |
| TdcC | LDAO | 3 | 1.00 | — | — |
| OG | 2 | 0.98 | — | — | |
| DM | 1 | 1.00 | 1 | 0.99 | |
| DDMa | 1 | 1.00 | 1 | 0.99 | |
| TyrP | LDAO | 3 | 0.99 | — | — |
| OG | 1 | 1.00 | 2 | 0.98 | |
| DM | 1 | 0.99 | 1 | 1.00 | |
| DDMa | 1 | 1.00 | 1 | 0.99 | |
| ChaA | LDAO | 1 | 0.98 | 1 | 0.97 |
| OG | 1 | 0.98 | 1 | 0.97 | |
| DMa | 1 | 0.99 | 1c | 0.98 | |
| DDM | 1 | 0.99 | 1 | 0.96 | |
| YrbG | LDAO | 4 | 0.98 | — | — |
| OG | 3 | 0.93 | — | — | |
| DM | 3c | 0.99 | 2d | 0.99 | |
| DDM | 1 | 0.99 | 2d | 0.82 | |
R2 is an indicator of how well the calculated Gaussian curve fits the experimental data.
The optimal detergent selected for solubilization and purification.
Solubilized protein levels were lower than needed for detection.
Major peak has a shoulder, still reanalyzed peak fraction 48 h post solubilization.
Calculated Gaussian Fit using mAu(λ = 485 nm), rather than mV, due to low protein abundance.
Figure 3.
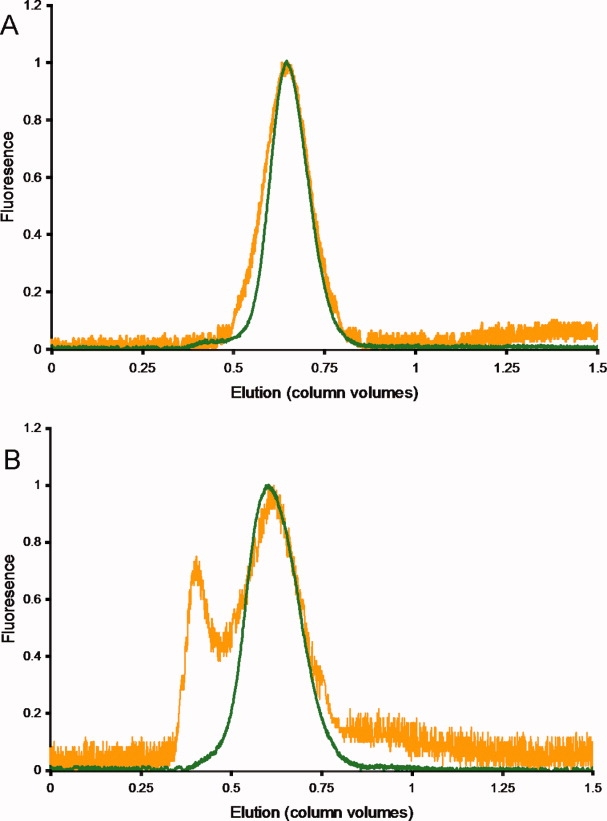
Detergent screening of DsdX via FSEC. DsdX was solubilized in (A) DM and (B) DDM and analyzed by fluorescence-detection size exclusion chromatography (FSEC). For the sake of comparison, the fluorescence traces were normalized against the highest intensity of the particular trace, giving a y-axis scale of 0 to 1. The traces in green were obtained just after solubilization and traces in orange were obtained 48 h post solubilization. When DsdX was solubilized in DM, the protein remained stable and monodisperse over the 48-h period, whereas DsdX in DDM became polydisperse over the 48-h period. [Color figure can be viewed in the online issue, which is available at http://www.interscience.wiley.com.]
Instability during and after isolation is a major obstacle preventing thorough structure–function characterization of membrane proteins. FSEC is as an excellent indicator for protein monodispersity and has proven to be an exceptional tool for precrystallization screening. However, it remained unknown whether these protein-detergent complexes remain stable over time. To investigate this parameter, we re-evaluated all monodisperse protein-detergent pairs 48 h after the first FSEC. Reanalysis via a second FSEC step proved to be critical as it revealed that four protein/detergent pairs, previously determined suitable, began forming aggregates after 48 h as identified by a shift to the void volume in the FSEC profile. Thus, we were able to eliminate four additional protein/detergent pairs (Mtr and SdaC in OG and DsdX and LldP in DDM) that were unable to maintain minimal protein stability [Fig. 3(B)]. In total, we were able to identify stable protein-detergent complexes for nine of the ten remaining protein targets, where YrbG was the sole protein unstable in all four detergents and will require further detergent screening. All nine proteins were found to have stable DM complexes, and seven of the proteins had stable DDM complexes as well. Cases where DM and DDM were both suitable detergents required a thorough examination of the elution profiles to qualitatively select the optimal detergent for solubilization and purification purposes. In total, five proteins were matched with DDM for optimal solubilization and the remaining four proteins were paired with DM (Table II).
Purification of membrane protein fusions
To confirm the reported expression levels and detergent selections are scalable for downstream structure-function applications, eight Cout proteins that had whole-cell fluorescence above 15 RFUs [Fig. 2(A)] were selected for large-scale expression and purification trials. The criterion of 15 RFUs represents the minimal expression level necessary for adequate isolation of a target protein in sufficient quantity for structure-function characterization (at least 1 mg L−1). The proteins were purified by immobilized metal ion affinity chromatography (IMAC) using the ideal detergent identified at the detergent screening stage (Table II); six of the eight fusion proteins were successfully purified. The two remaining proteins, Mtr and TdcC, precipitated upon elution using either DM or DDM which were previously identified as good detergent candidates. The reason for this behavior is unclear, however, it is possible that these proteins experienced detrimental lipid stripping during the extensive washes on the IMAC column, as has been reported for several other integral membrane proteins.23–27 The remaining six proteins were subjected to size-exclusion chromatography as a polishing step (Supporting Information Fig. 2) with overall yields of 2.9–11.4 mg L−1 culture (Table III). While these yields are certainly suitable to structure-function characterization, it is preferable to isolate the sample in a state that is as close to the native protein as possible. We have accomplished this by utilizing a protease cleavage site that was engineered for the purpose of removing the fusion tag.
Table III.
Analysis of Protein Yields
| Protein construct | Yield (mg protein per L culture) |
|---|---|
| ArsB-His8 | 0.1 |
| ArsB-HRV3C | 1.0 (29%) |
| ArsB-HRV3C-GpA-GFP-His8 | 3.5 |
| DsdX-His8 | 1.7 |
| DsdX-HRV3C | 0 (0.0%) |
| DsdX-HRV3C-GpA-GFP-His8 | 2.9 |
| GntU-His8 | 0.1 |
| GntU-HRV3C | 1.9 (30%) |
| GntU-HRV3C-GpA-GFP-His8 | 6.4 |
| LldP-His8 | 0.2 |
| LldP-HRV3C | 3.5 (31%) |
| LldP-HRV3C-GpA-GFP-His8 | 11.4 |
| SdaC-His8 | 0.1 |
| SdaC-HRV3C | 1.3 (21%) |
| SdaC-HRV3C-GpA-GFP-His8 | 6.2 |
| TyrP-His8 | 0.8 |
| TyrP-HRV3C | 3.8 (39%) |
| TyrP-HRV3C-GpA-GFP-His8 | 9.7 |
Values in parentheses are the recovery rates after cleavage removal of the GpA-GFP fusion tag.
Recovery of membrane protein from the fusion tag
The recent crystal structures of the sodium galactose transporter from Vibrio parahaemolyticus (vSGLT) 3DH4,28 revealed that the structures of the wild type protein and a vSGLT-GpA fusion resulted in virtually identical structures as determined by their superposition (RMSD of 1.1 Å). However, artificial fusion tags may be detrimental for both structure and function studies.29 To circumvent this limitation, the human rhinovirus 3c (HRV 3C) protease site was engineered in both pWarf vectors to liberate the membrane protein target from the fusion tag (Supporting Information Fig. 1). The HRV 3C protease was selected rather than tobacco etch virus protease (TEV) because it has been documented to have high efficiency and specificity at low temperatures (4°C).30 To test the viability of using HRV 3C for membrane protein recovery, the six purified Cout proteins reported above were digested with a fivefold molar excess of His-tagged HRV 3C for 24 h and then recovered by reverse IMAC. Analysis by SDS-PAGE indicated that five of the six fusion proteins were successfully cleaved with HRV 3C (Fig. 4). Similar to a previous study,14 the recovery rate after cleavage ranged from 21 to 39% (Table III), suggesting that while cleavage is a viable option for isolating pure, homogenous protein, it may not be the most efficient method. This issue was addressed by expressing the six proteins in the absence of the fusion tag.
Figure 4.
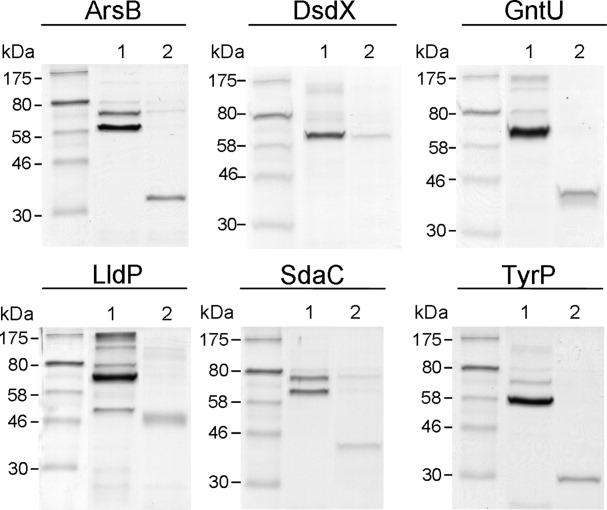
SDS-PAGE analysis of the HRV 3C protease reactions on the GpA-GFP-fusion proteins. The proteins were purified by IMAC, digested overnight with a fivefold molar excess of HRV 3C, recovered by reverse-IMAC, analyzed by SDS-PAGE and visualized with Coomassie Brilliant Blue R250. Thirty picomoles of each protein sample were loaded onto the gel. The gel lanes are: purified GFP-fusion protein (Lane 1), and recovered membrane protein (Lane 2). All proteins were successfully cleaved using HRV 3C except for DsdX.
Expression and purification of membrane proteins without the fusion tag
In an attempt to increase the recovery of membrane protein targets without a large fusion tag, expression and purification in the absence of the GpA-GFP fusion tag was initiated. The pWarf vector system was expanded to include the pWarf(n) vector that expresses the membrane protein with a C-terminal His8 tag [Supporting Information Fig. 1(c)]. The six target proteins successfully characterized with the GFP tag were examined for expression in the pWarf(n) vector. To ensure that optimal yields were achieved, we reoptimized the expression parameters and monitored expression by dot-blot analysis (Supporting Information Fig. 3). Surprisingly, the conditions identified using the GFP reporter were often not the best conditions for expression in the absence of the fusion tag, contrary to previous reports.3,4 In total, half of the proteins screened in the pWarf(n) vector required an adjustment to the expression parameters for optimal expression, and the most notable change was a shift to expression in the BL21(DE3) cell strain for five out of six target proteins (Supporting Information Table III). The expression conditions were analyzed using a three way ANOVA (Supporting Information) and the P-values for the variables alone and the interactions between variables are reported in Supporting Information Table IV. The ANOVA findings again show cell strain and growth medium are significant variables for screening protein overexpression as observed with GpA-GFP fusion expression. However, it must be noted that the specific conditions identified for each test protein have changed with the elimination of the GpA-GFP tag.
To determine the protein yields corresponding to the reoptimized expression conditions, the six test proteins were expressed and purified to homogeneity. The purification was carried out by IMAC using the previously identified optimal detergent and analyzed by SDS-PAGE (Supporting Information Figure 4). Expression levels significantly dropped when the membrane proteins were expressed in the pWarf(n) vector (Table III), as quantified by a modified Lowry assay that is compatible with both reducing agents and detergents (RC DC Assay, Bio-Rad). The sole exception was DsdX, which expressed to slightly lower levels than the GFP fusion. To ensure that these findings were not artificially altered due to protein assay incompatibility, a single test protein, TyrP (tagged with either GpA-GFP or His8), was purified and the protein concentration was measured by three techniques [BCA, Lowry, and Absorbance at 280 nm (A280) (Supporting Information Table V)]. As expected, the BCA, Lowry, and A280 measurements all gave similar values. These results indicate that HRV 3C recovery is the most effective method for obtaining protein suitable for structure-function characterization. However, in the event that HRV 3C is unsuccessful, expression reoptimization in the absence of the GpA-GFP tag is the best alternative.
Functional characterization of LldP-GpA-GFP
There may be concern as to whether overexpression with a large fusion tag impairs the function of the target protein, however previous studies have demonstrated membrane protein–GFP fusion overexpression can be accomplished while maintaining the protein's biological function.13 Although functional characterization of all test proteins is beyond the scope of this report, LldP-GpA-GFP was partially characterized to demonstrate the feasibility of generating functional protein using the GpA-GFP-fusion. The plasmid construct expressing LldP-GpA-GFP-His8 was transformed into the knockout E. coli strain ECL5106.31 From these cells, right-side out (RSO) vesicles were prepared and [14C]-l-lactate transport was measured (Fig. 5). The results would indicate that while the GpA-GFP fusion is attached to LldP, transport still occurs, further indicating that using a fusion tag for expression and purification is a suitable method for obtaining protein for crystallographic purposes.
Figure 5.
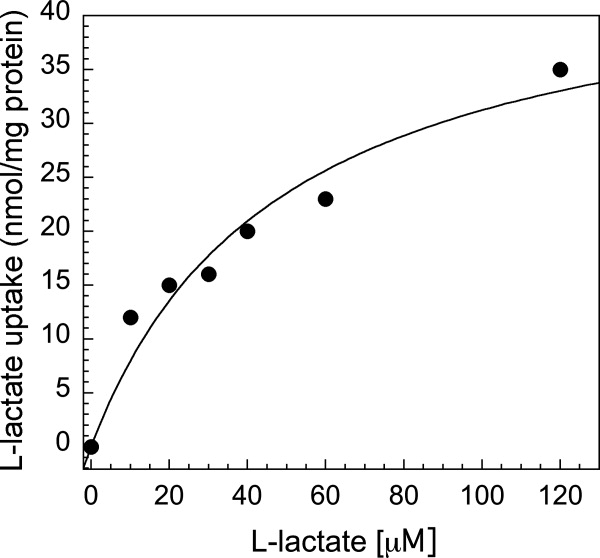
l-lactate transport via LldP-GpA-GFP. To test function of one of the eleven Cout proteins, the LldP-GpA-GFP-His8 construct was selected. The construct was expressed in the knockout strain, ECL 5106, and right-side-out vesicles were prepared, then transport of l-lactate measured. From the plot, the Km was calculated to be 48.6 μM l-lactate and the Vmax is 46.4 nmol l-lactate mg−1 protein per minute.
Discussion
Several studies have employed GFP as a reporter for membrane protein expression and detergent selection,3–6,9,10,14,15 but the technology has largely been restricted to proteins with intracellular C-termini (Cin) due to the GFP's inability to mature when localized to the E. coli periplasm.11 We have demonstrated that Glycophorin A (GpA)—a single spanning transmembrane helix—effectively converts Cout membrane proteins to become Cin proteins where the GFP can properly fold and fluoresce. Using the pWarf(+) vector, we have determined the expression levels of eleven Cout target proteins by monitoring whole-cell fluorescence and verified their previously predicted topology. Furthermore, we have identified the optimal detergent for both solubilization and stability by FSEC for nine of the eleven target proteins, where six of these proteins were successfully purified and subjected to site-specific proteolysis for removal of the fusion tag. LldP-GpA-GFP fusion was partially characterized demonstrating that the GpA-GFP tag does not prevent transport with this protein. In total, five target proteins completed the entire process, including the removal of the fusion tag, and are undergoing further structure-function characterization.
In addition to expanding the use of GFP reporting to proteins with extracellular C-termini, this study provides new observations regarding the importance of screening overexpression conditions and explores the use of statistical analysis via ANOVA to identify trends in expression conditions for increasing yields. Previous overexpression screens would focus primarily on cell strain, temperature, inducer concentration and/or time of induction;10 and while the findings presented here also support the screening of cell strain and time of induction, the ANOVA results provide evidence for preferential screening of growth medium over inducer concentration (Table IV). In correspondence with the ANOVA interaction findings, the majority of test proteins achieved the highest levels of expression in the enriched mediums (TB and/or CG) which can be explained by the cells' need for sustained nutrients during extended time periods of overexpression. GntP was the sole protein to have a four-way variable interaction. It was one of the lowest expressing proteins and likely required interactions of each variable to achieve slightly higher expression. These results support screening of cell strain, growth mediums, period of induction and, if resources permit, inducer concentration.
FSEC has proven to be an extremely useful precrystallization tool, as demonstrated by four recent membrane protein crystal structures which utilized this method.5–8 This report expands upon the current protocol by monitoring the protein's stability over time, a critical parameter for eliminating poor protein/detergent complexes. Additionally, we have implemented routine Gaussian peak fitting as a simple method to standardize the detergent selection process for all elution traces. The results indicate that DM and DDM are the most successful detergent candidates, where OG and LDAO could only maintain the stability for a single target protein (ChaA). Notably, DM was the sole detergent to stabilize two test proteins, exemplifying the need to screen multiple detergents even within a particular detergent family.
While large scale expression and purification of the GpA-GFP-fusion proteins leads to significant yields, removal of the GpA-GFP tag is time and resource extensive. Recovery of the membrane protein via HRV 3C protease yields, at best, 39%. However, expression in the absence of the GpA-GFP tag required re-optimization of the overexpression conditions for half the target proteins and the yields were strikingly lower than their GpA-GFP tag counterparts. It is possible that the GpA-GFP enhances expression as has been reported for the maltose-binding protein and the Mistic fusion protein,32,33 but the mechanism by which this occurs is unclear. Whether to optimize expression with or without the fusion can only be determined through trial and error; however, our findings support pursing expression of a target membrane protein with the GpA-GFP tag due to the added benefits of FSEC detergent screen and larger yields of the native membrane protein after protease cleavage.
The pWarf(+) vector system, containing the GpA-GFP fusion, has bridged the gap by facilitating the expression, detergent screening and stability testing for membrane proteins with extracellular C-termini (Cout) and furthermore provides a rapid method for screening C-terminal topology of membrane proteins.
Materials and Methods
Expression screening and optimization of membrane proteins using the pWarf vector system
Genes encoding each membrane protein were amplified by conventional PCR from E. coli K12 genomic DNA, and cloned into two modified pET28(a+) vectors: (A) pWarf(−) which has the HRV 3C protease recognition site (LEVLFQ↓GP), subsequent GFP and C-terminal His8 tag [Supporting Information Fig. 1(A)]; (B) pWarf(+) which has the HRV 3C protease site followed by the transmembrane segment of Glycophorin A (EITLIIFGVMAGVIGTILLISYGIRRLIK) and subsequent GFP and C-terminal His8 tag [Supporting Information Fig. 1(B)]. Complete vectors were sequenced and then transformed into three cells strains BL21(DE3), C41(DE3), and C43(DE3). In all cultures reported, cells were grown in the presence of 50 μg mL−1 kanamycin. For the expression screen, three colonies from each cell strain were used to inoculate 10 mL cultures in 50-mL conical tubes that were grown overnight at 37°C and 225 RPM in Luria Broth (LB) medium. The overnight cultures were diluted 100-fold into 50 mL fresh LB medium in 250-mL baffled flasks and incubated at 37°C and 225 RPM until the OD600 was 0.4–0.5, then the temperature was lowered to 25°C over 30 min and fusion protein expression was induced for 4 h with 0.5 mM isopropyl-β-d-thiogalactoside (IPTG). A 5 mL aliquot of cells was harvested at the time of induction, t = 0, and again after 4 h induction, t = 4. Cell pellets were stored overnight at −20°C. The following day cells were thawed on ice, resuspended in PBS buffer (OD600nm = 5) and 200 μL was transferred to a 96-well plate for measuring GFP fluorescence (excitation λ = 485 nm, emission λ = 512 nm) on a SpectraMax M5 multidetection reader (Molecular Devices, Sunnyvale, CA). The final cell resuspension was diluted 100-fold to a final volume of 200 μL in a second microtiter plate to measure the OD600nm for normalization.
An optimization screen was used to increase the level of expression by examining three growth media [LB, Terrific Broth (TB) and Circle Grow (CG) (MP Biomedicals, Solon, OH)], two IPTG concentrations (0.5 mM and 1.0 mM, as based on a compilation of previous reports4,10) and four induction periods (1, 2, 3, and 4 h). Ten milliliter overnight cultures were grown in LB medium at 37°C and 225 RPM. The overnight cultures were diluted 100-fold into 50 mL fresh media (LB, TB or CG) in 250 mL baffled flasks and incubated at 37°C and 225 rpm until the OD600 was appropriate (LB: 0.4–0.5; TB/CG: 1.6–1.8), then the temperature was lowered to 25°C over 30 min and expression was induced with the appropriate amount of IPTG (0.5 or 1.0 mM). Aliquots of 5 mL were removed at t = 0, 1, 2, 3, 4. Cell pellets were stored overnight at −20°C. Cells were thawed on ice, prepared and measured for fluorescence and OD600 in the same manner described previously.
All fluorescence data were processed using a method adapted from Waldo et al.34 To account for handling variability in cell resuspension, the fluorescent values were normalized against the OD600 measurement 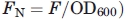 . Furthermore, to account for the inherent fluorescence of E. coli, all fluorescent values are reported as a relative fluorescence
. Furthermore, to account for the inherent fluorescence of E. coli, all fluorescent values are reported as a relative fluorescence 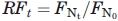 , where the normalized fluorescence at a given time point is divided by the normalized fluorescence of noninduced cells harboring the same plasmid. All values reported are RF values.
, where the normalized fluorescence at a given time point is divided by the normalized fluorescence of noninduced cells harboring the same plasmid. All values reported are RF values.
The data for each protein was examined using the STATA10 software package. Partial-factorial analysis was completed with the data available by grouping the time variable, where “T = 0” corresponds to t = 1 h and t = 2 h and “T = 1” corresponds to t = 3 h and t = 4 h; this step was needed to provide the replicates required to complete the analysis of variance (ANOVA) calculation and the assumption is that “T = 0” would be an average value at t = 1.5 h and “T = 1” would be an average value at t = 3.5 h. Upon completion of the ANOVA, the specific interactions were further analyzed by using a series of algorithms to dissect the interaction including (A) ANOVA, (B) the simple main effects (SME) algorithm35 and (C) the Tukey-Krammer pair-wise comparison algorithm.36 The specific sequence of tests for each protein are detailed in the Supporting Information.
Fluorescent size exclusion chromatography based detergent screen
The detergents n-dodecyl-N,N-dimethylamine-N-oxide (LDAO), n-octyl-β-d-glucopyranoside (OG), n-decyl-β-d-maltoside (DM), and n-dodecyl-β-d-maltoside (DDM) were screened (all from Anatrace, Maumee, OH). Each detergent was added to 0.5 mL of membrane suspension (0.2 g membranes/mL Buffer A, unless low expression, then 0.4 g membranes/mL Buffer A) (Buffer A: 50 mM Tris, pH 7.5, 150 mM NaCl) to a final concentration of 2% w/v, well in excess of the CMC to ensure solubilization,10,28 and samples were incubated for 1 h at 4°C with mild agitation, followed by centrifugation at 435,000g for 10 min and 4°C. Then, 250 μL of the supernatant was loaded onto a self-packed Superose 6 10/150 column (GE Health Life Sciences, Piscataway, NJ) that was pre-equilibrated in the respective detergent buffer (Buffer A with either: 0.046% LDAO, 1.06% OG, 0.176% DM, or 0.016% DDM) at a flow rate of 0.5 mL min−1. The eluant was monitored by GFP emission at 512 nm and absorbance at 280 nm. Fluorescent values corresponding to the FSEC trace were imported to OriginLab 7.5 (Northampton, MA) software.22 Traces were fitted with either single or multiple Gaussian functions to achieve an r2 coefficient of ≥ 0.97.
Purification of membrane-fusion proteins
Cells overexpressing the membrane-fusion proteins were grown in at least 2L cultures for large scale purification and were broken using the Emulsiflex C3 (ATA Scientific, Sutherland, Australia). All steps were carried out at 4°C. Cell debris was removed by low speed centrifugation (10,000g, 15 min), membranes were isolated by ultracentrifugation (302,000g, 1 h) and stored at −20°C. Later, 5 g of membranes were resuspended in Buffer A (0.2 g mL−1 buffer), solubilized by adding 2% of the appropriate detergent (identified in the detergent screen) and incubated for 1 h with gentle agitation. The solution was cleared by centrifugation (53,000g, 1 h). Purification was carried out using Buffer A and Buffer B (500 mM Imidazole added to Buffer A) containing the appropriate detergent (concentrations are the same as for FSEC). The solubilizate was loaded onto a preequilibrated (2% Buffer B) 10 mL Ni-NTA superflow resin column (Qiagen, Valencia, CA) at a flow rate of 1.0 mL min−1. The column was washed with 2% Buffer B for 20 column volumes (CV) and subsequently eluted via linear gradient from 2 to 100% Buffer B over 5 CV. Eluted fractions were then concentrated, centrifuged (12,000g, 30 min) and loaded onto a Superose 6 10/150 column with Buffer A for polishing. The samples were subjected to SDS-PAGE analysis (12% gels) where the gels were Coomassie Blue stained and protein concentration was determined using a Lowry based protein assay (RC DC Assay, Bio Rad, Hercules, CA).
Cleavage of the membrane protein from the GpA-GFP fusion
The HRV 3C reactions were set up with 10,000 pmoles of membrane-fusion protein and incubated for 24 h at 4°C with a 5:1 HRV 3C protease to protein molar ratio. The cleaved sample was incubated with IMAC nickel resin and 2% Buffer B (to prevent nonspecific binding) for 1 h and applied to a gravity flow column. The flow-through containing the membrane protein and elution containing the GpA-GFP were collected, concentrated and analyzed by SDS-PAGE (12% gel) where the gels were stained with Coomassie Blue. The yield of recovered protein was determined using a Lowry based protein assay (RC DC Assay, Bio Rad, Hercules, CA).
Expression and reoptimization screen for membrane proteins in pWarf(n)
Based on the GFP expression screen for each of the test proteins, expression was examined in the pWarf(n) vector. The conditions examined include 3 cell strains [(BL21(DE3), C41(DE3), C43(DE3)], 2 media (TB and CG, except LldP was LB and TB), 1 IPTG concentration (0.5 or 1.0 mM, depending on what was previously identified), and 2 time points (2 and 4 h). Aliquots of 10 mL were removed at t = 2 and 4 h. Cell pellets were stored overnight at −20°C. Expression was detected by dot-blot analysis, as adapted from Eshaghi et al.37 Cells were resuspended to 0.5 mg cells per ml lysis buffer (Lysis buffer: 50 mM Tris, pH 7.5, 150 mM NaCl, 2% DDM, 0.04 mg mL−1 DNase, 0.1 PMSF, 1 mg mL−1 lysozyme). The resuspension was incubated at 4°C for 1 h with gentle agitation. Samples were centrifuged (12,000g, 30 min) to clarify and 1.5 μL of supernatant was dotted onto nitrocellulose. The dotted samples were allowed to dry overnight. The His8 tagged proteins were probed using the Qiagen Tetra-His Antibody (Qiagen, Valencia, CA), BSA-free and the Pierce Rabbit Anti-Mouse peroxidase conjugated antibody (Thermo Scientific, Rockford, IL). The chemiluminescence signal was detected on film (Perkin Elmer, Waltham, MA) and spots were quantified using ImageJ image processing software.38
Purification of membrane proteins expressed in pWarf(n)
The membrane proteins expressed in pWarf(n) were purified similarly to the membrane-fusion proteins as described earlier in this report. The sole exception was an additional wash step added to the IMAC purification due to lower expression yields; the wash and elution scheme was (a) 2% Buffer B wash for 20 c/v (b) 10% Buffer B wash for 20 c/v and (c) elution with a linear gradient from 10–100% Buffer B. The samples were examined by SDS-PAGE analysis (12% gels) where the gels were stained with Coomassie Blue and protein concentration was determined using a Lowry based protein assay kit (RC DC Assay, Bio Rad, Hercules, CA). Furthermore, TyrP was quantified using BCA (Pierce, Rockford, IL) and A280 using an extinction coefficient generated using ProtParam.39
Functional characterization of LldP-GpA-GFP
E. coli ECL510631 transformed with the LldP-GFP-GpA-His8 construct and was grown in Luria-Bertani medium with kanamycin (100 μg mL−1). Overnight cultures were diluted 10-fold and allowed to grow for 2 h at 37°C before induction with 1 mM IPTG. After additional growth for 2–3 h at 37°C, cells were harvested by centrifugation and washed with 100 mM KPi (pH 7.5). RSO membrane vesicles were prepared from the 1.2 L culture expressing LldP-GFP-GpA-His8 by lysozyme/ethylenediaminetetraacetic acid treatment and osmotic lysis as described.40,41
Lactate transport in RSO membrane vesicles was assayed in the presence of 20 mM ascorbate/0.2 mM phenazine methosulfate (PMS) under oxygen with given concentrations of [14C]-l-lactate. The transport reaction was started by addition of [14C]-l-lactate into 50 μL (10 mg mL−1) RSO aliquots containing ascorbate/PMS. At given times the reaction mixture was spun through 2.5-mL Sephadex column equilibrated with 0.1 M KPi pH 5.5, 0.1 M LiCl, 0.01 MgSO4 for 30 s.42 The vesicle recovery was 25%. The eluted solution containing RSO membrane vesicles was subjected to liquid scintillation counter. The result was corrected for 100% RSO membrane vesicles.
Acknowledgments
The authors wish to thank: Dr. Ernest Wright for the preliminary work using GpA in connection with GFP for monitoring expression and for critical discussion in preparation of the manuscript; Dr. Ronald Kaback for assistance in the functional characterization of LldP; Dr. Phillip Ender and the UCLA Biostatistical department for statistical analyses and consulting; Drs. Vincent Chaptal and Thomas Vondriska for critical discussion in preparation of the manuscript; and Drs. Jan-Willem de Gier and David Drew for the kind gift of the original GFP fusion vector.10
References
- 1.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 2.Turk E, Kim O, le Coutre J, Whitelegge JP, Eskandari S, Lam JT, Kreman M, Zampighi G, Faull KF, Wright EM. Molecular characterization of vibrio parahaemolyticus vSGLT. J Biol Chem. 2000;275:25711–25716l. doi: 10.1074/jbc.M003127200. [DOI] [PubMed] [Google Scholar]
- 3.Drew DE, von Heijne G, Norlund P, de Gier JWL. Green fluorescent protein as an indicator to monitor membrane protein overexpression in Escherichia coli. FEBS Lett. 2001;507:220–224. doi: 10.1016/s0014-5793(01)02980-5. [DOI] [PubMed] [Google Scholar]
- 4.Kawate T, Gouaux E. Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure. 2006;14:673–681. doi: 10.1016/j.str.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 5.Sobolevsky AI, Rosconi MP, Gouaux E. X-ray structure, symmetry and mechanism of an AMPA-subtype glutamate receptor. Nature. 2009;462:745–756. doi: 10.1038/nature08624. PMID: 19946266 [Medline] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaffer PL, Goehring A, Shankaranarayanan A, Gouaux E. Structure and mechanism of a Na+ independent amino acid transporter. 2009;325:1010–1014. doi: 10.1126/science.1176088. PMID: 19608859 [Medline] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawate T, Michel JC, Birdsong WT, Gouaux E. Crystal structure of the ATP-gated P2X4 ion channel in the closed state. Nature. 2009;460:592–598. doi: 10.1038/nature08198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonzales EB, Kawate T, Gouaux E. Pore architecture and ion sites in acid-sensing ion channels and P2X receptors. Nature. 2009;460:599–604. doi: 10.1038/nature08218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drew D, Slotboom DJ, Friso G, Torsten R, Genevaux P, Rapp M, Meindl-Beinker NM, Lambert W, Lerch M, Daley DO, van Wijk KJ, Hirst J, Kunji E, de Gier JW. A scalable, GFP-based pipeline for membrane protein overexpression screening and purification. Protein Sci. 2005;14:2011–2017. doi: 10.1110/ps.051466205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drew D, Lerch M, Kunji E, Slotboom DJ, de Gier JW. Optimization of membrane protein overexpression and purification using GFP fusions. Nat Methods. 2006;3:303–313. doi: 10.1038/nmeth0406-303. [DOI] [PubMed] [Google Scholar]
- 11.Feilmeier BJ, Iseminger G, Schroeder D, Webber H, Phillips GJ. Green fluorescent protein functions as a reporter for protein localization in Escherichia coli. J Bacteriol. 2000;182:4068–4076. doi: 10.1128/jb.182.14.4068-4076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krogh A, Larsson B, von Heijne G, Sonnhammer ELL. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 13.Quick M, Wright EM. Employing Escherichia coli to functionally express, purify, and characterize a human transporter. Proc Natl Acad Sci USA. 2002;13:8597–8601. doi: 10.1073/pnas.132266599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drew D, Newstead S, Sonoda Y, Kim H, von Heijne G, Iwata S. GFP-based optimization scheme for the overexpression and purification of eukaryotic membrane proteins in Saccharomyces cerevisiae. Nat Protocols. 2008;3:784–798. doi: 10.1038/nprot.2008.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hammon J, Palanivelu DV, Chen J, Patel C, Minor DL., Jr A green fluorescent protein screen for identification of well-expressed membrane proteins from a cohort of extremophilic organisms. Protein Sci. 2009;18:121–133. doi: 10.1002/pro.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Newstead S, Kim H, von Heijne G, Iwata S, Drew D. High-throughput fluorescent-based optimization of eukaryotic membrane protein overexpression and purification in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2007;104:13936–13941. doi: 10.1073/pnas.0704546104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daley DO, Rapp M, Granseth E, Melén K, Drew D, von Heijne G. Global topology analysis of the Escherichia coli inner membrane proteome. Science. 2005;308:1321–1323. doi: 10.1126/science.1109730. [DOI] [PubMed] [Google Scholar]
- 18.Saier MH. Available at: http://www.tdcb.org/(2009, September 15) TCDB: transport Classification Database. Retrived from.
- 19.Oliveira C, Costa S, Teixeira JA, Domingues L. cDNA Cloning and functional expression of the α-d-galactose-binding lectin frutalin in Escherichia coli. Mol Biotechnol. 2009;43:212–220. doi: 10.1007/s12033-009-9191-7. [DOI] [PubMed] [Google Scholar]
- 20.Privé GG. Detergents for the stabilization and crystallization of membrane proteins. Methods. 2007;41:388–397. doi: 10.1016/j.ymeth.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Wiener MC. A pedestrian guide to membrane protein crystallization. Methods. 2004;34:364–372. doi: 10.1016/j.ymeth.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 22.Origin Lab. Version 7.5. Northampton, MA: OriginLab Corporation; 2006. [Google Scholar]
- 23.Abramson J, Smirnova I, Kasho V, Verner G, Kaback HR, Iwata S. structure and mechanism of the lactose permease of Eschericia coli. Science. 2003;301:610–615. doi: 10.1126/science.1088196. [DOI] [PubMed] [Google Scholar]
- 24.Boulter JM, Wang DN. Purification and characterization of human erythrocyte glucose transporter in decylmaltoside detergent solution. Protein Exp Purif. 2001;22:337–348. doi: 10.1006/prep.2001.1440. [DOI] [PubMed] [Google Scholar]
- 25.Kurisu G, Zhang H, Smith JL, Cramer WA. Structure of the cytochrome b6f complex of oxygenic photosynthesis: tuning the cavity. Science. 2003;302:1009–1014. doi: 10.1126/science.1090165. [DOI] [PubMed] [Google Scholar]
- 26.Lemieux MJ, Song J, Kim MJ, Huang Y, Villa A, Auer M, Li XD, Wang DN. Three-dimensional crystallization of the Escherichia coli glycerol-3-phosphate transporter: a member of the major facilitator superfamily. Protein Sci. 2003;12:2748–2756. doi: 10.1110/ps.03276603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nußberger S, Dörr K, Wang DN, Kühlbrandt W. Lipid-protein interactions in crystals of plant light-harvesting complex. J Mol Biol. 1993;234:347–356. doi: 10.1006/jmbi.1993.1591. [DOI] [PubMed] [Google Scholar]
- 28.Faham S, Watanabe A, Mercado Besserer G, Cascio D, Specht A, Hirayama BM, Wright EM, Abramson J. The crystal structure of a sodium galactose transporter reveals mechanistic insights into Na+/sugar symport. Science. 2008;321:810–814. doi: 10.1126/science.1160406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edwards AM, Arrowsmith CH, Hui R, Marino F, Savchenko A, Yamazaki K, Yee A. Producing Proteins. In: Chasman DI, editor. Protein structure: determination, analysis, and application for drug discovery. New York: Marcel Dekker, Inc; 2003. p. 14. [Google Scholar]
- 30.Walker PA, Leong LEC, Ng PWP, Tan SH, Waller S, Murphy D, Porter AG. Efficient and rapid affinity purification of proteins using recombinant fusion proteases. Biotechnology. 1994;12:601–605. doi: 10.1038/nbt0694-601. [DOI] [PubMed] [Google Scholar]
- 31.Núñez MF, Kwon O, Wilson TH, Aguilar J, Baldoma L, Lin ECC. Transport of l-Lactate, d-Lactate, and glycolate by the LldP and GlcA membrane carriers of Escherichia coli. Biochem Biophys Res Commun. 2002;290:824–829. doi: 10.1006/bbrc.2001.6255. [DOI] [PubMed] [Google Scholar]
- 32.Korepanova A, Moore JD, Nguyen HB, Hua Y, Cross TA, Gao F. Expression of membrane proteins from Mycobaterium tuberculosis in Escherichia coli as fusions with maltose binding protein. Protein Expr Purif. 2007;53:24–30. doi: 10.1016/j.pep.2006.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roosild TP, Greenwald J, Vega M, Castronovo S, Riek R, Choe S. NMR structure of mistic, a membrane-integrating protein for membrane protein expression. Science. 2005;307:1317–1321. doi: 10.1126/science.1106392. [DOI] [PubMed] [Google Scholar]
- 34.Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Rapid protein-folding assay using green fluorescent protein. Nat Biotechnol. 1999;17:691–695. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- 35.Ender PB. 2009. Available at http://www.ats.ucla.edu/stat/stata/ado/analysis/sme.hlp.htm Tests of simple main effects. UCLA: Academic Technology Services, Statistical Consulting Group.
- 36.Ender PB. 2009. Available at http://www.ats.ucla.edu/stat/stata/ado/analysis/sme.hlp.htm Tukey-kramer pairwise comparisons. UCLA: Academic Technology Services, Statistical Consulting Group.
- 37.Eshaghi S, Hedrén M, Nasser MIA, Hammarberg T, Thronell A, Norlund P. An efficient strategy for high-throughput expression screening of recombinant integral membrane proteins. Protein Sci. 2005;14:676–683. doi: 10.1110/ps.041127005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rasband WS. ImageJ. Bethesda, MD: US National Institutes of Health; 1997. –2005. [Google Scholar]
- 39.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The proteomics protocols handbook. New Jersey: Humana Press; 2005. pp. 571–607. [Google Scholar]
- 40.Kaback HR. Bacterial membranes. In: Kaplan NP, Jakoby WB, Colowick NP, editors. XXII. New York: Elsevier; 1971. pp. 99–120. Methods in enzymol. [Google Scholar]
- 41.Short SA, Kaback HR, Kohn LD. Localization of d-lactate dehydrogenase in native and reconstituted Escherichia coli membrane vesicles. J Biol Chem. 1975;250:4291–4296. [PubMed] [Google Scholar]
- 42.Jayaram H, Accardi A, Wu F, Williams C, Miller C. Ion permeation through a Cl--selective channel designed from a CLC Cl-/H+ exchanger. Proc Natl Acad Sci USA. 2008;105:11194–11199. doi: 10.1073/pnas.0804503105. [DOI] [PMC free article] [PubMed] [Google Scholar]