

Abstract
Nitric oxide (NO) signaling in mammals controls important processes such as smooth muscle relaxation and neurotransmission by the activation of soluble guanylate cyclase (sGC). NO binding to the heme domain of sGC leads to dissociation of the iron–histidine (Fe–His) bond, which is required for enzyme activity. The heme domain of sGC belongs to a larger class of proteins called H-NOX (Heme-Nitric oxide/OXygen) binding domains. Previous crystallographic studies on H-NOX domains demonstrate a correlation between heme bending and protein conformation. It was unclear, however, whether these structural changes were important for signal transduction. Subsequent NMR solution structures of H-NOX proteins show a conformational change upon disconnection of the heme and proximal helix, similar to those observed in the crystallographic studies. The atomic details of these conformational changes, however, are lacking in the NMR structures especially at the heme pocket. Here, a high-resolution crystal structure of an H-NOX mutant mimicking a broken Fe–His bond is reported. This mutant exhibits specific changes in heme conformation and major N-terminal displacements relative to the wild-type H-NOX protein. Fe–His ligation is ubiquitous in all H-NOX domains, and therefore, the heme and protein conformational changes observed in this study are likely to occur throughout the H-NOX family when NO binding leads to rupture of the Fe–His bond.
Keywords: nitric oxide activation, signal transduction, crystallography, Heme-Nitric oxide/Oxygen-binding proteins
Introduction
Nitric oxide (NO) signaling in mammals controls processes such as smooth muscle relaxation and neurotransmission via cGMP, which is produced upon activation of the enzyme soluble guanylate cyclase (sGC).1–3 In sGC, NO-mediated breakage of the Fe–His bond leads to increased catalytic activity. Recently, heme domains related to sGC were identified in prokaryotes and were termed Heme-Nitric oxide/OXygen binding (H-NOX) domains.4,5 These heme domains have also been referred to as H-NOBs.6 Prokaryotic H-NOX proteins fall into two distinct classes. The first consists of a stand-alone H-NOX protein most often found in a predicted operon with either a histidine kinase or, less frequently, with a GGDEF-diguanylate cyclase domain. The second class consists of H-NOX domains fused to methyl-accepting chemotaxis proteins.6,4,7 Homology to the sGC heme domain as well as genomic location adjacent to putative signaling proteins suggests that H-NOX domains likely serve as O2 and NO sensors in prokaryotes.4,7
Consistent with this hypothesis, the ferrous NO complex of the H-NOX from the facultative aerobe, Shewanella oneidensis (So H-NOX), which belongs to the first class of H-NOX proteins, inhibits autophosphorylation of a histidine kinase encoded in the same operon, whereas the ferrous unliganded protein has no effect on kinase activity.7 Similar to mammalian sGC, So H-NOX is high spin in the ferrous unliganded state, which then forms a transient six-coordinate complex upon binding NO that rapidly converts to a low-spin five-coordinate ferrous nitrosyl complex [Fig. 1(A)].8–10 The proximal Fe–His bond is severed upon formation of the five-coordinate NO species, and it is postulated that this results in an activated state through a conformational change induced by the movement of the proximal helix containing the histidine away from the heme cofactor when the Fe–His bond breaks [Fig. 1(A)].
Figure 1.
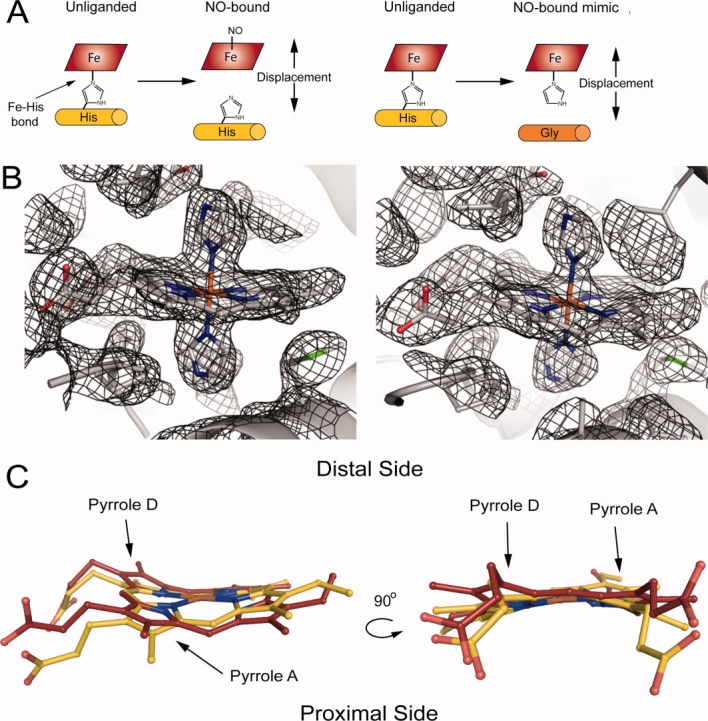
H102G mutant rationale and structural comparison of H102G (PDB ID: 3LAH and 3LAI) versus wild type (PDB ID: 1U56). (A) Nitric oxide (NO) binds in a five-coordinate state where the proximal Fe–His bond is broken, which leads to a displacement of the heme and proximal α-helix (α-helix F). Displacement of the heme and proximal helix in the axial histidine mutant. (B) 2Fo − Fc electron density map of the heme pockets in H102G crystal form F2 (PDB ID: 3LAI) for molecules A and B at 1.0σ contour levels. The imidazoles and hemes were omitted during the generation of the electron density map. (C) H102G heme cofactor (maroon) overlaid with wild type (gold). The heme cofactor in H102G adopts a significantly different conformation when compared with wild type. Pyrroles A and D move toward the distal pocket.
To date, several crystal structures of prokaryotic H-NOX domains have been solved.5,11,12 The H-NOX domain from Thermoanaerobacter tengcongensis (Tt H-NOX), which belongs to the second class of H-NOX domains, binds both NO and O2. A distal hydrogen-bonding network identified in the structure has been found to play a central role in O2 binding.5,13 Studies probing the importance of heme cofactor conformation showed that bending of the heme is correlated to N-terminal movement of the protein in Tt H-NOX.14 Although the correlation between heme conformation and protein structure is compelling, the role of this movement in the molecular mechanism of signal transduction was not clear.
To better understand the mechanistic basis of H-NOX activation, solution structures were solved that mimic the active five-coordinate NO and inactive (unliganded) So H-NOX.15 These structures suggest that removal of the Fe–His bond, mimicked by the axial Fe–His mutant (H103G), results in conformational changes in the heme cofactor and a N-terminal shift in the protein similar to those observed in P115A Tt H-NOX.14 Much insight into the structural changes resulting from mimicking “cleavage” of the Fe–His bond was gained from the NMR study. However, because of limitation in isotopic labeling, especially at the heme, the NMR structures do not allow for an in-depth, atomic analysis of the structural changes.
High-resolution crystallography, in contrast, can provide detailed information on all atoms in the protein and heme cofactor with high accuracy. Tt H-NOX is an ideal protein to study H-NOX structure because crystals typically diffract to ∼2 Å. Thus, the analogous axial histidine mutant, H102G, was made in Tt H-NOX. The goal of this study was to obtain insight into the molecular mechanism of H-NOX activation. The results with the axial ligand mutant should provide key structural information on H-NOX activation in atomic level detail. Information from the crystal structure will demonstrate whether heme cofactor conformation and structure correlation is a general mechanism with heme and proximal helix displacement, which mimics Fe–His bond dissociation, in H-NOX proteins.
Results and Discussion
Structure determination and analysis of Tt H-NOX H102G
The H102G Tt H-NOX mutant was expressed and purified as previously described4,16 and crystallized in several crystal forms. The crystal structure was solved by molecular replacement in two crystal forms (F1 and F2) and refined to 2.0 and 2.1 Å, respectively. The Rwork for crystal forms F1 and F2 was 21.5 and 20.7%, respectively, and the Rfree values were 25.1 and 25.5%, respectively (PDB ID: 3LAH and 3LAI). A total of two and three Fe(III)-bis-imidazole H102G molecules for crystals F1 and F2, respectively, were built in orthorhombic asymmetric unit cells. Crystallographic data and refinement statistics are summarized in Table I.
Table I.
Statistics of Crystallographic Data Collection and Refinement
| F1 | F2 | |
|---|---|---|
| Data collection | ||
| Space group | P212121 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 61.4, 88.7, 89.2 | 61.4, 86.5, 122.7 |
| α, β, γ (°) | 90 | 90 |
| Resolution (Å) | 44.6–2.0 | 50–2.1 |
| Rmerge (%) | 7.3 (43.5) | 6.9 (38.1) |
| I/σa | 24.8 (2.9) | 27.5 (4.3) |
| Completenessa (%) | 97.5 (80.1) | 100 (100) |
| Redundancy | 6.9 | 7.1 |
| Refinement | ||
| No. of reflections | 33172 | 36487 |
| Rwork/Rfreeb (%) | 21.5/25.1 | 20.7/25.5 |
| No. atoms | ||
| Protein | 3196 | 4604 |
| Heme | 86 | 129 |
| Imidazole molecules | 4 | 6 |
| Solvent molecules | 147 | 134 |
| B factors | ||
| Overall | 31 | 37 |
| RMS deviation | ||
| Bond lengths (Å) | 0.008 | 0.008 |
| Bond angles (°) | 0.867 | 1.043 |
The values in parentheses relate to highest resolution shells.
Rfree is calculated for a randomly chosen 5% of reflections.
H102G was compared to wild-type Fe(III) Tt H-NOX (PDB ID: 1U56 Molecule A) to maintain consistency in the oxidation states for structural analysis. Using the Difference Distance Matrix Program (Center for Structural Biology at Yale University, New Haven, CT), C-terminal residues 120–175 of H102G were aligned with wild type. N-terminal root mean square deviation (RMSD) from wild-type Tt H-NOX was calculated for H102G (Supporting Information Table S1). Figure 1(B) shows the electron density maps (2Fo − Fc) of H102G (crystal form F2 Molecule a A and B) at 1.0σ contour levels. The electron density map was generated with the imidazoles and hemes omitted. The electron density map clearly shows the disconnection of the proximal helix from the heme cofactor in the H102G mutant. Additionally, all atoms in the heme pocket have low B factors (∼20–40 Å2) relative to the rest of the protein (∼>40 Å2) and thus can be modeled with high confidence.
All five molecules in both crystal forms of the H102G H-NOX exhibit significant differences in conformation compared with wild-type Tt H-NOX, as illustrated by an alignment of H102G with wild type in Figure 2(A). The N-terminus in H102G is displaced from wild type with N-terminal RMSD values that range from 2.78 to 3.78 Å (Supporting Information Table S1). Importantly, the same movements are observed in two different crystal forms that vary in unit cell dimensions and asymmetric units, which act as a control to show that changes observed are not a result of a crystallographic artifact. Molecule B from H102G crystal F2 was used for subsequent analyses because it has the largest N-terminal RMSD from wild type.
Figure 2.
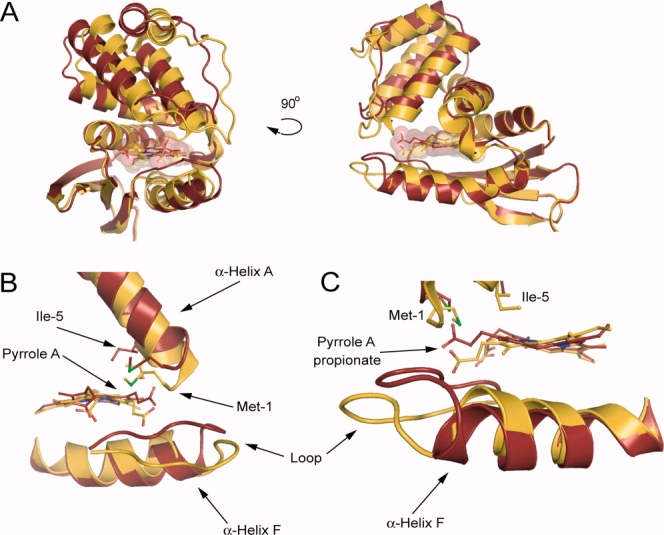
Effects of H102G on the overall structure and the local heme environment. (A) Overall structural comparison of H102G (maroon) versus wild type (gold). A significant N-terminal shift away from wild type is observed in H102G. Shown are F2 molecule B and molecule A for H102G and wild type, respectively. (B) Pyrrole A, Met-1, and Ile-5 move with the heme causing large conformational changes at the N-terminus. (C) The propionate group attached to pyrrole A moves above the heme plane along with an insertion of the loop following α-Helix F. The H102G mutant allows for displacement of α-helix F away from the heme cofactor.
Analysis of the heme-binding pocket
Although free porphyrin is planar in solution, protein-bound heme cofactors are not planar and adopt various conformations.17 The heme cofactor from wild-type Tt H-NOX contains one of the most distorted hemes reported to date.5 Biochemical and structural studies have revealed that heme distortion is correlated to changes in heme reactivity, ligand binding, and protein conformation.5,14,18–21,22,23 As observed in the structure reported here, disconnection of the heme and proximal helix in the H102G mutant results in a significantly relaxed heme cofactor conformation. A comparison of the heme cofactors shows that pyrroles A and D move away from the proximal side of the heme pocket toward the distal side in the H102G mutant compared with wild type [Fig. 1(C)].
Normal-coordinate analysis (NSD) was carried out on the heme cofactor in H102G for both crystal forms (Supporting Information Table S2). The major heme distortion modes in the wild-type protein are saddling and ruffling (Supporting Information Table S2). In Fe(III) wild-type molecule A, the NSD values for saddling and ruffling are −1.115 and −1.171, respectively. Dissociation of the heme from the proximal helix reduces saddling, whereas little change in ruffling is observed. Specifically, saddling in H102G ranges from −0.263 to 0.725. Heme ruffling, however, is similar to that of wild-type Tt H-NOX with a range between −0.980 and −1.216. Thus, the structure shows that only one distortion mode is affected upon removal of His-102, which mimics cleavage of the Fe–His bond.
Overall, the H102G crystal structure suggests that dissociation of the Fe–His bond leads to a general conformational change of the heme exemplified by a decrease in saddling. The resulting increased space in the heme pocket allows the heme to relax to a lower energy conformation. The H102G structure may illustrate the importance of the Fe–His bond in maintaining the unusually large deviation from planarity of the heme in Tt H-NOX. All H-NOX proteins possess proximal histidine ligation to the heme iron and strong evidence suggests that NO binding leads to breakage of this bond in most non-O2-binding H-NOX proteins.8,9 Thus, this type of heme conformational change may be a general feature in the H-NOX family that leads to an effect on downstream function.
Conformational change in the H102G mutant
Local rearrangements of heme pocket residues Met-1 and Ile-5 in H102G made possible by relaxation of the heme translate into large displacements of the N-terminus (greater than 6 Å) compared with wild-type Tt H-NOX [Fig. 2(A), Supporting Information Table S1]. Met-1 and Ile-5 move together with pyrrole A and the attached propionate group [Fig. 2(B,C)]. This propionate group moves above the plane of the heme toward the N-terminus and correlates with the observed N-terminal shift of the protein. Hydrogen-bonding contacts with the propionate groups, including those in the conserved YxSxR motif, have not appreciably changed from wild type.5 Both Met-1 and Ile-5 are part of α-helix A, which is at the beginning of the N-terminus and make van der Waals contact with the distal side of the heme. The helices that comprise the rest of the N-terminus move along with α-helix A about a pivot point between two glycines within α-helices D and G (residues 61–81 and 138–153, respectively), similar to those in So H-NOX.15
The H102G mutant also results in movement of α-helix F (residues 90–107) as observed in Figure 2(C). The liberated α-helix F moves away from the distal pocket toward the C-terminus of the protein. Along with the movement of the propionate group mentioned above, the loop between α-helix F and β-sheet A (residues 108–113) moves with the pyrrole A propionate group [Fig. 2(B,C)]. In the previous crystal structures of Tt H-NOX, the loop between α-helix F and β-sheet A was highly disordered and could not be modeled-in with high confidence. However, in the H102G structure, the loop is ordered and surprisingly moves with the N-terminus and heme propionate group.
Previously, the design of a flattened heme mutant (P115A) in Tt H-NOX has demonstrated that changes in the degree of heme distortion are coupled to N-terminal displacement.14 This proline makes tight van der Waals contact with the heme in wild-type Tt H-NOX, and the less crowded heme pocket in the mutant provides room for the heme to relax. The change in heme deformation was found to be correlated to movement in the N-terminal half of the protein. Here, we find that elimination of the α-helix F attachment to the heme cofactor via the H102G mutation results in similar structural changes. Thus, the elimination of the connection between the heme and α-helix F, which is suggested to occur upon five-coordinate NO formation, is consistent with our model linking heme distortion and N-terminal movement.
It is important to note that the distal imidazole makes a hydrogen bond with Tyr-140. The distance between the Fe and Tyr is increased by 1 Å; however, this is also observed in published structures (PDB ID: 3EEE and 3IQB) of mutants with planar hemes that are oxygen bound.14,24 If the histidine is responsible for the structural change alone, the 1 Å increase is not enough to account for the >6 Å movement at the N-terminus. Thus, the mechanism of the structural change is likely caused by heme flattening.
A conserved molecular mechanism for Fe–His bond dissociation
The high-resolution crystal structure reported here of H102G shows that changes in the conformation of the heme cofactor can be ascribed to a decrease in the amount of saddling. The N-terminus/heme coupled movement is similar to what is observed for the P115A mutant. That is, pyrrole A moves above the plane toward the distal side of the heme pocket, along with its attached propionate group, eliciting a shift in the N-terminal half of the protein through interactions with α-helix A. NMR structures of the analogous axial histidine mutant in So H-NOX also show a relaxation of the heme cofactor.15 Despite the fact that the resolution of these structures is limited because of restricted isotopic labeling of the heme, a flattening of the heme was observed, as well as a conformational change at the N-terminus.
We recently speculated that the Fe–His bond and the distorted heme may act as a “loaded spring” through maintaining the heme and protein in an inactive conformation in So H-NOX.15 Upon NO binding, the Fe–His bond is broken and the five-coordinate complex is formed, which leads to a kinase inhibitory form of So H-NOX. Here, we propose that the Tt H-NOX H102G mutation mimics NO binding through movement of the proximal helix away from the heme, allowing the heme to adopt a more relaxed conformation. When the Fe–His bond is broken, the heme may adopt a lower energetic conformation and stored potential energy is released, leading to the observed structural changes in the protein. The observation of similar conformational changes in two different homologues representing two classes of H-NOX domains suggests that the structural changes may be general with Fe–His bond dissociation and is possibly important for sensing and transmitting a ligand-binding signal.
Conclusions
The crystal structure of the axial histidine H102G mutant in Tt H-NOX, a mimic of the five-coordinate NO complex, was solved to 2.0 Å. The high-resolution structure showed that only one major heme distortion mode was affected by disconnection of the heme and proximal helix, which likely mimics Fe–His bond breakage, and atoms that were disordered in previous crystals structures are participating in the movement of the protein. Local changes at the heme cofactor and in the heme pocket lead to global conformational changes in the protein. These significant conformational changes caused by dissociation of the heme and proximal helix have now been observed in two different H-NOX homologues. Removal of the Fe–His bond may lead to heme and protein conformational changes, which likely represent a general mechanism for activation in the H-NOX family.
Materials and Methods
Protein purification
Mutagenesis was carried out using the QuikChange protocol from Stratagene. Expression and purification of Tt H-NOX H102G were carried out as previously described4,16 with the addition of 10 mM imidazole to all purification buffers and expression media. It has been demonstrated that heme binding in the axial histidine H-NOX mutants can be rescued with exogenous imidazole.16
H102G crystallization—F1 crystal form
Samples of Tt H-NOX H102G were exchanged into buffer containing 20 mM TEA (pH 7.5) and 10 mM imidazole (buffer A) and oxidized using 100 equiv of potassium ferricyanide [K3Fe(CN)6]. Potassium ferricyanide was removed using a PD10 column (Amersham Biosciences) preequilibrated with buffer A and H102G was then concentrated to 30 mg mL−1. Crystals were grown using sitting drop vapor diffusion by mixing 1 μL of the protein solution with 1 μL of the reservoir solution equilibrating against a 700 μL reservoir of 28% (w/v) PEG 3350 and 0.25 M Mg(CH3COO)2 at 20°C. Crystals began to appear within 24 h. Cryoprotection was achieved by transferring the crystals stepwise into mother liquor solutions containing increasing concentrations of glycerol ending with 15% (v/v) glycerol. Crystals were flash frozen in liquid nitrogen and stored for later use in data collection.
H102G crystallization—F2 crystal form
H102G samples were prepared as mentioned above. Small needle clusters were grown using hanging drop vapor diffusion by mixing 1 μL of the protein solution with 1 μL of reservoir solution, 20% (w/v) PEG 3350 and 0.2 M KCl, and equilibrated against 700 μL of the reservoir solution at 4°C. Large single crystals were grown overnight by mixing 1 μL of a 15 mg mL−1 protein solution with 1 μL of seeding solution. Seeding mother liquor was generated with the Seed Bead kit (Hampton Research). Crystals began to appear within 24 h. Cryoprotection was achieved as described above.
X-ray data collection, phasing, and refinement
X-ray data were collected using synchrotron radiation at beamline 5.0.3 at the Advanced Light Source, Lawrence Berkeley National Laboratory. Diffraction images were collected at 100 K with 5 s exposure times and 1° oscillation per frame. Data were processed using the HKL200025 suite, and molecular replacement was performed using Phaser26 with wild-type Tt H-NOX (PDB ID 1U55) as the search model. Model building was carried out using Coot27 and refined using Phenix.28 The final models for crystal forms F1 and F2 were refined to a final Rwork of 21.5 and 20.7% (Rfree of 25.1 and 25.5%) at 2.0 and 2.1 Å, respectively.
Acknowledgments
The authors thank Peter Zwart and supporting staff at ALS LBNL beamline 5.0.3. They thank members of the Marletta Lab for critical discussions and review of the manuscript. They also thank Jonathan A. Winger and members of the Kuriyan Lab for critical discussions and review of the manuscript.
Glossary
Abbreviations:
- Fe
iron
- His
histidine
- H-NOX
Heme-Nitric oxide/OXygen-binding protein/domain
- NO
nitric oxide
- O2
oxygen
- sGC
soluble guanylate cyclase
- So
Shewanella oneidensis
- Tt
Thermoanaerobacter tengcongensis
References
- 1.Denninger JW, Marletta MA. Guanylate cyclase and the •NO/cGMP signaling pathway. Biochim Biophys Acta. 1999;1411:334–350. doi: 10.1016/s0005-2728(99)00024-9. [DOI] [PubMed] [Google Scholar]
- 2.Russwurm M, Koesling D. NO activation of guanylyl cyclase. EMBO J. 2004;23:4443–4450. doi: 10.1038/sj.emboj.7600422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Derbyshire ER, Marletta MA. Biochemistry of soluble guanylate cyclase. Handb Exp Pharmacol. 2009;191:17–31. doi: 10.1007/978-3-540-68964-5_2. [DOI] [PubMed] [Google Scholar]
- 4.Karow DS, Pan D, Tran R, Pellicena P, Presley A, Mathies RA, Marletta MA. Spectroscopic characterization of the soluble guanylate cyclase-like heme domains from Vibrio cholerae and Thermoanaerobacter tengcongensis. Biochemistry. 2004;43:10203–10211. doi: 10.1021/bi049374l. [DOI] [PubMed] [Google Scholar]
- 5.Pellicena P, Karow DS, Boon EM, Marletta MA, Kuriyan J. Crystal structure of an oxygen-binding heme domain related to soluble guanylate cyclases. Proc Natl Acad Sci USA. 2004;101:12854–12859. doi: 10.1073/pnas.0405188101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iyer LM, Anantharaman V, Aravind L. Ancient conserved domains shared by animal soluble guanylyl cyclases and bacterial signaling proteins. BMC Genomics. 2003;4:5. doi: 10.1186/1471-2164-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Price MS, Chao LY, Marletta MA. Shewanella oneidensis MR-1 H-NOX regulation of a histidine kinase by nitric oxide. Biochemistry. 2007;46:13677–13683. doi: 10.1021/bi7019035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stone JR, Marletta MA. Spectral and kinetic studies on the activation of soluble guanylate cyclase by nitric oxide. Biochemistry. 1996;35:1093–1099. doi: 10.1021/bi9519718. [DOI] [PubMed] [Google Scholar]
- 9.Zhao Y, Brandish PE, Ballou DP, Marletta MA. A molecular basis for nitric oxide sensing by soluble guanylate cyclase. Proc Natl Acad Sci USA. 1999;96:14753–14758. doi: 10.1073/pnas.96.26.14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cary SP, Winger JA, Derbyshire ER, Marletta MA. Nitric oxide signaling: no longer simply on or off. Trends Biochem Sci. 2006;31:231–239. doi: 10.1016/j.tibs.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 11.Nioche P, Berka V, Vipond J, Minton N, Tsai AL, Raman CS. Femtomolar sensitivity of a NO sensor from Clostridium botulinum. Science. 2004;306:1550–1553. doi: 10.1126/science.1103596. [DOI] [PubMed] [Google Scholar]
- 12.Ma X, Sayed N, Beuve A, van den Akker F. NO and CO differentially activate soluble guanylyl cyclase via a heme pivot-bend mechanism. EMBO J. 2007;26:578–588. doi: 10.1038/sj.emboj.7601521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boon EM, Huang SH, Marletta MA. A molecular basis for NO selectivity in soluble guanylate cyclase. Nat Chem Biol. 2005;1:53–59. doi: 10.1038/nchembio704. [DOI] [PubMed] [Google Scholar]
- 14.Olea C, Boon EM, Pellicena P, Kuriyan J, Marletta MA. Probing the function of heme distortion in the H-NOX family. ACS Chem Biol. 2008;3:703–710. doi: 10.1021/cb800185h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erbil WK, Price MS, Wemmer DE, Marletta MA. A structural basis for H-NOX signaling by trapping a kinase inhibitory conformation of the H-NOX from S. oneidensis. Proc Natl Acad Sci USA. 2009;106:19753–19760. doi: 10.1073/pnas.0911645106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y, Schelvis JP, Babcock GT, Marletta MA. Identification of histidine 105 in the beta1 subunit of soluble guanylate cyclase as the heme proximal ligand. Biochemistry. 1998;37:4502–4509. doi: 10.1021/bi972686m. [DOI] [PubMed] [Google Scholar]
- 17.Shelnutt JA, Song XZ, Ma JG, Jia SL, Jentzen W, Medforth CJ. Nonplanar porphyrins and their significance in proteins. Chem Soc Rev. 1998;27:31–41. [Google Scholar]
- 18.Barkigia KM, Chantranupong L, Smith KM, Fajer J. Structural and theoretical-models of photosynthetic chromophores—implications for redox, light-absorption properties and vectorial electron flow. J Am Chem Soc. 1988;110:7566–7567. [Google Scholar]
- 19.Ravikanth M, Chandrashekar TK. Nonplanar porphyrins and their biological relevance—ground and excited-state dynamics. Coord Chem. 1995;82:105–188. [Google Scholar]
- 20.Roberts SA, Weichsel A, Qiu Y, Shelnutt JA, Walker FA, Montfort WR. Ligand-induced heme ruffling and bent no geometry in ultra-high-resolution structures of nitrophorin 4. Biochemistry. 2001;40:11327–11337. doi: 10.1021/bi0109257. [DOI] [PubMed] [Google Scholar]
- 21.Shokhireva T, Berry RE, Uno E, Balfour CA, Zhang H, Walker FA. Electrochemical and NMR spectroscopic studies of distal pocket mutants of nitrophorin 2: stability, structure, and dynamics of axial ligand complexes. Proc Natl Acad Sci USA. 2003;100:3778–3783. doi: 10.1073/pnas.0536641100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maes EM, Roberts SA, Weichsel A, Montfort WR. Ultrahigh resolution structures of nitrophorin 4: heme distortion in ferrous CO and NO complexes. Biochemistry. 2005;44:12690–12699. doi: 10.1021/bi0506573. [DOI] [PubMed] [Google Scholar]
- 23.Lee WC, Reniere ML, Skaar EP, Murphy ME. Ruffling of metalloporphyrins bound to IsdG and IsdI, two heme-degrading enzymes in Staphylococcus aureus. J Biol Chem. 2008;283:30957–30963. doi: 10.1074/jbc.M709486200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinert EE, Plate L, Whited CA, Olea C, Marletta MA. Determinants of ligand affinity and heme reactivity in H-NOX domains. Angew Chem Int Ed Engl. 2010;49:720–723. doi: 10.1002/anie.200904799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Macromol Crystallogr A. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 26.Mccoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, Mccoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]