

Summary
Background
Whether triglyceride-mediated pathways are causally relevant to coronary heart disease is uncertain. We studied a genetic variant that regulates triglyceride concentration to help judge likelihood of causality.
Methods
We assessed the −1131T>C (rs662799) promoter polymorphism of the apolipoprotein A5 (APOA5) gene in relation to triglyceride concentration, several other risk factors, and risk of coronary heart disease. We compared disease risk for genetically-raised triglyceride concentration (20 842 patients with coronary heart disease, 35 206 controls) with that recorded for equivalent differences in circulating triglyceride concentration in prospective studies (302 430 participants with no history of cardiovascular disease; 12 785 incident cases of coronary heart disease during 2·79 million person-years at risk). We analysed −1131T>C in 1795 people without a history of cardiovascular disease who had information about lipoprotein concentration and diameter obtained by nuclear magnetic resonance spectroscopy.
Findings
The minor allele frequency of −1131T>C was 8% (95% CI 7–9). −1131T>C was not significantly associated with several non-lipid risk factors or LDL cholesterol, and it was modestly associated with lower HDL cholesterol (mean difference per C allele 3·5% [95% CI 2·6–4·6]; 0·053 mmol/L [0·039–0·068]), lower apolipoprotein AI (1·3% [0·3–2·3]; 0·023 g/L [0·005–0·041]), and higher apolipoprotein B (3·2% [1·3–5·1]; 0·027 g/L [0·011–0·043]). By contrast, for every C allele inherited, mean triglyceride concentration was 16·0% (95% CI 12·9–18·7), or 0·25 mmol/L (0·20–0·29), higher (p=4·4×10−24). The odds ratio for coronary heart disease was 1·18 (95% CI 1·11–1·26; p=2·6×10−7) per C allele, which was concordant with the hazard ratio of 1·10 (95% CI 1·08–1·12) per 16% higher triglyceride concentration recorded in prospective studies. −1131T>C was significantly associated with higher VLDL particle concentration (mean difference per C allele 12·2 nmol/L [95% CI 7·7–16·7]; p=9·3×10−8) and smaller HDL particle size (0·14 nm [0·08–0·20]; p=7·0×10−5), factors that could mediate the effects of triglyceride.
Interpretation
These data are consistent with a causal association between triglyceride-mediated pathways and coronary heart disease.
Funding
British Heart Foundation, UK Medical Research Council, Novartis.
Introduction
Whether conditions characterised by increased circulating triglyceride concentration (so-called triglyceride-mediated lipid pathways) are causal in coronary heart disease is uncertain. Prospective epidemiological studies have tended to report positive associations between circulating triglyceride concentration and risk of coronary heart disease,1 but these associations disappear after controlling for HDL and LDL (or non-HDL) cholesterol.2 However, because of limited understanding of the relevant biological pathways, interpretation of such adjustment is difficult when making judgments about causality. If cholesterol in lipoproteins mediates any causal effect of triglyceride, then controlling for it could obscure potentially relevant associations between triglyceride concentration and coronary heart disease. In the absence of such mediating effects, however, failure to control for the confounding effects of such closely related lipids would exaggerate these associations.3 In principle, randomised trials of triglyceride-lowering agents should be able to resolve this uncertainty. In practice, however, available interventions (eg, fibrates, statins, nicotinic acid) cannot do so because they affect several major lipids (eg, triglyceride, LDL cholesterol, HDL cholesterol).4
Studies of genetic variants that specifically (or at least predominantly) affect triglyceride concentration provide another route to assist with the judgment of causality. These studies exploit the fact that genotypes are fixed at conception and should be an indicator of lifelong lipid concentrations (ie, Mendelian randomisation analysis).5 Genome-wide association studies have reported that polymorphisms in or near the apolipoprotein A5 (APOA5) gene are among the strongest known genetic determinants of triglyceride concentration.6 APOA5 is expressed in the liver and codes for apoAV—a 366 aminoacid protein that is predominantly associated with triglyceride-rich VLDL.7 Previous studies have reported associations between −1131T>C (rs662799)—a regulatory variant in the promoter region of APOA5—and triglyceride concentration (webappendix pp 11–12). However, available studies have been insufficiently powered to assess the magnitude and specificity of this association, or to establish its model of inheritance. Similar considerations apply to studies of −1131T>C and coronary heart disease (webappendix pp 11–12). Furthermore, available studies have been too small to assess reliably whether there is concordance between three separate associations: −1131T>C and triglyceride concentration; triglyceride concentration and risk of coronary heart disease; and −1131T>C and risk of coronary heart disease. If concordance between these associations were to be shown, then it would lend support to causality.
We assessed APOA5 −1131T>C in relation to triglyceride concentration, several other risk factors, and risk of coronary heart disease to assess the likelihood of a causal relation between triglyceride-mediated pathways and this disease by study of this one genetic variant.
Methods
Study design and rationale
This study had four inter-related components. First, to assess suitability of study of −1131T>C as a method for deconfounding, we investigated its associations with triglyceride concentration, several other major lipids (ie, HDL and LDL cholesterol, apolipoproteins AI and B), and non-lipid risk factors. Second, we studied −1131T>C in 20 842 cases of coronary heart disease and 35 206 controls. Third, we compared odds ratios (ORs) for coronary heart disease with genetically-raised triglyceride concentration versus hazard ratios (HRs) for this disease recorded with equivalent differences in triglyceride concentration. Fourth, to provide mechanistic insight, we analysed −1131T>C in relation to lipoprotein characteristics in 1795 participants.
Contributing studies
For genetic analyses, we included 73 252 individuals from 39 studies of −1131T>C (rs662799) identified through registry approaches8 and systematic searches of published work (webappendix pp 2, 10–13). According to a uniform protocol, all but one identified studies supplied data for: genotype frequencies by disease status (separately for myocardial infarction, non-overlapping coronary stenosis cases, and healthy controls); definition of coronary heart disease; population sampling and laboratory procedures; and mean (SD) of lipid measures by genotype in people without cardiovascular disease at time of measurement. Because one study involving 154 participants (or 0·2% of the total) was unable to provide tabular data, information for it was abstracted from published reports (webappendix p 2). Genotyping was done with the ITMAT-Broad-CARe 50K array,9 Taqman assay or similar platforms, and restriction fragment length polymorphism. Studies typically used standard lipid measurements (enzymatic assays for triglyceride; precipitation methods for HDL cholesterol; immunoassays for apolipoproteins). Lipoprotein particle concentration and diameter were assessed by nuclear magnetic resonance spectroscopy (LipoScience, Raleigh, NC, USA) in 1795 participants with no history of cardiovascular disease in the prospective EPIC-Norfolk Study.10 Myocardial infarction was defined according to WHO criteria11 and coronary stenosis by angiography (at least 50% of ≥one major coronary artery; webappendix p 2). Associations of −1131T>C with several non-lipid vascular risk factors (ie, blood pressure, smoking status, body-mass index, history of diabetes) were assessed in 13 331 participants in prospective studies with no history of cardiovascular disease at the baseline survey.
For assessment of associations of triglyceride association with incidence of coronary heart disease, we accessed individual participant data from 68 prospective studies of 302 430 participants without known cardiovascular disease at baseline. 12 785 outcomes of incident fatal coronary heart disease and non-fatal myocardial infarction (with use of WHO criteria) were recorded during 2·79 million person-years at risk.2 Because measurement error or within-person variability (ie, regression-dilution bias) in triglyceride concentration and other risk factors can lead to misestimation of risk,12 we calculated long-term average (usual) concentrations of triglyceride and other risk factors from serial measurements (median interval 4·7 years [IQR 2·7–6·1]) for 89 073 participants, as described previously.12 Serial measurements yielded age-adjusted and sex-adjusted regression dilution ratios of 0·63 (95% CI 0·60–0·67) for loge triglyceride, 0·69 (0·64–0·74) for HDL cholesterol, and 0·64 (0·57–0·71) for directly measured LDL cholesterol.2
Statistical analysis
We calculated estimates of association separately within each study before pooling across studies by random-effects meta-analysis (parallel analyses used fixed-effect models). Summary ORs for coronary heart disease and mean concentrations of lipid markers (and differences in mean concentrations compared with the common homozygotes on the original scale [ie, mmol/L for triglyceride, HDL cholesterol, and LDL cholesterol; g/L for apolipoproteins A and B]) were estimated. To enable comparisons across lipid measures, associations are also presented as percentage differences (calculated in reference to the weighted mean of each marker in common homozygotes). For studies that compared the same control group to patients with myocardial infarction and (non-overlapping) patients with angiographically documented coronary stenosis, we avoided any double counting by analysing cases of myocardial infarction and coronary stenosis separately before combining them into one coronary disease group.
We estimated HRs for coronary heart disease with triglyceride concentration with Cox proportional hazard regression models, using methods described previously.2,13 HRs were adjusted progressively for age, sex, systolic blood pressure, smoking status, history of diabetes, body-mass index, non-HDL cholesterol, and HDL cholesterol. We calculated HRs for coronary heart disease with differences in lipid concentration observed with −1131T>C to enable comparison with odds ratios for coronary heart disease with genetically-raised triglyceride concentration.
Heterogeneity was assessed by the I2 statistic,14 the Q statistic, and meta-regression of prespecified groupings of studies characteristics. Small-study effects were assessed with funnel plots and by comparing pooled results from studies of at least 500 patients with coronary heart disease (or, for gene–lipid investigations, at least 1000 healthy participants) with those from smaller studies. Analyses were done with Stata (version 11.0), two-sided p values, and 95% CIs.
Role of the funding source
The sponsors of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. NS and JD had full access to all the data and had final responsibility for the decision to submit for publication.
Results
Of the 73 252 participants with information about APOA5 −1131T>C, 47 784 (65%) were of European descent. The minor allele frequency of −1131T>C was 8% (95% CI 7–9) in people of European descent without coronary heart disease. −1131T>C was not significantly related to several non-lipid risk factors (webappendix p 3) or LDL cholesterol. For every C allele inherited, carriers had 3·5% (95% CI 2·6–4·6), or 0·053 mmol/L (0·039–0·068), lower mean HDL cholesterol; 1·3% (0·3–2·3), or 0·023 g/L (0·005–0·041), lower mean apolipoprotein AI; and 3·2% (1·3–5·1), or 0·027 g/L (0·011–0·043), higher mean apolipoprotein B (figure 1 and figure 2) than did non-carriers.
Figure 1.
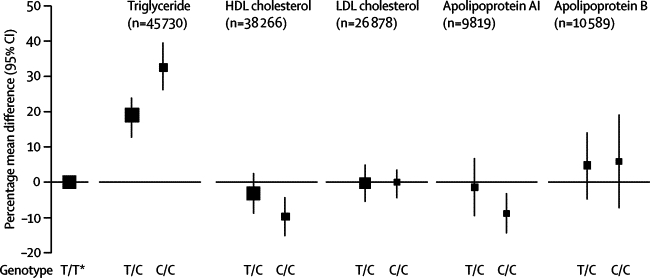
APOA5 −1131T>C genotypes and circulating lipid concentration
Size of data markers is proportional to the inverse of the variance of the weighted mean difference (the reference group is represented by a square with an arbitrary fixed size) and the vertical lines represent 95% CIs. To enable comparison of associations across lipids and apolipoproteins, associations are presented as percentage differences (calculated in reference to the weighted mean of each marker in common homozygotes). *Reference group.
Figure 2.
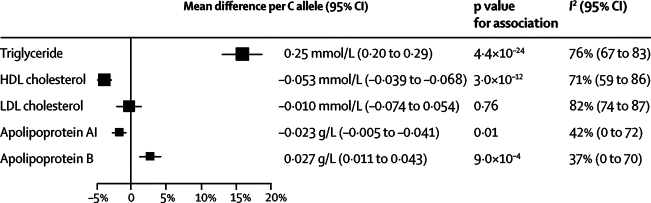
Association of APOA5 −1131T>C with circulating lipid concentration per C allele
Size of data markers is proportional to the inverse of the variance of the weighted mean difference, and the horizontal lines represent 95% CIs. To enable comparison of associations across lipids and apolipoproteins, associations are presented as percentage differences (calculated in reference to the weighted mean of each marker in common homozygotes).
−1131T>C was strongly related to triglyceride concentration, consistent with a per-allele model of inheritance. Carriers of two C alleles had 32·6% (95% CI 26·7–39·0), or 0·51 mmol/L (0·42–0·61), higher mean triglyceride than did non-carriers (figure 1). For every C allele inherited, carriers had 16·0% (12·9–18·7), or 0·25 mmol/L (0·20–0·29), higher mean triglyceride than did common homozygotes (p=4·4×10−24; figure 2). We noted significant heterogeneity in studies of triglyceride (I2=76%, 95% CI 67–83; p<0·0001), although little of it was explained by characteristics recorded here, including fasting status (webappendix p 5). We recorded no evidence of small-study effects (webappendix pp 5–7). In the subset of participants who had detailed information about lipoproteins, −1131T>C was significantly associated with higher VLDL particle concentration, smaller HDL particle size, higher LDL particle concentration, and smaller LDL particle size (table).
Table.
APOA5 −1131T>C genotypes and lipoprotein particle size and concentration in controls in the EPIC-Norfolk Study10
|
Genotype* |
β coefficient (95% CI)† | p value for trend | |||
|---|---|---|---|---|---|
| TT (n=1606) | TC (n=182) | CC (n=7) | |||
| LDL | |||||
| Particle size (nm) | 21·1 (0·6) | 21·0 (0·6) | 20·7 (0·4) | −0·13 (−0·05 to −0·21) | 0·002 |
| Particle concentration (nmol/L) | 1567 (418) | 1680 (461) | 1670 (435) | 105 (44 to 166) | 0·001 |
| VLDL | |||||
| Particle size (nm) | 50·8 (8·4) | 51·3 (7·9) | 59·6 (6·7) | 1·13 (−0·07 to 2·33) | 0·065 |
| Particle concentration (nmol/L) | 93·0 (31·2) | 106·1 (32·4) | 106·6 (23·0) | 12·2 (7·7 to 16·7) | 9·3×10−8 |
| HDL | |||||
| Particle size (nm) | 8·9 (0·5) | 8·8 (0·4) | 8·5 (0·3) | −0·14 (−0·08 to −0·20) | 7·0×10−5 |
| Particle concentration (μmol/L) | 34·0 (5·4) | 33·9 (5·7) | 35·8 (5·4) | 0·04 (−0·7 to 0·8) | 0·93 |
Data are mean (SD).
Calculated from linear regression of each lipoprotein marker on APOA5 genotype.
The OR for coronary heart disease was 1·40 (95% CI 1·12–1·75) in carriers of two C alleles compared with non-carriers (figure 3), with evidence of a per-allele model of inheritance. The OR for coronary heart disease was 1·18 (1·11–1·26; p=2·6×10−7) per C allele. We noted some heterogeneity in studies of coronary heart disease (I2=35%, 95% CI 0–59; p=0·03), although little of it was explained by characteristics recorded here, including type of coronary outcome (webappendix p 7). Exclusion of three studies that significantly (p<0·05) deviated from Hardy-Weinberg equilibrium among controls did not change the findings (data not shown). There were too few data for reliable analyses by ethnic origin, although we recorded no significant differences in analyses of white versus non-white participants (webappendix pp 5 and 7).
Figure 3.
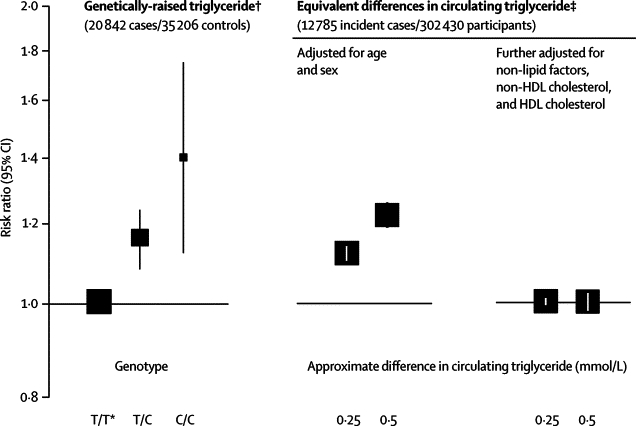
Association of APOA5 −1131T>C genotypes and equivalent differences in circulating triglyceride concentration with risk of coronary heart disease
Non-lipid factors adjusted for included smoking status, systolic blood pressure, body-mass index, and history of diabetes (webappendix p 5). Size of data markers is proportional to the inverse of the variance of the weighted mean difference (the reference group is represented by a square with an arbitrary fixed size) and the vertical lines represent 95% CIs. *Reference group. †Odds ratio for coronary heart disease associated with APOA5 −1131T>C. ‡Hazard ratio for coronary heart disease in prospective studies for differences in usual triglyceride concentration equal to those recorded with APOA5 −1131T>C (as reported in figure 1).
In 302 430 participants in prospective studies, the HR for coronary heart disease, adjusted for age and sex only, was 1·10 (95% CI 1·08–1·12) per 16% higher triglyceride concentration. The HR reduced to 1·00 (0·98–1·02) after further adjustment for several risk factors. Most of the attenuation in the HR was due to adjustment for HDL cholesterol and non-HDL cholesterol (webappendix pp 4 and 8). The OR for coronary heart disease associated with −1131T>C was concordant with the HR that was recorded with triglyceride concentration, provided that the HR was not adjusted for HDL cholesterol and non-HDL cholesterol (figure 3). The association between −1131T>C and HDL cholesterol could account for a modest component of the association between −1131T>C and coronary heart disease (webappendix p 9).
Discussion
We have reported a series of findings that are consistent with a causal role for triglyceride-mediated pathways in coronary heart disease. First, we have shown that −1131T>C—a regulatory variant in APOA5—is unrelated to several non-lipid risk factors or LDL cholesterol, and comparatively moderately related to HDL cholesterol and other major lipids. Second, we have shown that −1131T>C is strongly related to triglyceride concentration in a dose-dependent manner, with every C allele increasing triglyceride by about as much as having type 2 diabetes mellitus.15 Third, in an analysis of 20 842 cases and 35 206 controls, we have shown that −1131T>C is related to risk of coronary heart disease in an analogous dose-dependent manner, with about 18% higher risk per C allele. Fourth, in an analysis of 302 430 people, we have shown that risk of coronary heart disease with genetically raised triglyceride is concordant with risk of disease with equivalent differences in circulating triglyceride itself. Finally, we have shown that −1131T>C is associated with higher VLDL concentration and smaller HDL particle size—pathways through which triglyceride could affect risk of coronary heart disease.
These findings encourage further study of interventions that target triglyceride-mediated pathways.16–19 Because preliminary studies have suggested that responses to fibrates and statins might vary by APOA5 −1131T>C status,20–22 our data reinforce the need for more powerful such studies. Our results also have implications for observational studies. They suggest that, in attempts to understand the potential causal relevance of triglyceride to health outcomes, care is needed to avoid over-adjustment for potential mediating pathways. By contrast, such considerations do not apply to the assessment of prediction of coronary heart disease, for which a main consideration is inclusion of characteristics that maximise a model's predictive value. Previous analyses have suggested that, given knowledge of concentrations of HDL and total cholesterol, assessment of triglyceride concentration provides little incremental information for prediction of coronary heart disease.2
Our data are consistent with observations of premature atherosclerosis in patients with raised triglyceride concentration (eg, familial hypertriglyceridaemia, familial combined hyperlipidaemia, and remnant hyperlipidaemia).23–25 Disorders of high triglyceride concentration could remodel LDL and HDL particles, making them smaller and denser.26,27 For LDL, such changes could make particles more atherogenic.28 For HDL, the situation is more complex, but small dense HDL might be functionally impaired (or even proatherogenic).29 Hypertriglyceridaemia could be an indicator of increased concentration of proatherogenic VLDL and remnant lipoporoteins.30 These possibilities are consistent with our observation that −1131T>C is related to higher VLDL concentration and smaller HDL particle size.
APOA5 −1131T>C is in almost complete linkage disequilibrium with two other APOA5 polymorphisms in Europeans: +1891T>C (rs2266788) and −3A>G (rs651821).31 In-vitro studies suggest that these variants might act together because constructs of the haplotype carrying all three rare alleles are associated with about 50% lower gene expression than the wild type haplotype, resulting in decreased synthesis and incorporation into triglyceride-rich particles of apoAV.32 ApoAV activates lipoprotein lipase33 (an enzyme central to triglyceride catabolism) and acts as a ligand for receptor clearance of triglyceride-rich particles.34 However, although −1131T>C could directly mediate changes in triglyceride concentration, detailed studies are needed to identify the causative genetic loci and to improve understanding of their functional role.
Causal investigation with use of APOA5 −1131T>C should be more reliable than conventional observational studies because genetically-raised triglyceride is: determined at conception, avoiding reverse causality; unrelated to several non-lipid risk factors and comparatively moderately related to HDL cholesterol and other lipids, reducing the scope for confounding; and presumably indicative of lifelong concentrations. This final consideration might partly explain the possibly higher OR for coronary heart disease with genetically-raised triglyceride than the HR with equivalent differences in triglyceride concentration. Indeed, even estimation of long-term average lipid concentrations in midlife (as we did with serial measurements from 89 073 adults) might not necessarily approximate lifelong concentrations. Another potential explanation is that part of the association between −1131T>C and coronary heart disease indicates non-triglyceride-mediated pathways. We noted, for example, comparatively moderate associations between −1131T>C and HDL cholesterol. However, because −1131T>C is a regulatory variant of APOA5 that is predominantly associated with triglyceride concentration, the scope for pleiotropy is reduced. Future studies should aim to include several (unlinked) genetic variants as deconfounding methods, because if they produce congruent predictions with respect to the causal effect of triglycerides on coronary heart disease, then the scope for pleiotropy would be reduced still further.35
Although we used individual participant data for analyses of circulating triglyceride concentration and risk of coronary heart disease, we had access only to tabular genetic data, thereby preventing adjustment of associations between −1131T>C and coronary heart disease for lipids, or instrumental variables analysis. Since most participants were of European descent, further studies are needed in other races. Mechanistic studies are needed that include more extensive genotyping of APOA5 (and related loci) and lipoprotein assessment. Future studies should also assess whether triglyceride-related genotypes are related to liver function or accumulation of liver fat, since some triglyceride-modifying agents could promote hepatic dysfunction.36
Acknowledgments
Acknowledgments
The coordinating centre was supported by the British Heart Foundation, the UK Medical Research Council, and Novartis. A variety of sources have supported investigators, recruitment, follow-up, and laboratory measurements in the studies contributing to this report. Investigators of several of these studies have contributed to a list naming some of these funding sources, which can be found at http://www.phpc.cam.ac.uk/ceu/. S Shah assisted with provision of tabular data.
Contributors
Nadeem Sarwar and John Danesh drafted the report. Nadeem Sarwar, Adam S Butterworth, and Emanuele Di Angelantonio did the analyses. All members of the writing committee provided critical revisions. All investigators shared data and had opportunities to contribute to the interpretation of the results and critical revision of the report. The data management team undertook data collation and harmonisation. All members of the coordinating centre contributed to the collection, harmonisation, analysis, and interpretation of the data.
Triglyceride Coronary Disease Genetics Consortium and Emerging Risk Factors Collaboration
Writing committee Nadeem Sarwar, University of Cambridge, Cambridge UK; Manjinder S Sandhu, University of Cambridge, Cambridge, UK; Sally L Ricketts, University of Cambridge, Cambridge, UK; Adam S Butterworth, University of Cambridge, Cambridge, UK; Emanuele Di Angelantonio, University of Cambridge, Cambridge, UK; S Matthijs Boekholdt, Academic Medical Center, Amsterdam, Netherlands; Willem Ouwehand, University of Cambridge, Cambridge, UK; Hugh Watkins, University of Oxford, Oxford, UK; Nilesh J Samani, University of Leicester, Leicester, UK; Danish Saleheen, University of Cambridge, Cambridge, UK; Debbie Lawlor, University of Bristol, Bristol, UK; Muredach P Reilly, University of Pennsylvania, Philadelphia, PA, USA; Aroon D Hingorani, University College London, London, UK; Philippa J Talmud, University College London, London, UK; John Danesh, University of Cambridge, Cambridge, UK.
Triglyceride Coronary Disease Genetics Consortium investigators: BHF Family Study P S Braund, A S Hall, N J Samani, J Thompson; Bloodomics W März, W Ouwehand, S Sivapalaratnam, N Soranzo, M Trip; BWHHS D A Lawlor, J P Casas, S Ebrahim; EPIC-Norfolk B J Arsenault, S M Boekholdt, K T Khaw, S L Ricketts, M S Sandhu, N J Wareham; KORA H Grallert, T Illig; NPHSII S E Humphries, P J Talmud; PENNCATH D J Rader, J He, M P Reilly; PROCARDIS R Clarke, A Hamsten, J C Hopewell, H Watkins; PROMIS D Saleheen, P Frossard, P Deloukas, J Danesh; SAS S Ye, I A Simpson; TARFS A Onat, E Kömürcü-Bayrak; Verona Heart Study N Martinelli, O Olivieri, D Girelli; WHITE II A D Hingorani, M Kivimäki, M Kumari; other studies B E Aouizerat, L Baum, H Campos, R Chaaba, B S Chen, E Y Cho, D Evans, J Hill, L A Hsu, J A Hubacek, CQ Lai, J H Lee, K Klos, H Liu, L Masana, B Melegh, T Nabika, J Ribalta, E Ruiz-Narvaez, G N Thomas, B Tomlinson, C Szalai, H Vaverkova, Y Yamada, Y Yang.
Emerging Risk Factors Collaboration investigators: AFTCAPS R W Tipping; ALLHAT C E Ford, S L Pressel; ARIC C Ballantyne, A Brautbar; BHS M Knuiman; BRHS P H Whincup, S G Wannamethee, R W Morris; BRUN S Kiechl, J Willeit, P Santer, A Mayr; BUPA N Wald; BWHHS S Ebrahim, D A Lawlor; CaPS J W G Yarnell, J Gallacher; CASTEL E Casiglia, V Tikhonoff; CHS M Cushman, B M Psaty, R P Tracy (see http://www.chs-nhlbi.org for acknowledgments); COPEN A Tybjærg-Hansen, B G Nordestgaard, M Benn, R Frikke-Schmidt; CUORE S Giampaoli, L Palmieri, S Panico, D Vanuzzo, L Pilotto; DRECE A Gómez de la Cámara, J A Gómez-Gerique; DUBBO L Simons, J McCallum, Y Friedlander; EAS FGR Fowkes, A J Lee; EPESEBOS J Taylor, J M Guralnik, C L Phillips; EPESEIOW R Wallace, J M Guralnik, C L Phillips; EPESENCA D G Blazer, J M Guralnik, C L Phillips; EPESENHA J M Guralnik, C L Phillips; EPICNOR K-T Khaw; ESTHER H Brenner, E Raum, H Müller, D Rothenbacher; FIA J H Jansson, P Wennberg; FINE-FIN A Nissinen; FINE-IT C Donfrancesco, S Giampaoli; FINRISK-92, FINRISK-97 V Salomaa, K Harald, P Jousilahti, E Vartiainen; FRAMOFF R B D'Agostino, R S Vasan, M J Pencina; GLOSTRUP E M Bladbjerg, T Jørgensen, L Møller, J Jespersen; GOH R Dankner, A Chetrit, F Lubin; GOTOW C Björkelund, L Lissner, C Bengtsson; GRIPS P Cremer, D Nagel; HONOL B Rodriguez; HOORN J M Dekker, G Nijpels, C D A Stehouwer; IKNS S Sato, H Iso, A Kitamura, H Noda; KIHD J T Salonen, K Nyyssönen, T-P Tuomainen, S Voutilainen; LEADER T W Meade, J A Cooper; MRFIT L H Kuller, G Grandits; NHANES III R Gillum, M Mussolino; NHS E Rimm, S Hankinson, J A E Manson, J K Pai; NPHS II J A Cooper, K A Bauer; OSAKA S Sato, A Kitamura, Y Naito, H Iso; PRIME P Amouyel, D Arveiler, A Evans, J Ferrières; PROCAM H Schulte, G Assmann; PROSPER C J Packard, N Sattar, R G Westendorp, B M Buckley; QUEBEC B Cantin, B Lamarche, J-P Després, G R Dagenais; RANCHO E Barrett-Connor, D L Wingard, R Bettencourt; REYK V Gudnason, T Aspelund, G Sigurdsson, B Thorsson; RIFLE M Trevisan; SHHEC H Tunstall-Pedoe, R Tavendale, G D O Lowe, M Woodward; SHS B V Howard, Y Zhang, L Best, J Umans; SPEED Y Ben-Shlomo, G Davey-Smith; TARFS A Onat; TROMSØ I Njølstad, E B Mathiesen, M L Løchen, T Wilsgaard; ULSAM E Ingelsson, L Lind, V Giedraitis, K Michaëlsson; WHITE II E Brunner, M Shipley; WHS P Ridker, J Buring; WOSCOPS J Shepherd, S M Cobbe, I Ford, M Robertson; ZARAGOZA A Marin Ibañez; ZUTE E J M Feskens, D Kromhout.
Data management team M Walker, S Watson.
Coordinating centre R Collins, E Di Angelantonio, S Kaptoge, PL Perry, N Sarwar, A Thompson, S G Thompson, M Walker, S Watson, I R White, A M Wood, J Danesh.
Conflicts of interest
John Danesh reports receiving research grants from the British Heart Foundation; BUPA Foundation; Denka; diaDexus; European Union; Evelyn Trust; Fogarty International Centre; GlaxoSmithKline; Medical Research Council; Merck; National Heart, Lung, and Blood Institute; National Institute of Neurological Disorders and Stroke; Novartis; Pfizer; Roche; the Wellcome Trust; and UK Biobank. He has served on advisory boards for Merck and Novartis, for which he has received compensation. Aroon D Hingorani reports acting as a consultant for GlaxoSmithKline and as a scientific adviser to London Genetics, and has received honoraria for speaking at educational meetings related to cardiovascular risk. Danish Saleheen reports receiving grants from the Wellcome Trust. Hugh Watkins reports receiving grants from the British Heart Foundation. Philippa J Talmud reports receiving grants from the British Heart Foundation. Adam S Butterworth, Debbie Lawlor, Emanuele Di Angelantonio, Muredach P Reilly, Manjinder S Sandhu, Nadeem Sarwar, Nilesh J Samani, Sally L Ricketts, S Matthijs Boekholdt, and Willem Ouwehand declare that they have no conflicts of interest.
Correspondence to: Dr Nadeem Sarwar, Department of Public Health and Primary Care, University of Cambridge, Strangeways Research Laboratory, Wort's Causeway, Cambridge CB1 8RN, UK nadeem.sarwar@phpc.cam.ac.uk
Web Extra Material
References
- 1.Sarwar N, Danesh J, Eiriksdottir G. Triglycerides and the risk of coronary heart disease: 10 158 incident cases among 262 525 participants in 29 western prospective studies. Circulation. 2007;115:450–458. doi: 10.1161/CIRCULATIONAHA.106.637793. [DOI] [PubMed] [Google Scholar]
- 2.The Emerging Risk Factors Collaboration Major lipids, apolipoproteins and risk of vascular disease: individual data analysis of 302,430 participants from 68 prospective studies. JAMA. 2009;302:1993–2000. doi: 10.1001/jama.2009.1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Phillips A, Smith G Davey. How independent are “independent” effects? Relative risk estimation when correlated exposures are measured imprecisely. J Clin Epidemiol. 1991;44:1223–1231. doi: 10.1016/0895-4356(91)90155-3. [DOI] [PubMed] [Google Scholar]
- 4.Preiss D, Sattar N. Lipids, lipid modifying agents and cardiovascular risk: a review of the evidence. Clin Endocrinol. 2009;70:815–828. doi: 10.1111/j.1365-2265.2008.03490.x. [DOI] [PubMed] [Google Scholar]
- 5.Smith G Davey, Ebrahim S. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32:1–22. doi: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 6.Kathiresan S, Melander O, Guiducci C. Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008;40:189–197. doi: 10.1038/ng.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong K, Ryan RO. Characterization of apolipoprotein A-V structure and mode of plasma triacylglycerol regulation. Curr Opin Lipidol. 2007;18:319–324. doi: 10.1097/MOL.0b013e328133856c. [DOI] [PubMed] [Google Scholar]
- 8.Thompson A, Di Angelantonio E, Sarwar N. Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA. 2008;299:2777–2788. doi: 10.1001/jama.299.23.2777. [DOI] [PubMed] [Google Scholar]
- 9.Keating BJ, Tischfield S, Murray SS. Concept, design and implementation of a cardiovascular gene-centric 50 K SNP array for large-scale genomic association studies. PLoS One. 2008;3:e3583. doi: 10.1371/journal.pone.0003583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El Harchaoui K, van der Steeg WA, Stroes ES. Value of low-density lipoprotein particle number and size as predictors of coronary artery disease in apparently healthy men and women: the EPIC-Norfolk Prospective Population Study. J Am Coll Cardiol. 2007;49:547–553. doi: 10.1016/j.jacc.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 11.Tunstall-Pedoe H, Kuulasmaa K, Amouyel P. Myocardial infarction and coronary deaths in the World Health Organization MONICA Project. Registration procedures, event rates, and case-fatality rates in 38 populations from 21 countries in four continents. Circulation. 1994;90:583–612. doi: 10.1161/01.cir.90.1.583. [DOI] [PubMed] [Google Scholar]
- 12.Fibrinogen Studies Collaboration Correcting for multivariate measurement error by regression calibration in meta-analyses of epidemiological studies. Stat Med. 2009;28:1067–1092. doi: 10.1002/sim.3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The Emerging Risk Factors Collaboration. Statistical methods for the time-to-event analysis of individual participant data from multiple epidemiological studies. Int J Epidemiol (in press). [DOI] [PMC free article] [PubMed]
- 14.Higgins JP, Thompson SG, Deeks JJ. Measuring inconsistency in meta-analyses. BMJ. 2003;327:557–560. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sattar N, McConnachie A, Shaper AG. Can metabolic syndrome usefully predict cardiovascular disease and diabetes? Outcome data from two prospective studies. Lancet. 2008;371:1927–1935. doi: 10.1016/S0140-6736(08)60602-9. [DOI] [PubMed] [Google Scholar]
- 16.Barter P, Ginsberg HN. Effectiveness of combined statin plus omega-3 fatty acid therapy for mixed dyslipidemia. Am J Cardiol. 2008;102:1040–1045. doi: 10.1016/j.amjcard.2008.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuda D, Tomoda H. DGAT inhibitors for obesity. Curr Opin Investig Drugs. 2007;8:836–841. [PubMed] [Google Scholar]
- 18.Hussain MM, Bakillah A. New approaches to target microsomal triglyceride transfer protein. Curr Opin Lipidol. 2008;19:572–578. doi: 10.1097/MOL.0b013e328312707c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ladenson PW, Kristensen JD, Ridgway EC. Use of thyroid hormone analogue eprotirome in statin-treated dyslipidemia. N Engl J Med. 2010;362:906–916. doi: 10.1056/NEJMoa0905633. [DOI] [PubMed] [Google Scholar]
- 20.Hegele RA, Pollex RL. Apolipoprotein A-V genetic variation and plasma lipoprotein response to fibrates. Arterioscler Thromb Vasc Biol. 2007;27:1224–1227. doi: 10.1161/ATVBAHA.107.144980. [DOI] [PubMed] [Google Scholar]
- 21.Cardona F, Guardiola M, Queipo-Ortuño MI. The −1131T>C SNP of the APOA5 gene modulates response to fenofibrate treatment in patients with the metabolic syndrome: a postprandial study. Atherosclerosis. 2009;206:148–152. doi: 10.1016/j.atherosclerosis.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 22.Hubacek JA, Adamkova V, Prusikova M. Impact of apolipoprotein A5 variants on statin treatment efficacy. Pharmacogenomics. 2009;10:945–950. doi: 10.2217/pgs.09.17. [DOI] [PubMed] [Google Scholar]
- 23.Gaddi A, Cicero AF, Odoo FO. Practical guidelines for familial combined hyperlipidemia diagnosis: an up-date. Vasc Health Risk Manag. 2007;3:877–886. [PMC free article] [PubMed] [Google Scholar]
- 24.Garg A, Simha V. Update on dyslipidemia. J Clin Endocrinol Metab. 2007;92:1581–1589. doi: 10.1210/jc.2007-0275. [DOI] [PubMed] [Google Scholar]
- 25.Brunzell JD. Clinical practice. Hypertriglyceridemia. N Engl J Med. 2007;357:1009–1017. doi: 10.1056/NEJMcp070061. [DOI] [PubMed] [Google Scholar]
- 26.Ginsberg HN. New perspectives on atherogenesis: role of abnormal triglyceride-rich lipoprotein metabolism. Circulation. 2002;106:2137–2142. doi: 10.1161/01.cir.0000035280.64322.31. [DOI] [PubMed] [Google Scholar]
- 27.Sarwar N, Sattar N. Triglycerides and coronary heart disease: have recent insights yielded conclusive answers? Curr Opin Lipidol. 2009;20:275–281. doi: 10.1097/MOL.0b013e32832dd4dc. [DOI] [PubMed] [Google Scholar]
- 28.Packard CJ. Triacylglycerol-rich lipoproteins and the generation of small, dense low-density lipoprotein. Biochem Soc Trans. 2003;31:1066–1069. doi: 10.1042/bst0311066. [DOI] [PubMed] [Google Scholar]
- 29.Ansell BJ, Fonarow GC, Fogelman AM. The paradox of dysfunctional high-density lipoprotein. Curr Opin Lipidol. 2007;18:427–434. doi: 10.1097/MOL.0b013e3282364a17. [DOI] [PubMed] [Google Scholar]
- 30.Nordestgaard BG, Benn M, Schnohr P. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298:299–308. doi: 10.1001/jama.298.3.299. [DOI] [PubMed] [Google Scholar]
- 31.Palmen J, Smith AJ, Dorfmeister B. The functional interaction on in vitro gene expression of APOA5 SNPs, defining haplotype APOA52, and their paradoxical association with plasma triglyceride but not plasma apoAV levels. Biochim Biophys Acta. 2008;1782:447–452. doi: 10.1016/j.bbadis.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 32.Merkel M, Loeffler B, Kluger M. Apolipoprotein AV accelerates plasma hydrolysis of triglyceride-rich lipoproteins by interaction with proteoglycan-bound lipoprotein lipase. J Biol Chem. 2005;280:21553–21560. doi: 10.1074/jbc.M411412200. [DOI] [PubMed] [Google Scholar]
- 33.Dichlberger A, Cogburn LA, Nimpf J. Avian apolipoprotein A-V binds to LDL receptor gene family members. J Lipid Res. 2007;48:1451–1456. doi: 10.1194/jlr.C600026-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Nilsson SK, Christensen S, Raarup MK. Endocytosis of apolipoprotein AV by members of the low density lipoprotein receptor and the Vps10p domain receptor families. Biol Chem. 2008;283:25920–25927. doi: 10.1074/jbc.M802721200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith G Davey, Timpson N, Ebrahim S. Strengthening causal inference in cardiovascular epidemiology through Mendelian randomization. Ann Med. 2008;12:524–541. doi: 10.1080/07853890802010709. [DOI] [PubMed] [Google Scholar]
- 36.Joy TR, Hegele RA. Microsomal triglyceride transfer protein inhibition-friend or foe? Nat Clin Pract Cardiovasc Med. 2008;5:506–508. doi: 10.1038/ncpcardio1251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.