

Abstract
The Hematopoietic- and neurologic-expressed sequence 1 (Hn1) gene encodes a small protein that is highly conserved among species. Hn1 expression is upregulated in regenerating neural tissues, including the axotomized adult rodent facial motor nerve and dedifferentiating retinal pigment epithelial cells of the Japanese newt. It is also expressed in numerous tissues during embryonic development as well as in regions of the adult brain that exhibit high plasticity. Hn1 has also been reported as a marker for human ovarian carcinoma and it is expressed in high-grade human gliomas. This study was directed toward understanding the function of Hn1 in a murine melanoma cell line. Hn1 mRNA and protein were identified in B16.F10 cells and in tumors formed from these cells. Inhibition of Hn1 protein expression with siRNA increased melanogenesis. Hn1-depleted cells expressed higher levels of the melanogenic proteins tyrosinase and Trp2 and an increased interaction between actin and Rab27a. The in vitro cell growth rate of Hn1-depleted cells was significantly reduced due to G1/S cell cycle arrest. This was consistent with a reduction in the phosphorylation of retinoblastoma protein as well as lower levels of p27 and increased expression of p21. Decreased expression of c-Met, the receptor for hepatocyte growth factor, was also detected in the Hn1-depleted cells, however HGF-dependent stimulation of phosphorylated-ERK was unaffected. Hn1 depletion also led to increased basal levels of phosphorylated p38 MAPK, while basal ERK phosphorylation was reduced. Moreover, Hn1-depleted cells had reduced expression of transcription factors MITF and USF-1, and increased expression of TFE3. These data, coupled with reports on Hn1 expression in regeneration and development, suggest that Hn1 functions as a suppressor of differentiation in cells undergoing repair or proliferation.
Keywords: Dedifferentiation, P38 MAPK, MITF, TFE3
1. Introduction
The regulation of melanocyte and melanoma cell proliferation and differentiation is a complex phenomenon. The rapidly invasive and metastatic nature of melanomas distinguishes them from their precursors, the skin melanocytes. Differentiated melanocytes contain lysosome-like organelles called melanosomes, which store the melanogenic enzymes tyrosinase, Trp1 and Trp2 that are necessary for the synthesis of the pigment melanin (Lin and Fisher, 2007). Melanosomes are transported by microtubules from the perinuclear region of the cell toward the periphery where they bind Rab27a, a small GTPase that facilitates their association with actin filaments in order to secrete melanin onto the surrounding keratinocytes of the epidermis (Hume et al., 2001). These melanogenic and secretory properties, combined with cell cycle arrest, are characteristic features of differentiated melanocytes. Melanocyte differentiation can be induced by phosphorylated p38 and prevented by ERK phosphorylation (Englaro et al., 1998; Saha et al., 2006; Wu et al., 2000). Understanding the various factors that distinguish melanoma tumorigenesis from melanocyte differentiation is a useful goal in developing novel therapies against melanomas.
The hematopoietic- and neurologic-expressed sequence 1 (Hn1) gene encodes a small protein of unique amino acid sequence whose function has not yet been elucidated (Tang et al., 1997). The only other mammalian-expressed gene that shares high sequence similarity with Hn1 is called Hn1-like (HN1L) and its function is also unknown (Zhou et al., 2004). In rodents, Hn1 is widely expressed in numerous tissues during embryonic development. In the adult brain, Hn1 expression is restricted to regions of high plasticity, including the hippocampus, cortex and cerebellum. Hn1 is upregulated in nervous tissues that regenerate, including axotomized adult rodent facial motor and vagal nerves (Zujovic et al., 2005). In addition, an ortholog of Hn1 in the Japanese newt is induced in dedifferentiating retinal pigment epithelial cells that arise subsequent to surgical removal of the neural retina (Goto et al., 2006). Hn1 has also been reported as one of four genes that distinguish human ovarian carcinoma from healthy ovarian epithelial tissue (Lu et al., 2004). Additionally, intracranial implantation of Hn1-depleted murine GL261 glioma cells into mice results in tumor volumes that are significantly smaller than those established from Hn1-expressing cells (Laughlin et al., 2009). The reported expression of Hn1 in models of development, regeneration, plasticity and cancer suggests that this gene is critical for maintaining a stage in cellular development and growth that precedes differentiation. The high conservation of this gene across multiple species suggests that its function may be critical for survival.
This study was undertaken to evaluate Hn1 expression and function in a murine model of melanoma. Here, we report the identification of Hn1 mRNA and protein in the murine B16.F10 melanoma cell line and in tumors formed from these cells. The original B16 cell line was derived from a spontaneous melanoma on the ear of a C57BL/6 mouse (Stephenson and Stephenson, 1970). The B16.F10 cells were established from this cell line and selected for their high metastatic potential (Cranmer et al., 2005; Fidler, 1973). The cells exhibit a high rate of proliferation but express proteins that are phenotypically associated with melanocytes. Using a highly efficient siRNA to inhibit the expression of Hn1 in these cells, we determined that this gene functions to regulate melanogenesis and cell growth.
2. Materials and methods
2.1. Cell culture
B16.F10 cells (from ATCC) were grown in DMEM (Gibco BRL, Grand Island, NY) with 10% FBS, 1% Penicillin-Streptomycin (1000 units/ml of penicillin, 1000 μg/ml or streptomycin) (Gibco BRL, Grand Island, NY), and 1% sodium pyruvate (0.11 mg/ml) (Sigma-Aldrich, St. Louis, MO) at 37 °C and 5% CO2.
2.2. Northern blot analyses
Total RNA was isolated from B16.F10 cells using TRIzol Reagent according to the manufacturer's recommended procedure (Life Technologies, Grand Island, NY). RNA (20 μg/lane) was electrophoresed through denaturing 1.2% agarose and subjected to Northern blot analysis (Church and Gilbert, 1984). The nylon membrane was hybridized with a 32P-radiolabeled cDNA generated from a 450-bp murine Hn1 protein coding DNA sequence, which was 32P-radiolabeled by the random primer method to a specific activity of 1.2 × 109 dpm/μg.
2.3. Development of anti-Hn1 antibodies
Mouse Hn1 was expressed in E. coli using the pET22a and pATH11 expression vectors. The pATH11 vector produces Hn1 fused to the C-terminus of E. coli Trp-E, while pET22a produces a His-tagged protein. The Trp-E fusion protein was purified by excision of the appropriate gel band from a 6 M urea extract of a bacterial inclusion body preparation as described (Harris et al., 1991). The His-tagged protein was affinity purified using a nickel-chelating column chromatography. Mice (Balb/c) and rabbits (New Zealand white) were injected with the Trp-E-Hn1 fusion protein and then boosted with His-tagged Hn1. Sera were tested by Western blotting for activity on Trp-E-Hn1 and His-tagged Hn1. Strong reactivity with both proteins indicated good reactivity with Hn1. Serum was collected from rabbits and affinity purified on His-tagged Hn1 coupled to cyanogen bromide activated Sepharose 4B. Spleen cells from a mouse with a strong Hn1-specific immune response were fused to the SP0 myeloma line to produce hybridoma cells, which were screened initially by ELISA on His-tagged Hn1, and then by Western blotting and immunocytochemistry. Two clones, 3C4 and 3G6, were selected for further study. Both are IgG2a class with kappa light chains and appeared to be similar or identical in their properties. Clone 3C4 was used in the present studies.
2.4. Development of siRNAs against Hn1
Using siDirect (http://design.RNAi.jp), three small interfering RNA (siRNA) sequences were identified that predicted maximum Hn1-target specificity to degrade the murine Hn1 mRNA. These siRNA sequences were initially tested for their ability to inhibit expression of murine Hn1 in a co-transfection paradigm using HEK293 cells, which lack endogenous Hn1. The sequences were individually cloned into a vector (pH1rSC) containing the H1 promoter that allows for packaging into recombinant self--complementary (double stranded) adeno-associated virus (AAV) (McCarty et al., 2001). The most efficient siRNA-expressing plasmid (Hn1-siRNA) was generated by cloning the following complimentary oligonucleotides into the AscI and NheI sites of pH1rSC: 5′-CGC GGG GAG AAG GTG ATA TGC ATT TCA AGA CAA TGC ATA TCA CCT TCT CCC TTT TTG GAA A-3′ and 5′-CTA GTT TCC AAA AAG GGA GAA GGT GAT ATG CAT TGT CTT GAA ATG CAT ATC ACC TTC TCC C-3′. A second highly efficient Hn1-specific siRNA (Hn1-siRNA-B) was also developed and used to determine if off-target effects were associated with the Hn1-siRNA described above. The sequences of the two complimentary oligonucleotides used to generate siRNA-B were: 5′-CGC GCT GTG AGG AAG AAC AAG ATT TCA AGA CAA TCT TGT TCT TCC TCA CAG TTT TTG GAA A-3′ and 5′-CTA GTT TCC AAA AAC TGT GAG GAA GAA CAA GAT TGT CTT GAA ATC TTG TTC TTC CTC ACA G-3′. A scrambled-siRNA was also developed for use as a control for the impact of expressing a double-stranded hairpin RNA. The sequences of the two oligonucleotides used to generate this construct were: 5′-CGC GGG TCG AAT TCG ATA TCC TAT TCA AGA CAT AGG ATA TCG AAT TCG ACC TTT TTG GAA A-3′ and 5′-CTA GTT TCC AAA AAG GTC GAA TTC GAT ATC CTA TGT CTT GAA TAG GAT ATC GAA TTC GAC C-3′. The three plasmids were packaged into AAV6, a serotype of AAV that was determined to be the most efficient (>90%) at transducing B16.F10 cells. High titer virus was produced and purified according to previously published protocols (Zolotukhin et al., 2002).
B16.F10 cells were seeded on 24- or 6-well cell culture plates. One day later, representative wells of cells were counted and transduced at a multiplicity of infection (M.O.I.) of 5000 (unless indicated as 3000) with serum-free media containing AAV6 expressing Hn1-siRNA, scramb1ed-siRNA, control-AAV6 (containing the H1 vector alone), or no virus. All dishes were incubated for three hours at 37 °C in 5% CO2, after which the volumes were doubled with media containing 20% FBS, 1% P/S and 1% sodium pyruvate. The viral vector transduction was repeated one day later unless otherwise indicated. Western blot, cell cycle, and immunocytochemical analyses were carried out two or three days after the first viral transduction. For evaluating a potential non-specific impact of the viral transduction on melanin production, the control virus was also used at 30,000 M.O.I.
2.5. Western blot analyses
B16.F10 protein lysates were subjected to SDS-PAGE, transferred onto PVDF membranes and probed with the following antibodies. The murine monoclonal anti-Hn1 antibody was used at 1:500 and the rabbit polyclonal anti-Hn1 antibody at 1:1000. The antibodies for tyrosinase (Pep7), Trp1 (Pep1), and Trp2 (Pep8), from Dr. Vincent Hearing, National Institutes of Health, were each used at 1:2000. The monoclonal anti-Rab27a antibody from Drs. Miguel Seabra and Alistair Hume, Imperial College London, was used at 1:1000. The monoclonal anti-actin antibody (1:1000) and polyclonal anti-β-catenin antibody (1:8000) were purchased from Sigma-Aldrich, St. Louis, MO. Antibodies for p21 (1:1000), p27 (1:600), cyclins A and E (1:1000), and USF-1 (1:200) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The cyclin D1 antibody (1:1000) was purchased from Neomarkers (Lab Vision Corporation, Fremont, CA) and the antibody for total Rb (1:1000) was purchased from Pharmingen (BD Biosciences, San Jose, CA). The antibodies for the various phosphorylated forms of Rb, total and phosphorylated p38, total and phosphorylated ERK, total and phosphorylated Akt, and c-Met (all used at 1:1000) were purchased from Cell Signaling Technology (Danvers, MA). The C5 monoclonal antibody for MITF (Abcam Inc., Cambridge, MA) was used at 1:2000. The antibody against TFE3 (1:500) was purchased from BD Pharmingen, San Jose, CA. The secondary antibodies used were either HRP-conjugated sheep anti-mouse from Sigma-Aldrich or goat anti-rabbit from Cell Signaling Technology.
2.6. Quantification of melanin
B16.F10 cells were plated on 24-well dishes and transduced the next day with either scrambled- or Hn1-siRNA AAV6 at an M.O.I. of 5000 as indicated above or no virus. Starting two days later, and daily for the next three consecutive days, media (600 μl) from triplicate wells of cells from each treatment were collected separately into 1.5 ml tubes. The remaining cells were counted using a hemacytometer to determine the number of cells per well. PBS (400 μl) was added to each tube containing medium for a total volume of 1 ml. The solution was made 0.8 mM in NaOH by the addition of 0.8 μl of 1 M NaOH. Absorbance measurements (500 nm) were made and the content of melanin was calculated by comparison to a melanin standard curve (0–40 μg) generated using synthetic melanin (Sigma-Aldrich, St. Louis, MO).
2.7. HGF stimulation of phosphorylated-ERK (p42/44 MAPK)
B16.F10 cells were plated on 12-well dishes and transduced the next day, at an M.O.I. of 3000, with either the control-AAV6 or the Hn1-siRNA AAV6. The following day, the cells were serum-starved for approximately 16 h, after which time they were stimulated with murine hepatocyte growth factor (HGF) at concentrations of 0, 10, or 100 ng/ml under serum-free conditions for 10 min at 37 °C. The cells were rinsed with ice-cold PBS and collected with lysis buffer containing protease inhibitors. The samples were analyzed by Western blot using anti-phosphorylated ERK, anti-total ERK, and mouse monoclonal anti-Hn1 antibodies.
2.8. In vitro cell proliferation assay
B16.F10 cells were seeded on 6-well dishes at a density of 10,000 cells per well. The following day, cells were transduced with control-AAV6, Hn1-siRNA-AAV6, or no virus. Two days after transduction, triplicate wells of cells were collected by trypsinization and counted manually using a hemacytometer. Cell counts were obtained every 24 or 48 h.
2.9. Cell cycle analysis
Cells were plated on 6-well dishes and treated with AAV6 expressing either scrambled or Hn1-siRNA or no virus. Two and three days later, cells (approximately 75% confluent) were trypsinized and collected in a 1.5 ml tube using DMEM with serum. An aliquot was used to count cells manually using a hemacytometer. The remaining cells were pelleted by centrifugation at 1000g for 5 min and the medium was aspirated carefully. For 500,000 cells, 500 μl of propidium iodide solution (containing 25 μg propidium iodide, 0.5 mg sodium citrate, 0.5 μl Triton X-100, 5 μg/ml RNAse A, and water) was added and gently pipetted up and down ten times. Tubes were incubated at room temperature in the dark for two hours. Cells were transferred to polystyrene round-bottom tubes (Falcon) on ice and analyzed by Flow cytometry on a FACSort machine.
2.10. B16.F10 tumor preparation
B16.F10 cells were harvested from 100 mm plates by trypsinization, washed one time in sterile phosphate buffered saline (sPBS), and finally suspended in sPBS. Cells (300,000 in 9 μl) were implanted subcutaneously into isoflurane-anesthetized female C57BL/6 mice (ages six to eight weeks). Two weeks after cell implantation, mice were given an injection of sodium pentobarbital (32 mg/kg) and euthanized by transcardial perfusion with 0.9% saline followed by buffered 4% paraformaldehyde (PFA). Skin tumors were removed, post-fixed for 1 h with 4% PFA, sectioned, cryoprotected overnight with 30% sucrose, and frozen in iso-pentane cooled by liquid N2. Cryosections (20 μm) of the tumor were prepared and thaw-mounted on Superfrost/Plus slides (Fisher Scientific, Pittsburgh, PA) and finally subjected to either in situ hybridization or immunohistochemical procedures.
2.11. In situ hybridization analysis
Hn1 hybridization probes (Zujovic et al., 2005) were used for in situ hybridization (ISH) analysis as previously described (Harrison et al., 2003). Sections were hybridized separately with antisense and sense riboprobes and then apposed to film and then dipped in LM-1 emulsion. After exposure for one week, slides were developed, fixed and counterstained with hematoxylin and eosin (H&E).
2.12. Immunofluorescent staining
Tissue sections of subcutaneous B16.F10 tumors were permeabilized using 0.5% Triton X-100 for 10 min and blocked with 3% BSA for 20 min. The affinity purified polyclonal anti-Hn1 antibody was used at 1:50 in PBS for 1 h at room temperature. Sections were rinsed in PBS three times for 10 min and an anti-mouse Alexa Fluor 594 red fluorescent secondary antibody (Sigma-Aldrich, St. Louis, MO) was added at 1:2000 in PBS for 1 h at room temperature. The sections were counterstained with DAPI at 1:333 in PBS.
B16.F10 cells cultured in vitro were plated on glass coverslips in 24-well cell culture dishes and transduced as described above. Three days after the first viral transduction, cells were fixed with 4% PFA for 30 min at 4 °C and stained. The antibodies used were the polyclonal anti-Hn1 (undiluted), Pep7 (Tyrosinase), Pep1 (Trp1) and Pep8 (Trp2), all at 1:200, polyclonal anti-Rab27a (1:100), and phalloidin-488 (Invitrogen, San Francisco, CA) at 1:40. Immunofluorescence was analyzed using a confocal microscope (Leica, Microsystems Heidelberg GmbH, Version 2.61, Build 1538).
2.13. Immunoprecipitation analysis
B16.F10 cells plated on two 150 mm dishes were transduced twice with either Hn1- or scrambled-siRNA AAV6. Two days later cells were rinsed with cold PBS and collected using a buffer containing 20 mM Hepes, pH 7.6, 5% glycerol, 0.1% Triton X-100, 0.1% β-Mercaptoethanol, 1 mM EDTA, 1 mM EGTA, 20 mM tetrasodium pyrophosphate, 1 mM Na3VO4, 145 mM KCl, and supplemented with protease inhibitors. The cell lysate was collected, subjected to sonication on ice, and centrifuged at 16,000g for 5 min at 4 °C. The supernatants were transferred to new tubes and pre-cleared by incubating on a shaker overnight at 4 °C with 50 μl recombinant Protein G Sepharose beads (Invitrogen, San Francisco, CA). The tubes were subjected to centrifugation at 800g at 4 °C for 5 min and again at 16,000g for 1 min at room temperature. The pre-cleared lysates were then supplemented with one percent (mass of antibody/mass of lysate protein) polyclonal rabbit anti-actin antibody (Sigma-Aldrich, St. Louis, MO) and incubated on a shaker for 2 h at room temperature. Protein G-Sepharose beads (50 μl) were added and the reactions were incubated on a shaker overnight at 4 °C. The beads in each tube were subjected to five washes, consisting of centrifuging at 16,000g for 1 min at room temperature, aspirating the supernatant, and re-suspending the pellet in 1 ml Tris buffered saline with Tween (TBS-T). The final pellets were resuspended in 80 μl of SDS sample buffer and subjected to SDS-PAGE followed by Western blot analysis using the monoclonal murine anti-actin and anti-Rab27a antibodies.
2.14. Statistical analysis
For analysis of statistical significance, the data were subjected to student's t-test (two-tailed distribution and two-sample equal variance test). Unless otherwise indicated, experiments were conducted at least three times.
3. Results
3.1. Hn1 mRNA and protein are expressed in B16.F10 murine melanoma cells and tumors
Northern blot analysis of extracts from B16.F10 cells identified two Hn1 mRNA species of 0.7 and 1.4 kb (Fig. 1A), both of which are large enough to encode the 154 amino acid protein. Hn1 protein in B16.F10 cells cultured in vitro was also detected by Western blot analysis using either the mouse monoclonal or rabbit polyclonal anti-murine Hn1 antibodies. Both antibodies used for Western blot analysis identified a single protein band of equal molecular weight in the B16.F10 cells (Figs. 1B and 2A). The apparent molecular mass of the protein was 25 kDa, which is slightly larger than the calculated molecular mass based on the primary amino acid sequence. The in vitro findings were complemented by in situ hybridization (ISH) and immunohistochemical analyses of B16.F10 tumors established in murine C57BL/6 dermal tissue (Fig. 1C–H). Strong hybridization signals were evident within the tumor mass, indicating high levels of Hn1 mRNA (Fig. 1C). Hybridization with sense riboprobes established the specificity of the anti-sense riboprobe (Fig. 1D). These skin melanomas, visualized by light microscopy (Fig. 1E) also expressed Hn1 protein as detected by immunohistochemical analysis using the rabbit polyclonal anti-Hn1 antibody (Fig. 1F).
Fig. 1.
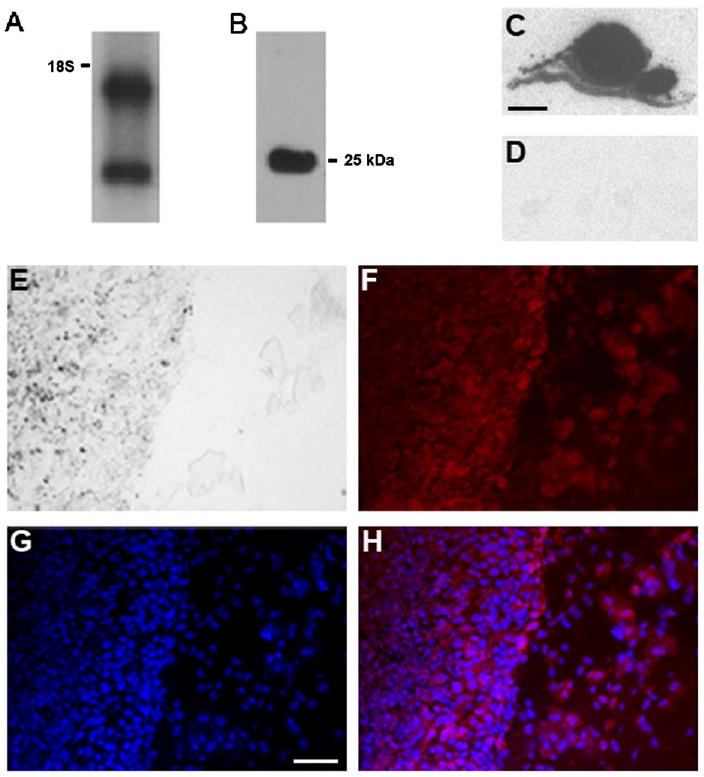
Hn1 is expressed in B16.F10 murine melanoma cells and tumors. Northern (A) and Western blot (B) analyses of B16.F10 cell extracts showing Hn1 mRNA and protein expression. The mouse monoclonal anti-Hn1 antibody was used for Western blot analysis. The position of the 18S ribosomal RNA and calculated molecular mass of Hn1 protein (25 kDa) are shown. (C, D) In situ hybridization analysis of Hn1 mRNA within a B16.F10 skin tumor section. (C) A section was hybridized with an Hn1 anti-sense riboprobe; (D) an adjacent section was hybridized with a sense riboprobe. Scale bar: 2 mm. (E) Light microscopy of a murine B16.F10 skin tumor, tumor tissue (left) and surrounding normal tissue (right). (F) Hn1-expressing cells determined by anti-Hn1 immunohistochemistry using the rabbit polyclonal anti-Hn1 antibody. (G) DAPI-stained cell nuclei. (H) A merged image of (F) and (G). Scale bar: 50 μm.
Fig. 2.
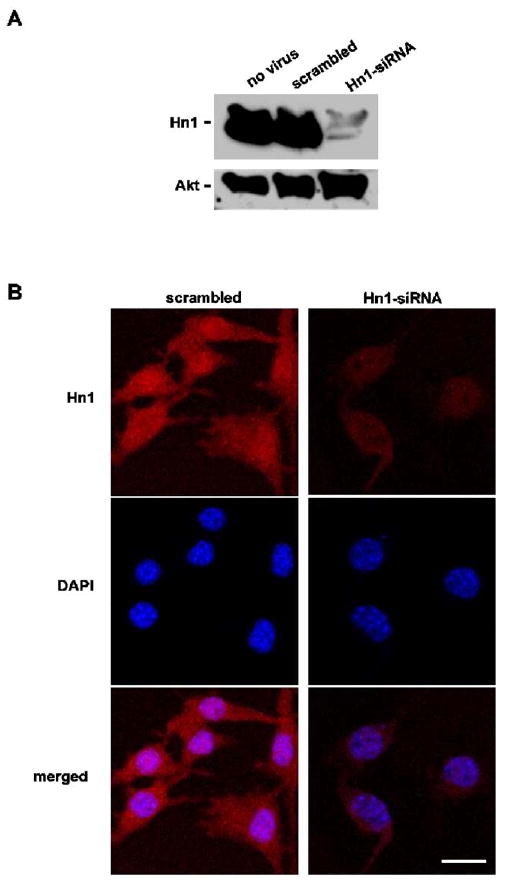
Depletion of Hn1 protein in B16.F10 cells by an anti-Hn1 siRNA. (A) Western blot analysis of B16.F10 cells treated with scrambled-siRNA and Hn1-siRNA adeno-associated viruses at an M.O.I. of 5000 using the rabbit anti-Hn1 polyclonal antibody. Untreated cells are also shown. (B) Confocal immunofluorescence microscopy (CIM) of Hn1 protein in cells with and without Hn1. The rabbit polyclonal anti-Hn1 antibody was used for immunocytochemical analyses and DAPI was used as the nuclear stain. Scale bar: 16 μm.
3.2. Hn1 depletion by a specific Hn1-siRNA and localization of Hn1 protein to the nucleus and cytoplasm of B16.F10 cells
To study the effects of depleting Hn1 in B16.F10 cells, a recombinant AAV serotype 6 (AAV6) was engineered to express a silencing RNA sequence (siRNA) specifically targeted to degrade murine Hn1 mRNA. To test the efficiency of this siRNA, B16.F10 cells were transduced with AAV6 containing either an anti-Hn1 siRNA or a non-specific scrambled-siRNA at a M.O.I. of 5000. Cell lysates collected two days after viral treatment were subjected to Western blot analysis using the rabbit polyclonal anti-Hn1 antibody (Fig. 2A). The Hn1-siRNA very efficiently inhibited the expression of Hn1 protein in the B16.F10 cells. Lysates from scrambled-siRNA-treated and untreated cells had similar levels of Hn1 protein. Immunohistochemical analysis of cultured B16.F10 cells determined that Hn1 protein resides throughout the cell, with a prominent nuclear localization (Fig. 2B); the anti-Hn1 immunoreactivity was diminished in cells treated with the Hn1-siRNA.
3.3. Effect of Hn1 depletion on melanogenesis and melanin secretion
Hn1-depleted B16.F10 cells synthesized and secreted more melanin. This phenomenon was not observed after using a M.O.I. of the control virus ten times greater than what was used for the Hn1-siRNA-AAV6. The qualitative increase in melanin secretion (Fig. 3A) was confirmed by quantification of melanin levels in the media. A significant increase in the levels of melanin in the medium from Hn1-depeleted cells was evident by three days after viral vector treatment (Fig. 3B). The increase in melanin was also visible by light microscopy within the cells cultured in vitro, suggesting that Hn1-depletion not only affected the secretion of melanin, but also its production (Fig. 3C).
Fig. 3.
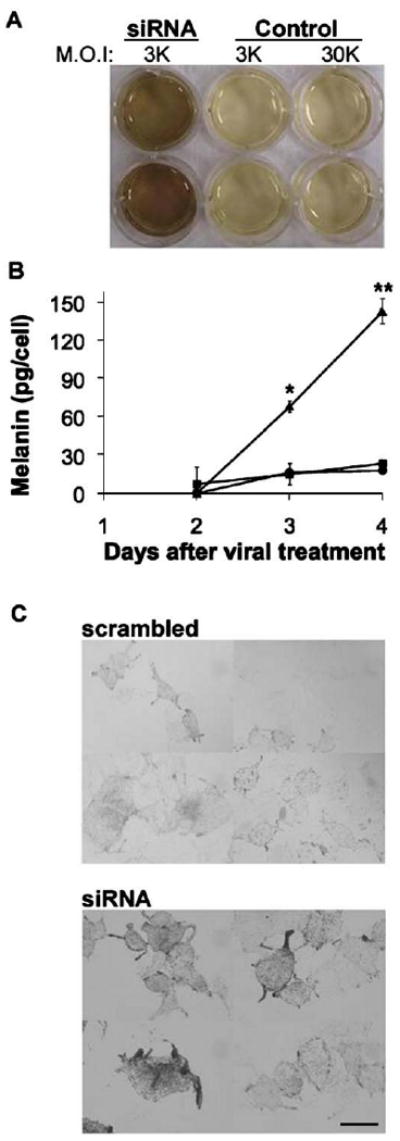
Hn1 depletion in B16.F10 cells increases melanin secretion. (A) Qualitative analysis of melanin secreted by B16.F10 cells treated with Hn1-siRNA virus at and M.O.I. of 3000 or control virus at M.O.I.s of 3000 or 30,000. The higher M.O.I. of the control virus did not impact melanin secretion. (B) Quantitative analyses of melanin secretion by B16.F10 cells treated with Hn1-siRNA (triangles), scrambled-siRNA (squares), or no virus (circles). Shown are the means of triplicate samples ±SEM (*p<0.01; **p<0.00001) of a representative experiment conducted three independent times. (C) Light microscopy of Hn1-expressing (scrambled) and -depleted (siRNA) B16.F10 cells. Scale bar: 16 μm.
The expression levels of several melanogenic proteins, including tyrosinase and the related proteins, Trp1 and Trp2, were analyzed in cells treated with Hn1- or scrambled-siRNAs to determine if changes in these proteins could account for the increased production of melanin by the Hn1-depleted cells. Immunohistochemical analyses using rabbit polyclonal antibodies specific to tyrosinase, Trp1 and Trp2 indicated that tyrosinase and Trp2 expression were upregulated in cells lacking Hn1; the level of Trp1 was unchanged (Fig. 4A). These observations were confirmed by Western blot analyses using the same antibodies (Fig. 4B). The upregulation of tyrosinase was also seen when Hn1 was depleted with the Hn1-siRNA-B (data not shown).
Fig. 4.
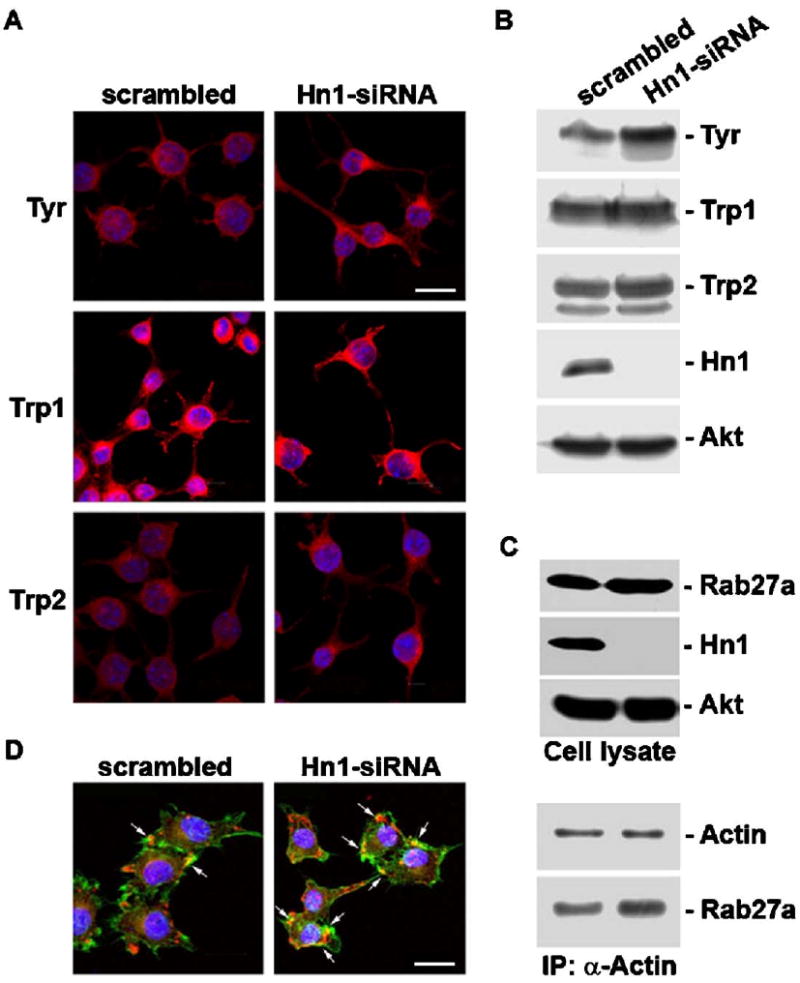
Hn1-depleted B16.F10 cells have increased levels of Tyrosinase and Trp2 and an enhanced association of Rab27a and actin. (A) CIM and (B) Western blot analyses of B16.F10 cells treated with the scrambled or Hn1-siRNA showing Tyrosinase (Tyr), Trp1 and Trp2 protein localization and levels. An anti-total Akt antibody was used as the loading control for the Western blot. (C) Upper panels depict Western blot analyses of total cell extracts (cell lysate) of B16.F10 cells with and without Hn1 using antibodies against Rab27a, Hn1 and total Akt, the loading control. Lower panels show the final pellets from an immunoprecipitation reaction of Hn1-expressing and -depleted B16.F10 cell lysates using an anti-actin polyclonal antibody and analyzed by Western blot analysis using the monoclonal anti-actin and anti-Rab27a antibodies. (D) CIM of Rab27a expression in B16.F10 cells with and without Hn1; Rab27a (red), actin (green), DAPI-stained nuclei (blue); arrows indicate colocalization of Rab27a and actin. Scale bars: 16 μm. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The effect of Hn1 depletion on a process associated with melanosomal transport and secretion was also evaluated. Rab27a, a small GTPase that forms a complex with myosin Va and melanophilin, is essential for melanin secretion by enabling the association of melanosomes with actin filaments (Hume et al., 2001, 2007). The expression and localization of Rab27a was analyzed in cells with and without Hn1. While there was no difference in total levels of Rab27a, the association of Rab27a with actin was increased in the absence of Hn1 (Fig. 4C). Immunohistochemical analysis showed enhanced co-localization of actin and Rab27a in the Hn1-depleted cells (Fig. 4D).
3.4. Hn1 depletion induces a G1/S cell cycle arrest
The in vitro proliferation rates of cells with and without Hn1 were compared. Hn1-depleted cells grew significantly slower than cells treated with either the scrambled-siRNA or no virus (Fig. 5A); the difference was visible by three days after viral treatment. A cell cycle analysis of cells two and three days after siRNA treatment, revealed a significant G1/S arrest in the Hn1-depleted cells (Fig. 5B). Compared to Hn1-expressing cells, Hn1-depleted cells had a higher fraction in G1 phase and a corresponding lower percentage of cells in the S phase. A variety of cell cycle-regulating proteins were then evaluated by Western blot analyses (Fig. 5C). While the total level of retinoblastoma protein (Rb) did not change, lower levels of phosphorylated forms of Rb were present in cells lacking Hn1, in particular phospho-Ser795. Consistent with the decreased reactivity of the anti-phospho-Rb antibodies, the anti-total Rb antibody detected two forms of Rb in Hn1-depeleted cells, with the lower band representing less phosphorylated forms of the protein. The level of cyclin D1 was increased in Hn1-depeleted cells while both cyclin A and cyclin E were unaltered by the lack of Hn1. The expression of p21 was elevated, whereas p27 levels were reduced in the absence of Hn1.
Fig. 5.
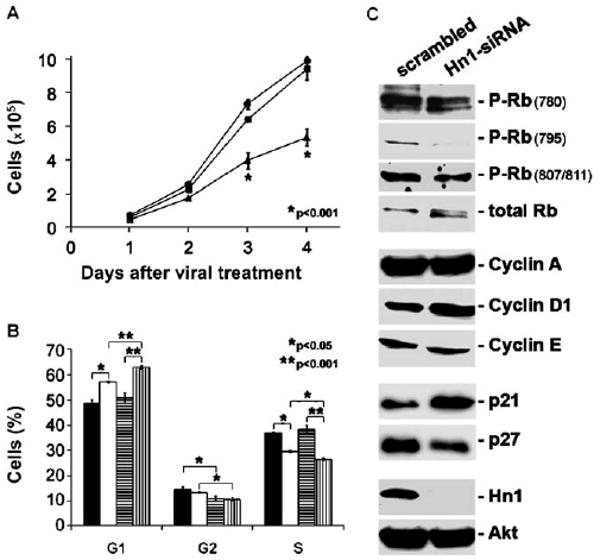
Hn1 depletion induces B16.F10 cell cycle arrest. (A) Growth rates of B16.F10 cells treated with Hn1-siRNA (triangles), scrambled-siRNA (squares), or no virus (circles). Shown are the means±SEM (*p<0.001) of triplicate samples from a representative experiment conducted six independent times. (B) Cell cycle analysis of Hn1-expressing and Hn1-depleted cells 2 and 3 days after siRNA treatment. Two-day scrambled (black), two-day siRNA (white), three-day scrambled (horizontal line pattern), three-day siRNA (vertical line pattern); (*p<0.05; **p<0.001). (C) Western blot analyses of extracts from Hn1-expressing and Hn1-depleted B16.F10 cells using antibodies specific to a variety of cell cycle proteins. An anti-total Akt antibody was used as the loading control.
The expression and phosphorylation states of various cell growth signaling molecules including c-Met, Akt, and the MAPKs ERK (p42/44) and p38 were also evaluated in B16.F10 cells with and without Hn1 using Western blot analyses (Fig. 6). Expression of c-Met was reduced in cells lacking Hn1 while total and phosphorylated Akt levels were unchanged (Fig. 6A). Despite the c-Met reduction, the ability of its ligand, hepatocyte growth factor (HGF), to stimulate ERK phosphorylation was not altered (Fig. 6B). However, reduced basal levels of ERK phosphorylation were observed while levels of phosphorylated p38 were highly increased in the Hn1-depleted cells (Fig. 6C); the increase in p38 phosphorylation was also seen when Hn1 was depleted with Hn1-siRNA-B (data not shown). The increased phosphorylation of p38 was independent of serum content in the culture media (data not shown). Moreover, conditioned media from Hn1-depleted cells had no effect on the basal levels of phosphorylated p38 in naïve B16.F10 cells (data not shown).
Fig. 6.

Effect of Hn1 depletion on growth signaling molecules. (A) Western blot analyses of B16.F10 cells with and without Hn1 using anti-c-Met, -phosphorylated Akt (P-Akt), -total Akt and -Hn1 antibodies. (B) Western blot analyses of hepatocyte growth factor (HGF) stimulation of ERK phosphorylation in B16.F10 cells with and without Hn1. (C) Western blot analyses of B16.F10 cells treated with scrambled, Hn1-siRNA or no virus using anti-phosphorylated ERK (P-ERK), -total ERK, -phosphorylated p38 (P-p38), -total p38, and -Hn1 antibodies.
3.5. Hn1 depletion decreases MITF and USF-1 expression and increases TFE3 expression
Because of the collective changes seen in p38 and ERK phosphorylation, p21, p27 and melanogenic protein expression, it was hypothesized that MITF would also be upregulated in the Hn1-depleted cells (Carreira et al., 2005, 2006). However, Western blot analysis of Hn1-depeleted cells revealed a marked decrease in MITF expression (Fig. 7). The expression of β-catenin, which can also regulate MITF expression, did not differ between the Hn1-depleted and -expressing cells. The levels of two other transcription factors, USF-1 and TFE3, were also evaluated. While USF-1 expression decreased slightly, levels of TFE3 were elevated in the Hn1-depleted cells.
Fig. 7.
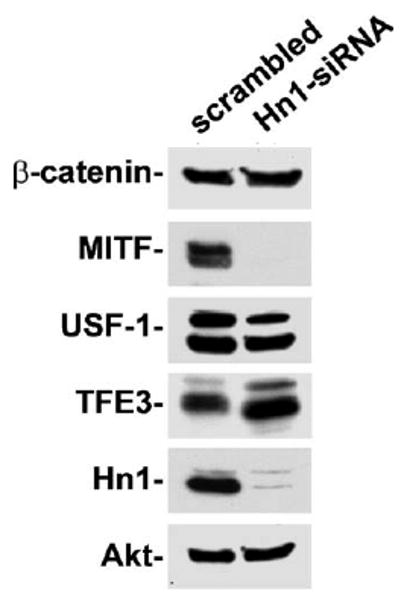
Effect of Hn1 depletion on transcription factors that can regulate melanocyte differentiation. The expression levels of transcription factors MITF, USF-1 and TFE3, as well as β-catenin, were evaluated in B16.F10 cells with and without Hn1 by Western blot analyses.
4. Discussion
Published information on Hn1 has largely been confined to studies of the expression and localization of this gene in various tissues. Here we provide additional data that indicate that B16.F10 melanoma cells and tumors express Hn1 mRNA and protein. Moreover, the main findings reported here establish that inhibition of Hn1 expression in these cells suppresses cell proliferation and increases melanogenesis. These data, coupled with the previous reports demonstrating Hn1 expression in developing tissues, regenerating tissues, ovarian carcinoma, and high grade gliomas suggest that this protein plays an important role in processes associated with transitions in cell differentiation.
The pigmented property of the B16.F10 cells was especially useful at identifying a role of Hn1 in this cancer cell line. The increase in melanin production, evident within the cells and the culture medium, was easily observed shortly after depleting Hn1 protein. The enhanced melanogenesis in the Hn1-depleted cells can be explained by the increased expression of the melanogenic enzymes tyrosinase and Trp2. The higher levels of secreted melanin detected in the medium of Hn1-depleted cells also suggested that processes associated with melanosomal transport were affected by the loss of Hn1. In order to secrete melanin onto the surrounding cells of the epidermis, melanosomes are transported from the perinuclear region of the cell towards the periphery where they must bind actin filaments via a Rab27a–myosin Va–melanophilin complex (Hume et al., 2001, 2007). The enhanced interaction of Rab27a and actin in the Hn1-depleted cells is therefore an additional mechanism contributing to the increased secretion of melanin. The shift to a more pronounced melanogenic phenotype in the Hn1-depleted cells suggests that Hn1 normally functions to inhibit mechanisms associated with the transition towards the more differentiated melanocyte phenotype.
Further evidence in support for a role of Hn1 in regulating differentiation of these mouse melanoma cells is apparent from the growth inhibition observed in B16.F10 cells lacking Hn1 protein. This is consistent with in vivo studies of murine gliomas established from Hn1-depleted GL261 cells, which form significantly smaller tumors than Hn1-expressing cells, even though GL261 cells in vitro grow at equal rates whether or not they express Hn1 protein (Laughlin et al., 2009). The difference between the in vitro growth of GL261 and B16.F10 cells with and without Hn1 could be due to environmental and immune factors that influence in vivo GL261 tumor growth, or to the slower in vitro growth rate of GL261 cells compared to B16.F10 cells. A more prolonged analysis of proliferation of the GL261 cells may be required in order to see significant differences in the in vitro growth rates of Hn1-expressing and -inhibited GL261 cells.
Depletion of Hn1 results in a G1/S cell cycle arrest that is consistent with the observed reduction in phosphorylation of retinoblastoma (Rb) protein, elevated cyclin D1, increased p21 and reduced levels of p27. The Hn1-depleted cells also exhibit reduced levels of basal ERK phosphorylation and increased levels of phosphorylated p38. ERK is activated in up to 90% of human melanomas and its sustained activity in melanocytes leads to proliferation (Wu et al., 2000). Moreover, pharmacological inhibition of the ERK pathway triggers B16.F10 cell differentiation (Englaro et al., 1998). The diminished c-Met expression in the Hn1-depleted cells may be one factor that contributes to the reduced basal phosphorylation of ERK. However, HGF-stimulation of ERK phosphorylation through this receptor was unaltered, suggesting that the change in c-Met is probably not sufficient to impact an HGF-dependent growth effect.
The regulation of melanogenesis and the cell cycle in melanoma is a complex phenomenon, controlled in large part by the microphthalmia transcription factor (MITF), which is expressed in cells of melanocytic origin (Carreira et al., 2005). MITF expression is positively regulated by p38 phosphorylation (Saha et al., 2006). Conversely, ERK phosphorylation elicits transient activation of MITF followed by its proteasomal degradation (Saha et al., 2006; Wu et al., 2000). In mouse melanoma cells, the consequences of ERK activation on MITF activity are predominately inhibitory (Wellbrock et al., 2008). The collective changes seen in p38 and ERK phosphorylation, p21, p27 and melanogenic protein expression, suggested that MITF expression would be increased in the Hn1-depleted cells (Carreira et al., 2005, 2006). However, the opposite effect was observed. For this reason, the expression of β-catenin, which can also regulate MITF expression, was evaluated, but levels of this protein did not change in the absence of Hn1 (Widlund et al., 2002). An alternative downstream target of p38 is the ubiquitously expressed related transcription factor, upstream stimulating factor 1 (USF-1) (Corre and Galibert, 2005; Corre et al., 2004). USF-1 is necessary for embryonic development and the UV-induced melanogenic response, and it is involved in regulating cell cycle transitions. Loss of its transcriptional activity has been detected in other cancer models including breast cancer cell lines, demonstrating its importance for cell proliferation control (Corre and Galibert, 2005). Similar to results with MITF, USF-1 expression decreased in Hn1-depleted cells. These data suggest that the growth and melanogenic effects of Hn1 depletion are independent of MITF and USF-1.
Another transcription factor evaluated was TFE3, which can also activate tyrosinase expression. Basal expression of TFE3 is very high in B16 cells. It has high sequence identity with MITF and binds the same promoter motif. In B16 cells, however, TFE3 binding to this motif is prevented by an unidentified mechanism. If TFE3 were functional, the effect of MITF overexpression on tyrosinase and Trp1 would be reduced, since their basal expression from the active TFE3 would already be significantly elevated (Verastegui et al., 2000). The transcription factor of the same family, TFEC, can bind TFE3 and prevent its transcriptional activation (Zhao et al., 1993). TFE3 gene and protein fusions are very commonly found in some cancers (Teixeira, 2006; Wu et al., 2005). Furthermore, TFE3 overexpression in myeloblasts causes proliferation arrest and macrophage differentiation. It strongly induces terminal differentiation in hematopoietic stem cells (Zanocco-Marani et al., 2006). TFE3 expression was evaluated in B16.F10 cells with and without Hn1 and it was found to be elevated in the Hn1-depleted cells, indicating that this is likely the mechanism by which Hn1 inhibition impacted melanoma cell differentiation. Additional studies will be required to determine if and how Hn1 can regulate cell differentiation via TFE3. Given the nuclear and cytoplasmic localization of the Hn1 protein in B16.F10 cells, mechanisms that include direct interactions with these transcription factors or indirect regulation of either signaling to the nucleus, e.g via modulating MAPK pathways, or cytoplasmic-nuclear shuttling of proteins are all possibilities worthy of further exploration.
The expression of Hn1 during development, nerve regeneration and cancer collectively points to a role for this gene in processes involved in either undifferentiated or dedifferentiated states of cells. During development, cells divide and migrate to their specific locations prior to differentiating. The presence of Hn1 in brain regions involved in learning and memory, such as hippocampal and cortical neurons, suggests a role for this gene in the phenomenon of plasticity. Indeed, in the injury responses that lead to motoneuron and retinal regeneration, neurons and retinal pigment epithelial (RPE) cells must halt specific functions associated with a differentiated phenotype in order to undergo repair and regenerate. These periods of cellular dedifferentiation are also marked by significant increases in the expression of Hn1 (Zujovic et al. 2005; Goto et al., 2006). If Hn1 is to be a possible target for anti-cancer or neuro-regenerative therapies, it will be necessary to determine its precise mechanism of action. On one hand, inhibition of Hn1 may promote successful suppression of cancer growth, while alternatively in situations requiring tissue regeneration its activity may need to be enhanced.
Acknowledgments
We thank Ms. Irene Zolotukhin for manufacturing the AAVs. We are also grateful to Dr. Vincent Hearing for providing the antibodies against Tyrosinase, Trp1 and Trp2 and to Drs. Miguel Seabra and Alistair Hume for providing the anti-Rab27a antibodies. These studies were supported in part by a grant from the National Institutes of Health to J.K.H. (AI058256).
Footnotes
Publisher's Disclaimer: This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues.
Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited.
In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier's archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
References
- Carreira S, Goodall J, Aksan I, La Rocca SA, Galibert MD, Denat L, Larue L, Goding CR. Mitf cooperates with Rb1 and activates p21Cip1 expression to regulate cell cycle progression. Nature. 2005;433:764–769. doi: 10.1038/nature03269. [DOI] [PubMed] [Google Scholar]
- Carreira S, Goodall J, Denat L, Rodriguez M, Nuciforo P, Hoek KS, Testori A, Larue L, Goding CR. Mitf regulation of Dia1 controls melanoma proliferation and invasiveness. Genes Dev. 2006;20:3426–3439. doi: 10.1101/gad.406406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church GM, Gilbert W. Genomic sequencing. Proc Natl Acad Sci USA. 1984;81:1991–1995. doi: 10.1073/pnas.81.7.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corre S, Galibert MD. Upstream stimulating factors: highly versatile stress-responsive transcription factors. Pigment Cell Res. 2005;18:337–348. doi: 10.1111/j.1600-0749.2005.00262.x. [DOI] [PubMed] [Google Scholar]
- Corre S, Primot A, Sviderskaya E, Bennett DC, Vaulont S, Goding CR, Galibert MD. UV-induced expression of key component of the tanning process, the POMC and MC1R genes, is dependent on the p-38-activated upstream stimulating factor-1 (USF-1) J Biol Chem. 2004;279:51226–51233. doi: 10.1074/jbc.M409768200. [DOI] [PubMed] [Google Scholar]
- Cranmer LD, Trevor KT, Bandlamuri S, Hersh EM. Rodent models of brain metastasis in melanoma. Melanoma Res. 2005;15:325–356. doi: 10.1097/00008390-200510000-00002. [DOI] [PubMed] [Google Scholar]
- Englaro W, Bertolotto C, Busca R, Brunet A, Pages G, Ortonne JP, Ballotti R. Inhibition of the mitogen-activated protein kinase pathway triggers B16 melanoma cell differentiation. J Biol Chem. 1998;273:9966–9970. doi: 10.1074/jbc.273.16.9966. [DOI] [PubMed] [Google Scholar]
- Fidler IJ. Selection of successive tumour lines for metastasis. Nat New Biol. 1973;242:148–149. doi: 10.1038/newbio242148a0. [DOI] [PubMed] [Google Scholar]
- Goto T, Hisatomi O, Kotoura M, Tokunaga F. Induced expression of hematopoietic- and neurologic-expressed sequence 1 in retinal pigment epithelial cells during newt retina regeneration. Exp Eye Res. 2006;83:972–980. doi: 10.1016/j.exer.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Harris J, Ayyub C, Shaw G. A molecular dissection of the carboxyterminal tails of the major neurofilament subunits NF-M and NF-H. J Neurosci Res. 1991;30:47–62. doi: 10.1002/jnr.490300107. [DOI] [PubMed] [Google Scholar]
- Harrison JK, Luo D, Streit WJ. In situ hybridization analysis of chemokines and chemokine receptors in the central nervous system. Methods. 2003;29:312–318. doi: 10.1016/s1046-2023(02)00354-7. [DOI] [PubMed] [Google Scholar]
- Hume AN, Collinson LM, Rapak A, Gomes AQ, Hopkins CR, Seabra MC. Rab27a regulates the peripheral distribution of melanosomes in melanocytes. J Cell Biol. 2001;152:795–808. doi: 10.1083/jcb.152.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume AN, Ushakov DS, Tarafder AK, Ferenczi MA, Seabra MC. Rab27a and MyoVa are the primary Mlph interactors regulating melanosome transport in melanocytes. J Cell Sci. 2007;120:3111–3122. doi: 10.1242/jcs.010207. [DOI] [PubMed] [Google Scholar]
- Laughlin KM, Luo D, Liu C, Shaw G, Warrington KH, Jr, Qiu J, Yachnis AT, Harrison JK. Hematopoietic- and neurologic-expressed sequence 1 expression in the murine GL261 and high-grade human gliomas. Pathol Oncol Res. 2009 Jan 15; doi: 10.1007/s12253-008-9147-4. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY, Fisher DE. Melanocyte biology and skin pigmentation. Nature. 2007;445:843–850. doi: 10.1038/nature05660. [DOI] [PubMed] [Google Scholar]
- Lu KH, Patterson AP, Wang L, Marquez RT, Atkinson EN, Baggerly KA, Ramoth LR, Rosen DG, Liu J, Hellstrom I, Smith D, Hartmann L, Fishman D, Berchuck A, Schmandt R, Whitaker R, Gershenson DM, Mills GB, Bast RC., Jr Selection of potential markers for epithelial ovarian cancer with gene expression arrays and recursive descent partition analysis. Clin Cancer Res. 2004;10:3291–3300. doi: 10.1158/1078-0432.CCR-03-0409. [DOI] [PubMed] [Google Scholar]
- McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8:1248–1254. doi: 10.1038/sj.gt.3301514. [DOI] [PubMed] [Google Scholar]
- Saha B, Singh SK, Sarkar C, Bera R, Ratha J, Tobin DJ, Bhadra R. Activation of the Mitf promoter by lipid-stimulated activation of p38-stress signalling to CREB. Pigment Cell Res. 2006;19:595–605. doi: 10.1111/j.1600-0749.2006.00348.x. [DOI] [PubMed] [Google Scholar]
- Stephenson EM, Stephenson NG. Karyotype analysis of the B16 mouse melanoma with reassessment of the normal mouse idiogram. J Natl Cancer Inst. 1970;45:789–800. [PubMed] [Google Scholar]
- Tang W, Lai YH, Han XD, Wong PM, Peters LL, Chui DH. Murine Hn1 on chromosome 11 is expressed in hemopoietic and brain tissues. Mamm Genome. 1997;8:695–696. doi: 10.1007/s003359900540. [DOI] [PubMed] [Google Scholar]
- Teixeira MR. Recurrent fusion oncogenes in carcinomas. Crit Rev Oncog. 2006;12:257–271. doi: 10.1615/critrevoncog.v12.i3-4.40. [DOI] [PubMed] [Google Scholar]
- Verastegui C, Bertolotto C, Bille K, Abbe P, Ortonne JP, Ballotti R. TFE3, a transcription factor homologous to microphthalmia, is a potential transcriptional activator of tyrosinase and TyrpI genes. Mol Endocrinol. 2000;14:449–456. doi: 10.1210/mend.14.3.0428. [DOI] [PubMed] [Google Scholar]
- Wellbrock C, Rana S, Paterson H, Pickersgill H, Brummelkamp T, Marais R. Oncogenic BRAF regulates melanoma proliferation through the lineage specific factor MITF. PLoS ONE. 2008;3:e2734. doi: 10.1371/journal.pone.0002734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widlund HR, Horstmann MA, Price ER, Cui J, Lessnick SL, Wu M, He X, Fisher DE. β-Catenin-induced melanoma growth requires the downstream target Microphthalmia-associated transcription factor. JCB. 2002;158:1079–1087. doi: 10.1083/jcb.200202049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Hemesath TJ, Takemoto CM, Horstmann MA, Wells AG, Price ER, Fisher DZ, Fisher DE. c-Kit triggers dual phosphorylations, which couple activation and degradation of the essential melanocyte factor Mi. Genes Dev. 2000;14:301–312. [PMC free article] [PubMed] [Google Scholar]
- Wu J, Brinker DA, Haas M, Montgomery EA, Argani P. Primary alveolar soft part sarcoma (ASPS) of the breast: report of a deceptive case with xanthomatous features confirmed by TFE3 immunohistochemistry and electron microscopy. Int J Surg Pathol. 2005;13:81–85. doi: 10.1177/106689690501300112. [DOI] [PubMed] [Google Scholar]
- Zanocco-Marani T, Vignudelli T, Gemelli C, Pirondi S, Testa A, Montanari M, Parenti S, Tenedini E, Grande A, Ferrari S. Tfe3 expression is closely associated to macrophage terminal differentiation of human hematopoietic myeloid precursors. Exp Cell Res. 2006;312:4079–4089. doi: 10.1016/j.yexcr.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Zhao GQ, Zhao Q, Zhou X, Mattei MG, de Crombrugghe B. TFEC, a basic helix-loop-helix protein, forms heterodimers with TFE3 and inhibits TFE3-dependent transcription activation. Mol Cell Biol. 1993;13:4505–4512. doi: 10.1128/mcb.13.8.4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Wang J, Zhang Y, Zhong C, Ni J, Wang L, Guo J, Zhang K, Yu L, Zhao S. Cloning, expression and subcellular localization of HN1 and HN1L genes, as well as characterization of their orthologs, defining an evolutionarily conserved gene family. Gene. 2004;331:115–123. doi: 10.1016/j.gene.2004.02.025. [DOI] [PubMed] [Google Scholar]
- Zolotukhin S, Potter M, Zolotukhin I, Sakai Y, Loiler S, Fraites TJ, Jr, Chiodo VA, Phillipsberg T, Muzyczka N, Hauswirth WW, Flotte TR, Byrne BJ, Snyder RO. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158–167. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]
- Zujovic V, Luo D, Baker HV, Lopez MC, Miller KR, Streit WJ, Harrison JK. The facial motor nucleus transcriptional program in response to peripheral nerve injury identifies Hn1 as a regeneration-associated gene. J Neurosci Res. 2005;82:581–591. doi: 10.1002/jnr.20676. [DOI] [PubMed] [Google Scholar]