

Abstract
Superparamagnetic nanoparticles (NPs) have been attractive for medical diagnostics and therapeutics due to their unique magnetic properties and their ability to interact with various biomolecules of interest. The solution phase based chemical synthesis provides a near precise control on NP size, and monodisperse magnetic NPs with standard deviation in diameter of less than 10% are now routinely available. Upon controlled surface functionalization and coupling with fragments of DNA strands, proteins, peptides or antibodies, these NPs can be well-dispersed in biological solutions and used for drug delivery, magnetic separation, magnetic resonance imaging contrast enhancement and magnetic fluid hyperthermia. This Perspective reviews the common syntheses and controlled surface functionalization of monodisperse Fe3O4-based superparamagnetic NPs. It further outlines the exciting application potentials of these NPs in magnetic resonance imaging and drug delivery.
1. Introduction
Superparamagnetic (SPM) nanoparticles (NPs) are a typical class of ferromagnetic materials. The magnetic moment of the NPs as a whole is free to fluctuate in response to thermal energy while the individual atomic moments maintain their ordered state relative to each other and there is no remanence in this rapidly changing magnetic state.1 Fig. 1 shows the magnetization curve, also called the hysteresis loop, of a group of SPM NPs. Normally, these NPs magnetized under an external magnetic field (H) have a saturation magnetization (Ms). This Ms under the fixed magnetic field is, however, NP size-dependent as the magnetization directions of the SPM NPs are subject to thermal fluctuation, leading to the magnetization randomization. To achieve the same Ms value, a stronger magnetic field is needed. Due to the same thermal fluctuation, the SPM NPs have no remnant magnetization value, i.e. without external magnetic field, the net moment of the particles is randomized to zero.
Fig. 1.
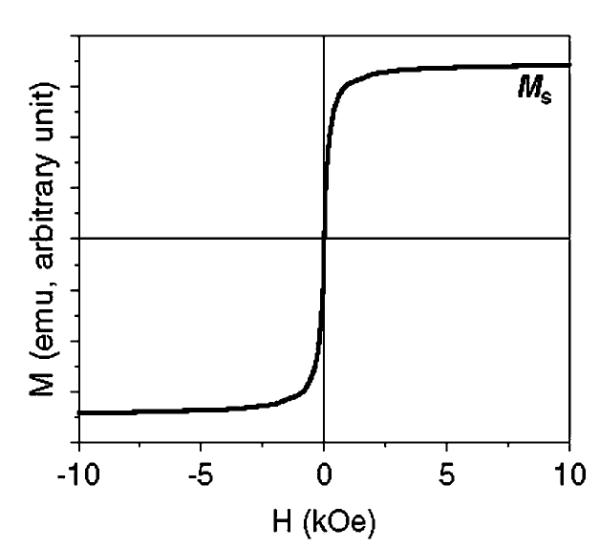
Schematic illustration of a hysteresis loop of SPM NPs.
The SPM NPs have been considered as attractive magnetic probes for biological imaging and therapeutic applications.2 In normal biological conditions, these SPM NPs are not subject to strong magnetic interactions in the dispersion due to the randomization of their magnetization and are readily stabilized in physiological conditions. Under an external magnetic field, however, they exhibit a magnetic signal far exceeding that from any of the known biomolecules and cells. This makes SPM NPs readily identified by a magnetic sensing device from the ocean of biomolecules. At a core diameter at less than 20 nm and overall hydrodynamic diameter at less than 50 nm, these NPs have the size that is comparable to the nuclear pore size (∼50 nm) and is much smaller than a cell (normally 10–30 μm). Once coupled with a target agent, they can serve as a nano-vector and interact specifically with biomolecules of interest through well established biological interactions, providing controllable means of magnetically tagging bio-identity. Under the normal range of magnetic field strengths used in magnetic resonance imaging (MRI) scanners (usually higher than 1 T), these SPM NPs in the targeted area can be magnetically saturated, establishing a substantial locally perturbing dipolar field that leads to a marked shortening of proton relaxation (T2 relaxation) in MRI process and giving a “darker” image of the targeted area over the biological background. Furthermore, under an alternating magnetic field with controlled field amplitude and field reversal frequency, magnetization of the SPM NPs attached to the bio-identity can be switched back and forth. This magnetization re-orientation may result from either a physical rotation of the particle (Brownian relaxation), which creates friction between the NP and its surrounding liquid medium, or internal magnetization switching from one direction to another (Néel relaxation). In both cases, these SPM NPs function as a heater to heat the area they target to. This magnetic field induced NP heating has been known as magnetic fluid hyperthermia and has been studied extensively for future cancer therapy.3
To succeed in biomedical applications, SPM NPs should be monodisperse so that each individual NP has nearly identical physical and chemical properties for controlled bio-distribution, bio-elimination and contrast effects. These SPM NPs should also have high magnetic moment, and can chemically withstand physiological conditions without property degradation in the detection or treatment time period. The NPs should be small with a hydrodynamic size of <50 nm so that they can have extravasation ability and are stable against uptake by the reticuloendothelial system (RES).4 More importantly, the NPs can be modified so that they are capable of binding specifically to biomolecules of interest through biological interactions.
Magnetite (Fe3O4) NPs are thus far the most attractive SPM NPs used for biomedical applications due to their chemical stability and biocompatibility.3c,5 To obtain uniform SPM Fe3O4 NPs with controlled chemical and magnetic properties, various synthetic methods, especially high temperature solution phase syntheses, have been developed. The synthetic efforts have also led to numerous surface modifications and various kinds of bio-compatible SPM Fe3O4 NPs have been produced.1a,6 The syntheses have been extended to the SPM NPs of MFe2O4 (M = Mn, Co, or Ni)7 and high moment Fe,8 CoFe9 to maximize contrast effects in MRI. Some bifunctional NPs of the dumb bell-like Au-Fe3O4 or Ag-Fe3O4 are also prepared so that both optically and magnetically active NPs can be incorporated into one nanostructure to achieve the desired NP targeting and the sensitive NP detection.10 This Perspective highlights the recent developments in the synthesis and surface modification of SPM Fe3O4-based NPs as well as their potential applications in diagnostics and therapeutics.
2. SPM NP synthesis
SPM NPs can be synthesized by a variety of methods ranging from traditional co-precipitation of metal salts in basic solution, high temperature organic phase decomposition, and chemical vapour deposition. SPM NPs used in biomedical applications are often synthesized by the co-precipitation of ferrous and ferric ions at 1 to 2 ratio in an alkaline medium.3c,6a In recent years, high-temperature organic phase reductive decomposition of metal salt or organometallic precursors has been applied to produce monodisperse SPM NPs. With proper surface functionalization, these SPM NPs have shown great potential for diagnostic and therapeutic applications.
2.1 MFe2O4 NPs
Magnetic ferrite MFe2O4 NPs, especially magnetite Fe3O4 NPs, are widely studied due to their chemical and magnetic stability. This oxide represents a well-known and important class of iron oxide materials where oxygen forms an fcc packing, and M2+ and Fe3+ occupy either tetrahedral or octahedral interstitial sites. By adjusting the chemical identity of M2+, the magnetic configurations of MFe2O4 can be molecularly engineered to provide a wide range of magnetic properties.
MFe2O4 NPs are commonly made by hydrolysis/condensation of M2+ and Fe3+ ions by a base, usually NaOH, or NH3·H2O, in an aqueous solution,11 or in reverse micelles.12 Although this co-precipitation method is suitable for mass production of magnetic MFe2O4 ferro-fluids, it requires careful adjustment of the pH value of the solution for particle formation and stabilization, and it is difficult to control sizes and size distributions, particularly for particles smaller than 20 nm. An alternative approach to make monodisperse iron oxide NPs is via high temperature organic phase decomposition of Fe(CO)5 in the presence of (CH3)3NO, air or 3-chloro-peroxybenzoic acid.13
More conveniently, MFe2O4 NPs are synthesized by reductive decomposition of metal acetylacetonates or carboxylates in an organic phase.7 For example, monodisperse Fe3O4 NPs are prepared by high temperature (up to 305 °C) reductive decomposition of Fe(acac)3 in the presence of a long chain 1,2-hydrocarbon diol, oleic acid and oleylamine.14 MFe2O4 NPs (with M = Co, Ni, Mn, etc.) are made by simply adding a different metal acetylacetonate precursor to the mixture of reactants used for Fe3O4 synthesis.7 The size of the NPs is controlled by varying the reaction temperatures or changing the concentrations of metal precursors. Alternatively, with the smaller NPs as seeds, larger NPs up to 20 nm in diameter can be synthesized by seed mediated growth.
Fe3O4 NPs can also be made by chemical conversion of FeO NPs.15 FeO NPs are synthesized by heating the mixture of Fe(acac)3, oleic acid and oleylamine. When treated under atmospheric pressure and air at 120 °C for 90 min, the assynthesized FeO NPs are converted to Fe3O4 NPs. Using this method, large Fe3O4 NPs up to 100 nm in diameter can be made.
By combining decomposition/oxidation of Fe(CO)5 and reductive decomposition of iron oleate complex, 1 nm size control of iron oxide NPs can be achieved.13a,16 Monodisperse NPs with particle sizes of 6, 7, 9, 10, 12, 13 and 15 nm have been produced. The monodispersity of the NPs can be readily seen in the representative TEM images of 7, 8, 9 nm NPs in Fig. 2.
Fig. 2.

TEM images of (a) 7, (b) 8 and (c) 9 nm iron oxide NPs showing the one nanometre level increments in diameter. Reproduced with permission from ref. 16c.
Large-scale synthesis of iron oxide NPs is achieved through high temperature decomposition of iron oleate.16b In the synthesis, iron chloride reacts with sodium oleate to form a waxy iron-oleate complex that is subject to further thermal decomposition at 320 °C in 1-octadecene, leading to the formation of monodisperse iron oxide NPs. Another method that has the potential for large scale synthesis of magnetic NPs is via a liquid-solid-solution (LSS) phase transfer.17 The chemistry for the synthesis is illustrated in Fig. 3. It involves the reaction of metal precursor at the interfaces of metal linoleate (solid), ethanol—linoleic acid liquid phase (liquid) and water—ethanol solutions (solution) at different designated temperatures. A phase transfer process occurs spontaneously across the interface of the solid and the solution. The NPs generated at the interface are coated with a layer of linoleic acid, resulting in a spontaneous phase-separation and the formation of hydrophobic NPs that are easily collected at the bottom of the container.
Fig. 3.
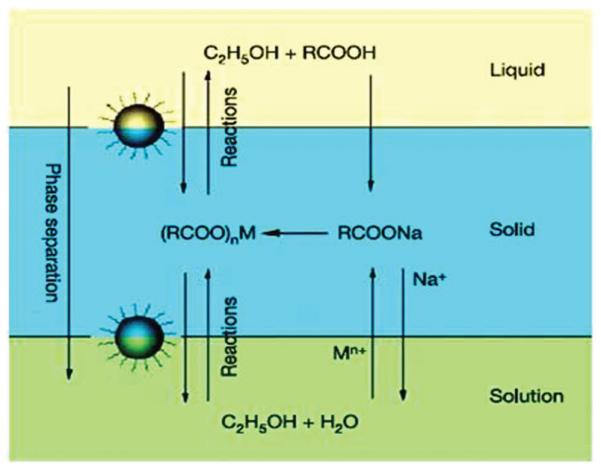
Schematic illustration of the liquid—solid—solution (LSS) phase transfer synthesis of various NPs. Reproduced with permission from ref. 17.
Recently, the ultra-small Fe3O4 NPs ranging from 2.5–5 nm were made by thermal decomposition of Fe(CO)5 in benzyl ether at 300 °C followed by room temperature air oxidation. Different from the previous synthesis methods, this preparation used a small molecule 4-methylcatechol (4-MC) as the surfactant and the sizes of the NPs were tuned by the MC—Fe ratio.18 More importantly, the 4-MC coated Fe3O4 NPs can be directly conjugated with a peptide, c(RGDyK), via the Mannich reaction, rendering the biocompatible SPM NPs with a hydrodynamic diameter of around 8 nm, suitable for target-specific delivery and imaging applications.
2.2. Fe3O4-stabilized SPM Fe NPs
Magnetic metallic NPs (Fe, Co and CoFe) have high magnetization value, ∼160 emu g-1 for Co, ∼218 emu g-1 for Fe, ∼245 emu g-1 for CoFe, making them ideal candidates for highly sensitive magnetic applications. However, unlike iron oxide NPs, these metallic NPs are chemically unstable, prone to fast oxidation once exposed to ambient conditions.
Monodisperse 7 nm Fe NPs (Fig. 4a) are made by reduction of iron(II) bis(trimethylsilyl)amide (Fe{N[Si(CH3)3]2}2) in the presence of hexadecylamine and oleic acid or hexadecylammonium chloride at 150 °C in mesitylene with dihydrogen as the reducing agent.19 To stabilize Fe NPs against fast oxidation, crystalline Fe3O4 is used to coat Fe NP surface.8 In a recent stabilization demonstration, monodisperse Fe NPs are first produced by decomposition of Fe(CO)5 in octadecene (ODE) at 180 °C and then subject to controlled oxidation by an oxygen transfer agent (CH3)3NO at 250 °C. This controlled oxidation gives core/shell structured Fe/Fe3O4 in which Fe3O4 has inverse spinel structure and acts as an anti-oxidation layer (Fig. 4b). The thickness of the shell is tuned by controlling the amount of (CH3)3NO added into the reaction mixture. After this treatment, the chemical and dispersion stability of the particles are improved dramatically. For Fe/Fe3O4 NPs containing 4 nm (radius) Fe core and 2.5 nm Fe3O4 shell, their saturation magnetization reaches 123.5 emu g-1 [Fe] ([Fe] = Fe + Fe3O4). Taking into account of the lower magnetization caused by the presence of the Fe3O4, the saturation magnetization of the Fe core are close to the bulk value of 218 emu g-1.
Fig. 4.

TEM images of (a) the 7 nm Fe NPs (adapted from ref. 19) and (b) the 2.5 nm/5 nm Fe/Fe3O4 NPs (adapted from ref. 8).
High moment FeCo NPs are synthesized by reductive thermal decomposition of Fe(CO)5 and Co(η3-C8H13)(η4-C8H12) or Co(N(SiMe3)2)2 in the presence of 1 equiv. hexadecylamine and 1 equiv. oleic acid.9 CoFe NPs can also be made by polyol reduction of Co(acac)2 and Fe(acac)3 under a gas mixture of 93% Ar + 7% H2 at 300 °C.20 The 20 nm NPs have a magnetization value of 207 emu g-1. The stability of the CoFe NPs can be enhanced by embedding them into a carbon matrix.21 In the synthesis, Fe and Co salts are first absorbed onto the high surface-area silica powder by impregnation in a methanol solution. The metal-loaded silica is dried at 800 °C under H2 and then subject to methane chemical vapour deposition (CVD) for carbon deposition on FeCo. Once cooled down to room temperature, the silica is etched away in HF, leaving pure FeCo/graphitic carbon NPs that show dramatic stability against deep oxidation and are promising for bioimaging applications.
2.3. Fe3O4-based bifunctional NPs
Bifunctional NPs are those containing two different nanoscale functionalities within one integrated identity. Dumb bell-like and core/shell NPs are two representative bifunctional systems that have shown great potential for biomedical applications.
The dumb bell-like Au-Fe3O4 NPs are prepared via the decomposition of iron pentacarbonyl, Fe(CO)5, at 300 °C over the surface of the pre-formed Au NPs followed by oxidation in air, as illustrated in Fig. 5a.10b The Au NPs can be either synthesized in situ by injecting HAuCl4 solution into the reaction mixture or pre-made in the presence of oleylamine. The size of the Au NPs is tuned by controlling the temperature at which the HAuCl4 solution is added, or by controlling the HAuCl4—oleylamine ratio. The size of the Fe3O4 NPs is controlled by amount of Fe(CO)5 added in the reaction mixture. Fig. 5b shows the TEM image of the Au-Fe3O4 NPs with Fe3O4 at around 14 nm and Au at 8 nm. Fig. 5c is a typical high-resolution TEM (HRTEM) image of a dumbbell-like NP with Fe3O4 at 12 nm and Au at 8 nm. In the structure, a Fe3O4 (111) plane grows onto an Au (111) plane, giving the dumbbell-like structure. These dumbbell-like NPs show a plasmonic absorption at around 530 nm and have a saturation magnetization of 80 emu g-1—a value close to the pure Fe3O4 NPs.10b
Fig. 5.

(a) Schematic illustration of the growth of Au-Fe3O4 NPs. (b) TEM and (c) HRTEM images of the Au-Fe3O4 NPs. Reproduced with permission from ref. 10b.
Different from Au-Fe3O4 NPs, the Ag-Fe3O4 NPs are made by controlled nucleation of Ag on the pre-formed Fe3O4 NPs.10a In the synthesis, the as-prepared Fe3O4 NPs dispersed in organic solution and AgNO3 dissolved in water are mixed and agitated by ultra-sonication. The sonication provides the energy required for the formation of a micro emulsion with the Fe3O4 NPs assembling at the liquid/liquid interface. Fe(II) on the NPs acts as catalytic center for the reduction of Ag+ and nucleation/growth of Ag NPs, as illustrated in Fig. 6. The partial exposure of the NPs to the aqueous phase causes the formation of Ag-Fe3O4 NPs. The NPs show a plasmonic absorption from Ag NPs and the same magnetic hysteresis behaviour as Fe3O4 NPs.
Fig. 6.
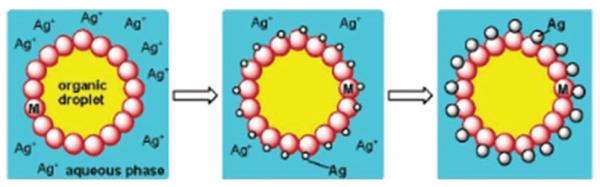
Schematic illustration of the formation of Ag-Fe3O4 NPs in a micellar structure by ultra-sonication of a heterogeneous solution with as-prepared Fe3O4 NPs in the organic phase and AgNO3 in water. Reproduced with permission from ref. 10a.
Core/shell NPs are another group of NPs that can incorporate multifunctionality into one structure. One recent example is Fe3O4/Au or Fe3O4/Au/Ag NPs.22 In the synthesis, the pre-made Fe3O4 NPs are mixed with a solution of HAuCl4 and oleylamine. HAuCl4 is reduced under this condition, forming a thin layer of Au shell over the Fe3O4 surface (Fig. 7a). The surface of the particles is then treated with sodium citrate and cetyltrimethylammonium bromide (CTAB). NPs treated this way are water soluble and can serve as seeds to grow more Au or Ag on their surface. The thicker coating is achieved by mixing the seeding Fe3O4/Au NPs with HAuCl4 or AgNO3 and ascorbic acid in the presence of CTAB, and incubating the mixture at 30 °C (Fig. 7b). The control on shell thickness allows the tuning of plasmonic properties of the core/shell NPs to be either red-shifted (to 560 nm with more Au coating) or blue-shifted (to 501 nm with more Ag coating).
Fig. 7.
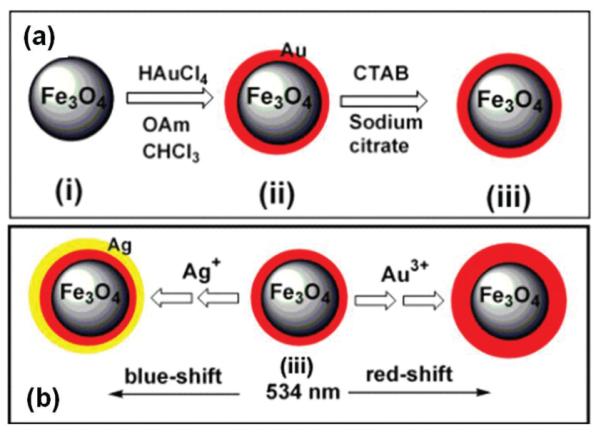
(a) Schematic illustration of surface coating of Fe3O4 NPs (i) with Au to form hydrophobic Fe3O4/Au NPs (ii) and hydrophilic Fe3O4/Au NPs (iii). (b) Schematic illustration of the formation of Fe3O4/Au and Fe3O4/Au/Ag and the control on the plasmonic properties. Reproduced with permission from ref. 22.
3. NP surface functionalization
The SPM NPs prepared above are normally stabilized by the surfactants like oleic acid or oleylamine or their combination. The formation of metal carboxylate and/or metal—amine bonds at the interface leaves NPs surrounded with a layer of hydrocarbon, making them hydrophobic and only soluble in non-polar or weakly polar organic solvents. For NPs to be useful in a biological system, they must be water soluble and stable at various pH values ranging from 5–9, at salt concentrations at hundreds of mM, and at various cell culture temperatures. They must also achieve target-specific binding in biological systems.
Surfactant addition and surfactant exchange are two general approaches for NP surface functionalization, as illustrated in Fig. 8.1b Surfactant addition is achieved through the adsorption of amphiphilic molecules that contain both a hydrophobic segment and a hydrophilic component. The hydrophobic segment forms a double layer structure with the original hydrocarbon chain, while hydrophilic groups expose to the outside of the NPs, rendering them water-soluble. The NP biocompatibility can be further improved by using biodegradable amphiphilic polymers originally developed for drug delivery applications.23 One of the most widely utilized and successful polymers has been the functionalized phospholipids.21,24 As various phospholipids or amphiphilic polymers are commercially available, this addition method offers a convenient approach to functionalize NPs with biotin, -COOH, -SH and/or -NH2, facilitating their conjugation with fragments of DNA, proteins, peptides, or antibodies.
Fig. 8.
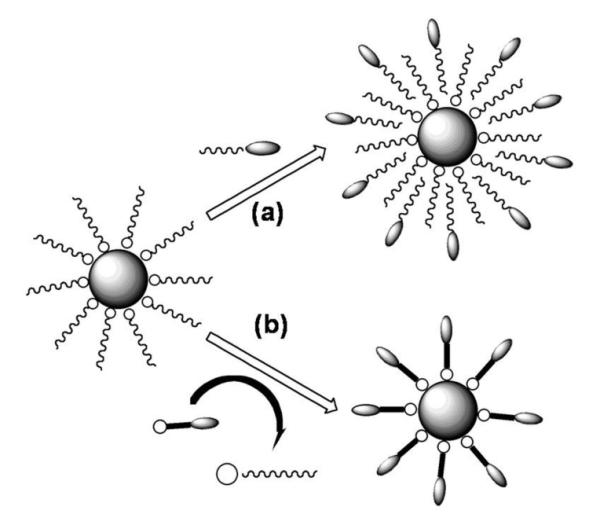
Schematic illustration of NP surface functionalization via (a) surfactant addition and (b) surfactant exchange.
Surfactant exchange is the direct replacement of the original surfactant with a new bifunctional surfactant. This bifunctional surfactant has one functional group capable of binding to the NP surface tightly via a strong chemical bond and the second functional group at the other end with a polar character so that the NPs can be dispersed in water or be further functionalized. Various monomeric species, such as dimercaptosuccinic acid, dopamine and peptides have been applied for this NP functionalization purpose.25
The functionalization described above offer the SPM NPs with robust colloidal and bio-stability. The targeting agents coupled with these NPs allow facile bio-recognition event via strong biological interactions. These, plus their multifunctional magnetic and optical properties, make SPM NPs ideal for diagnostic and therapeutic applications.
4. SPM NPs for biomedical applications
For SPM NPs to be useful for biomedical applications, they should be first stabilized against the absorption of plasma proteins and non-specific uptake by reticular-endothelial system (RES), like macrophage cells.4 Due to their large surface area, when exposed to a physiological environment, these NPs tend to interact with plasma proteins, causing size increase that often results in serious agglomeration. They may be also considered as an intruder by the innate immune system and be readily recognized and engulfed by the macrophage cells. In both cases, the particles will be removed from the blood circulation and lose their function, leading to dramatic reduction in efficiency in NP-based diagnostics and therapeutics. To inhibit the plasma coating and to escape from the RES for longer circulation times, the NPs are usually coated with a layer of hydrophilic and biocompatible polymer such as dextran, dendrimers, polyethylene glycol (PEG), or polyethylene oxide (PEO).26
As an amphiphilic polymer and a non-specific interaction reducing reagent,26e polyethylene glycol (PEG) was used recently to functionalize monodisperse 9 nm Fe3O4 NPs.4f In this study, PEG is anchored on the Fe3O4 NPs through a covalent bond as illustrated in Fig. 9a. These PEG coated Fe3O4 NPs are incubated with the RAW 264.7 cells—one kind of mouse macrophage cell line, at three different concentrations: 0.1 mg Fe mL-1, 0.01 mg Fe mL-1 and 0.001 mg Fe ml-1. The results from the 0.01 mg Fe mL-1 of PEG-coated samples are shown in Fig. 9b. It can be seen that the dextran coated NPs give the highest uptake, followed by PEG600 coated NPs, of which the uptake is about 30–50% of that from the dextran coated ones. For PEG3000, PEG6000 and PEG20000 coated NPs, their uptake are comparative with the background, indicating negligible uptake of these NPs by the macrophage cells.4f
Fig. 9.

(a) Schematic of ligand exchange reaction on the surface of Fe3O4 NPs (b) Macrophage uptake assay of the Fe3O4 NPs in (a) with initial Fe concentration at 0.01 mg Fe mL-1. Reproduced with permission from ref. 4f.
To act as a sensitive probe for cell imaging, a NP must be taken up by cells to “stain” the cells. As in normal biological transport process, this uptake can be either passive or active, or both. Passive uptake utilizes diffusion concept and is often concentration driven and has no targeting capability. Active uptake, on the other hand, involves receptor-mediated endocytosis. As fast grown cells, especially tumour cells, often over-express certain receptors of folic acid, sugars, peptides, proteins, or antibodies. NPs coupled with these molecules tend to be recognized by these cells and endocytosed for internalization, achieving target-specific binding.
4.1. SPM NPs as contrast agents in MRI
MRI is one of the most powerful non-invasive imaging techniques utilized in clinical medicine. It’s based on the principle that protons align and precess along an applied magnetic field. Upon applying a transverse radiofrequency pulse, these precessed protons are perturbed from the magnetic field direction. The subsequent process, through which the pulsing field is turned off to allow protons to return to their original state, is referred to as relaxation. Two independent relaxation processes, longitudinal relaxation (T1-recovery) and transverse relaxation (T2-decay), are utilized to generate a bright and a dark MR image respectively. The longitudinal relaxation is primarily a measure of the dipolar coupling of the proton moments to their surroundings. Transverse relaxation is driven by the loss of phase coherence in the precessing protons due to their magnetic interaction with each other and with other fluctuating moments in the tissue. Upon accumulation in tissues, SPM NPs are magnetically saturated in the normal range of magnetic field strengths in MRI scanner and establish a substantial locally perturbing dipolar field, which leads to a marked shortening of T2* along with a less marked reduction of T1. Thus SPM NPs are a good candidate for T2 contrast agent to provide a dark image and the contrast enhancement is proportional to the magnetization magnitude.3,27
As the magnetization value of a SPM NP at a certain magnetic field is dependent on the size and magnetocrystalline anisotropy of the NP, SPM NPs with different sizes and structures have been prepared and compared for their contrast effects in MRI.25a In a recent comprehensive study on ferrite NPs for MRI application,7d the NPs were fabricated by a high-temperature, non-hydrolytic reaction between divalent metal chloride (MCl2) and iron tris-2,4-pentadionate in the presence of oleic acid and oleylamine as surfactants. The hydrophobic ligand was exchanged with 2,3-dimercaptosuccinic acid (DMSA). The DMSA-coated NPs show high colloidal stability at a salt (NaCl) concentration of 250 mM, across a wide pH range (pH 6–10) and in serum. 12 nm MnFe2O4 NPs have the highest mass magnetization value of 110 (emu per mass of magnetic atoms). This value is reduced to 101, 99 and 85 (emu per mass of magnetic atoms) for Fe3O4, CoFe2O4 and NiFe2O4, respectively. The spin—spin relaxation time (T2)-weighted MR images for each sample at 1.5 T are consistent with the magnetization results with MnFe2O4 NPs show the strongest MR contrast effect with a relaxation value reaching 358 mM-1 s-1, much larger than 218, 172, 152 and 62 mM-1 s-1 for Fe3O4, CoFe2O4, NiFe2O4 NPs, respectively. This size and structure dependent relaxation of MFe2O4 NPs can be seen in Fig. 10, from which one can conclude that larger NPs have large contrast effect, but at the same size, MnFe2O4 NPs have the largest contrast enhancement due to the small magnetocrystalline anisotropy and easy magnetization reversal in MnFe2O4 structure.
Fig. 10.
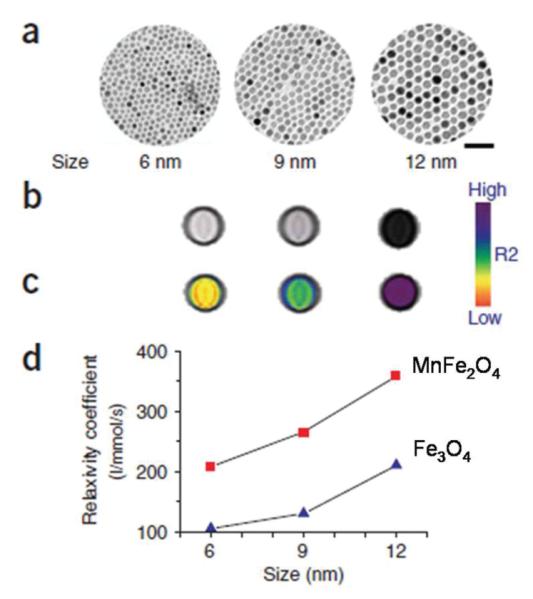
Size-dependent MR contrast effect of MnFe2O4 and Fe3O4 NPs. (a) TEM images of MnFe2O4 NPs (scale bar, 50 nm), (b) T2-weighted MR images, (c) colour maps of 6, 9 and 12 nm MnFe2O4 NPs, and (d) a plot of NP size versus R2 relaxation. Reproduced with the permission from ref. 7d.
The cancer detection sensitivity of these ferrite NPs are further evaluated. In a recent test, the MnFe2O4 NPs were coupled with the cancer-targeting Herceptin, an antibody specifically binding to the HER2/neu marker over-expressed on the surface of breast and ovarian cancers.28 Various cell lines with different levels of HER2/neu over-expression: Bx-PC-3, MDA-MB-231, MCF-7 and NIH3T6.7 (relative HER2/neu expression levels are 1, 3, 28 and 2,300, respectively) were used for the test. With the MnFe2O4-Herceptin conjugates, the detection of the Bx-PC-3 cell line occurred with a noticeable MR contrast. As the relative HER2/neu expression level increased to 3, 28 and 2,300, the MR contrast increased consistently for the MDA-MB-231, MCF-7 and NIH3T6.7 cell lines, respectively. In contrast, when Fe3O4-Herceptin conjugates were used, the only MR-detectable cell line was NIH3T6.7. Fig. 11 shows the colour coded MRI of a mouse implanted with the cancer cell line NIH3T6.7 and treated with 50 mg of NP-Herceptin. It can be seen that the tumor treated with the MnFe2O4-Herceptin NPs show colour changes from red to blue in the colour-coded MR images (Fig. 11a-c). In contrast, those treated with the Fe3O4-Herceptin NPs at the same dosage have no apparent change in the colour-coded MR images (Fig. 11d-f). These indicate that the high MR sensitivity of MnFe2O4-Herceptin conjugates enables the MR detection of tumours.7d
Fig. 11.
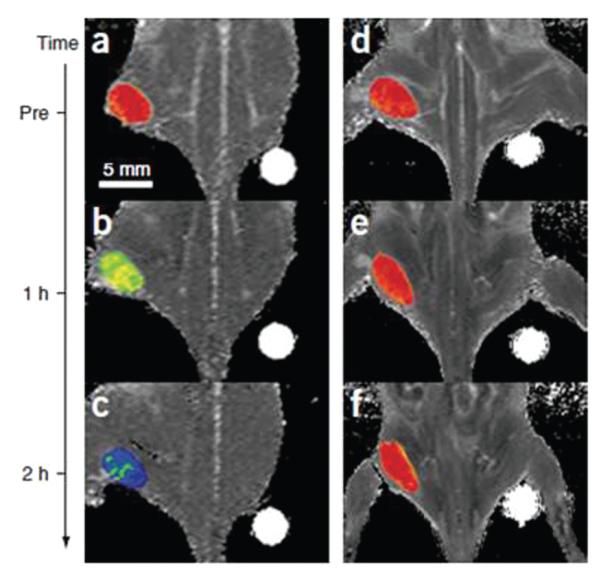
Colour maps of T2-weighted MR images of a mouse implanted with the cancer cell line NIH3T6.7 at different time points after injection of MnFe2O4-Herceptin (a—c) and Fe3O4-Herceptin (d—e) conjugates (pre-injection (a, d); and 1 h (b, e) or 2 h (c, f) after injection). Reproduced with permission from ref. 7d.
Ultra-small Fe3O4 NPs (<10 nm in hydrodynamic diameter) are also active for tumour-specific detection. In a recent demonstration, the Fe3O4 NPs with 4.5 nm core size were synthesized by thermal decomposition of Fe(CO)5 followed by air oxidation. The synthesis used a small ligand 4-methylcatechol (4-MC) as the surfactant and this 4-MC can be directly conjugated with c(RGDyK) peptide via the Mannich reaction, rendering the particles stable in physiological environment. The targeting ability of the c(RGDyK)-Fe3O4 NPs to integrin αvβ3 in vivo is evaluated by T2-weighted fast spin-echo MR imaging with mice bearing U87MG tumours. The r2 relaxation of the c(RGDyK)-Fe3O4 NPs is measured to be 165 mM-1 s-1, that is larger than that of the commercial Feridex NPs (104 mM-1 s-1) with similar core size.18 The tumour MR signal intensity decreases significantly after injection of c(RGDyK)-Fe3O4 NPs (Fig. 12a and b). Such signal reduction is significantly inhibited in the presence of a blocking dose of c(RGDyK) peptide (10 mg kg-1) (Fig. 12c), proving the specificity of the particle targeting. To chemically characterize the preferred Fe3O4 NP uptake, Prussian blue staining is employed to visualize the disposition of the c(RGDyK)-Fe3O4 NPs in the tissue samples obtained after the MR scan at 4 h time point. Prominent blue spots on the U87MG tumour slices are observed (Fig. 12d) and the blue spots are significantly reduced in the presence of free c(RGDyK) (Fig. 12e), further confirming that the accumulation of c(RGDyK)-Fe3O4 NPs is specifically mediated by integrin αvβ3 binding.
Fig. 12.
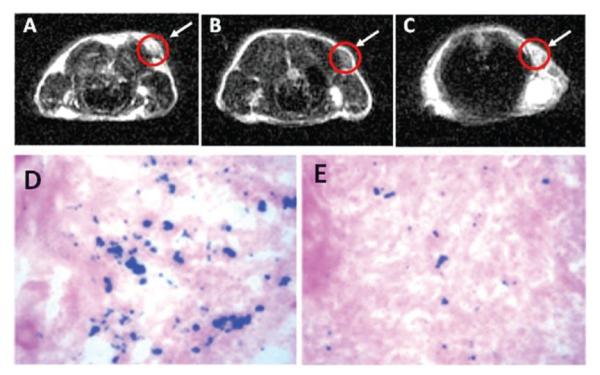
MRI of the cross section of the U87MG tumours implanted in a mouse: (a) without NPs, (b) with the injection of 300 μg of c(RGDyK)-Fe3O4 NPs, and (c) with the injection of the blocking dose of c(RGDyK) then c(RGDyK)-Fe3O4 NPs; and Prussian staining of U87MG tumors in the presence of (d) c(RGDyK)-Fe3O4 NPs and (e) blocking dose of c(RGDyK) and then c(RGDyK)-Fe3O4 NPs. Reproduced with permission from ref. 18.
Bifunctional Au-Fe3O4 NPs are also tested for imaging applications.29 In this structure, Au NPs are plasmonicly active and Fe3O4 NPs are magnetically active. The different NP surface also facilitates the controlled functionalization of each NP. As synthesized, the Au-Fe3O4 NPs are coated with a layer of oleate and oleylamine. The NPs are made biocompatible by replacing the hydrophobic coating with a hydrophilic one. Fig. 13a illustrates the surface functionalization of the 8–20 nm Au—Fe3O4 NPs. The original oleate/oleylamine coating on Fe3O4 is replaced by a catechol unit present in dopamine molecule that is linked to polyethylene glycol (PEG, Mr = 3000), and oleylamine around the Au NP is exchanged by HS-PEG-NH2 (Mr = 2204) with HS attaching to Au. The epidermal growth factor receptor antibody (EGFRA) is linked to the PEG via EDC/NHS chemistry on the Fe3O4 side. Fig. 13b is the TEM image of the NPs after surface modification. The antibody is used to target A431 cells that are known to over-express the epidermal growth factor receptor (EGFR).30
Fig. 13.
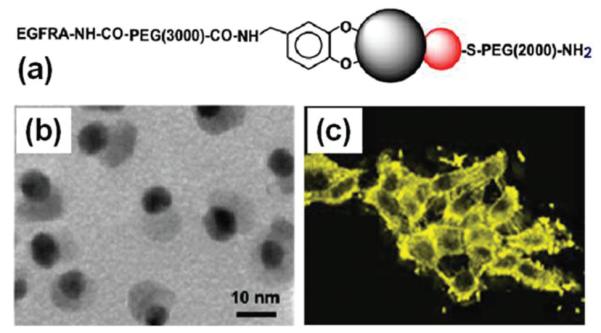
(a) Schematic illustration of surface functionalization of the Au-Fe3O4 DBNPs, (b) TEM image of the 8–20 nm Au-Fe3O4 NPs after surface modification, (c) Reflection images of the 8–20 nm Au-Fe3O4 labelled A431 cells. Reproduced with permission from ref. 29.
The preferred binding between EGFR and EGFRA enables the Au-Fe3O4 to be populated on the surface or within the cytoplasm of A431 cells. MRI analyses reveals that A431 cells labeled with 8–20 nm Au—Fe3O4 NPs shorten the T2 relaxation of the water molecules with relaxation r2 = 80.4 mM-1 s-1. A431 cells labelled with 8–20 nm Au-Fe3O4 NPs can also be visualized with a scanning confocal microscope at 594 nm, as shown in Fig. 13c. The signal detected is from Au NPs in the Au-Fe3O4 structure and reflects the typical morphology of epithelial cells. It can be seen that the signal is much stronger in the region of cell—cell contact due to the preferred binding between EGFR and EGFRA. The study finds that the Au NP-based optical probe is very stable and shows no signal loss after three days. This is extremely important for long-term NP tracking in cellular environments. The detection limit for the 8–20 nm dumb bell is 90 pM Au.
4.2. SPM NPs for drug delivery
SPM NPs described here are ideal platforms for drug delivery. On targeted delivery, SPM NPs have distinct advantages over the other polymer based delivery systems: (1) the pathway of the drug can be readily tracked in the biological systems through SPM NPs by MRI; (2) the drug-NPs can be guided or held in place by an external magnetic field; and (3) under an alternate magnetic field, the SPM NPs act as a heater and can trigger controlled drug release.
In the delivery study, therapeutic drugs are normally coupled to SPM NPs via a covalent bond. Hydrophobic drugs can also be adsorbed onto NP surface to be stored in the NP coating layer to preserve their activity.31 Ideally, the drug-NPs are introduced in the biological systems and concentrated in the targeted area by an active targeting. Drug release can proceed by simple diffusion or through enzymatic activity or the changes in physiological conditions such as pH or temperature.32 Here, we present two examples on pH controlled drug release from Fe3O4 NPs.
Methotrexate (MTX), a chemotherapeutic drug that can target many cancer cells whose surfaces are over expressed by folate receptors, can be conjugated with Fe3O4 NPs through an amide bond, as shown in Fig. 14.33 In the conjugation process, the NPs are first modified with (3-aminopropyl)-trimethoxysilane and subsequently conjugated with MTX through amidation between the carboxylic acid end groups on MTX and the amine groups on the particle surface. Drug release experiments show that MTX can be cleaved from the NPs under low pH conditions mimicking the intracellular conditions in the lysosome. Cellular viability studies in human breast cancer cells (MCF-7) and human cervical cancer cells (HeLa) further demonstrate that such chemical cleavage of MTX is very effective inside the target cells through the action of intracellular enzymes. The intracellular trafficking of NP-MTX can be monitored through MRI. The results show that the MTX-Fe3O4 NPs target cells with folate receptors and inhibit their growth.
Fig. 14.
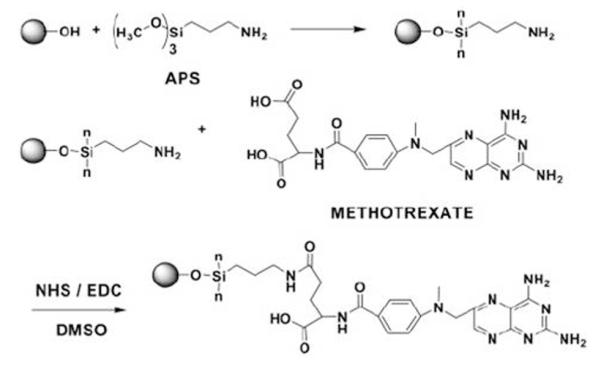
Surface modification of Fe3O4 NPs with MTX. Reproduced with permission from ref. 33.
Chromones are a group of naturally occurring compounds containing core structure of benzopyranone and have shown to be antifungal, antiviral, antihypertensive, and anticancer agents.34 However, their low solubility and short blood circulation time limit their usage for efficient therapeutic applications. Recently, 6-hydroxy-chromone-carbaldehyde was coupled to Fe3O4 NPs via a Schiff-base bond, as shown in Fig. 15a.35 In this structure, chromone binds to Fe3O4 through PEG and dopamine. The conjugation enhanced the chromone solubility greatly from less than 2.5 for free chromone to 633 μg mL-1 for the conjugate.
Fig. 15.
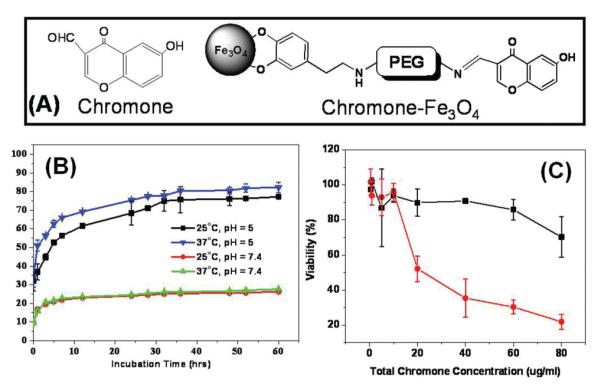
(a) Structure of chromone and the schematic illustration of the coupling between chromone and a Fe3O4 NP. (b) The percentage of the chromone released with the incubation time under different pH’s and temperatures in PBS + 10% FBS. (c) Viability of HeLa cells in the presence of the same chromone amount for free chromone (black curve) and chromone-Fe3O4 NPs (red curve). Adapted from ref. 35.
The Schiff-base bond is biodegradable via hydrolysis and the process can be accelerated at low pH conditions.36 Fig. 15b shows the percentage of chromone released from chromone-Fe3O4 NPs at different pH’s and temperatures. It can be seen that few chromone is detached from chromone-Fe3O4 NPs in the PBS with pH = 7.4 at both 25 °C and 37 °C culture conditions. However, in the PBS of pH = 5, the conjugate exhibits a drastic increase in free chromone and the higher incubation temperature (37 °C) results in higher chromone concentration in the solution.
The increased solubility of chromone present in chromone-Fe3O4 NPs leads to the enhanced uptake of chromone-Fe3O4 NPs by HeLa cells and this enhanced uptake results in high toxicity to HeLa cells. Fig. 15c is the HeLa cell viability data in the presence of the same amount of chromone added to the cell culture medium. It can be seen that chromone-Fe3O4 NPs are highly toxic with majority of the cells destroyed at ∼40 μg chromone per mL. This high toxicity of chromone-Fe3O4 NPs arises from their enhanced uptake by the HeLa cells and the controlled release of free chromone in the low pH cellular environment. The IC50, the half maximal (50%) inhibitory concentration (IC), of chromone-Fe3O4 NPs can be extracted from Fig. 15c to be ∼21 μg mL-1.
5. Conclusions
Recent research progress have indicated that monodisperse SPM NPs can be readily synthesized and functionalized for biomedical applications. First, the monodipserse NPs have been made with controlled magnetic properties and chemical stability. Second, the initial research shows that the SPM NPs can be made stable in biological environment to escape from RES. Third, various targeting agents, especially tumour specific antibodies, have been coupled to SPM NPs for testing their specific targeting in biological environments. Fourth, SPM NPs functionalized with targeting agents are excellent contrast agent for MRI and the contrast effects can be optimized by controlling NP size, structure and coating thickness. Fifth, therapeutic drugs can be coupled to the NP surface with enhanced stability and solubility in biological systems and can be released in a pH controlled manner. All these experimental results reveal that it is now possible to explore fully the great potential of monodisperse SPM NPs for early medical diagnostic and therapeutic applications. With effective targeting agent and highly sensitive NP probes, one is able to study in detail site-specific targeting, cell uptake and NP-drug pathway within biological systems. With the uniform control in NP physical and chemical properties, one can also study in a more precise way the biodistribution and bioelimination of the drug-NP conjugates in biological circulations. This understanding will finally allow the therapeutic and toxic effects of various drugs to be carefully evaluated. In a word, SPM NPs are ideal platforms for future success in diagnostic medicine and therapy.
Footnotes
Work at Brown University was supported by NIH/NCI 1R21CA12859.
References
- 1(a).Sun SH. Adv. Mater. 2006;18:393. [Google Scholar]; (b) Xu CJ, Sun SH. Polym. Int. 2007;56:821. [Google Scholar]; (c) Jun YW, Seo JW, Cheon JW. Acc. Chem. Res. 2008;41:179. doi: 10.1021/ar700121f. [DOI] [PubMed] [Google Scholar]
- 2(a).Gupta AK, Gupta M. Biomaterials. 2005;26:3995. doi: 10.1016/j.biomaterials.2004.10.012. [DOI] [PubMed] [Google Scholar]; (b) Sun C, Lee JSH, Zhang MQ. Adv. Drug Deliv. Rev. 2008;60:1252. doi: 10.1016/j.addr.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Weissleder R, Pittet MJ. Nature. 2008;452:580. doi: 10.1038/nature06917. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jun YW, Lee JH, Cheon JW. Angew. Chem., Int. Ed. 2008;47:5122. doi: 10.1002/anie.200701674. [DOI] [PubMed] [Google Scholar]
- 3(a).Pankhurst QA, Connolly J, Jones SK, Dobson J. J. Phys. D: Appl. Phys. 2003;36:R167. [Google Scholar]; (b) Mornet S, Vasseur S, Grasset F, Duguet E. J. Mater. Chem. 2004;14:2161. [Google Scholar]; (c) Corot C, Robert P, Idee JM, Port M. Adv. Drug Deliv. Rev. 2006;58:1471. doi: 10.1016/j.addr.2006.09.013. [DOI] [PubMed] [Google Scholar]; (d) Ito A, Shinkai M, Honda H, Kobayashi T. J. Biosci. Bioeng. 2005;100:1. doi: 10.1263/jbb.100.1. [DOI] [PubMed] [Google Scholar]
- 4(a).Oberdorster G, Oberdorster E, Oberdorster J. Environ. Health Perspect. 2005;11:823. doi: 10.1289/ehp.7339. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kong G, Braun RD, Dewhirst MW. Cancer Res. 2000;60:4440. [PubMed] [Google Scholar]; (c) Ishida O, Maruyama K, Sasaki K, Iwatsuru M. Int. J. Pharm. 1999;190:49. doi: 10.1016/s0378-5173(99)00256-2. [DOI] [PubMed] [Google Scholar]; (d) Hu Y, Xie J, Tong YW, Wang CH. J. Contr. Release. 2007;118:7. doi: 10.1016/j.jconrel.2006.11.028. [DOI] [PubMed] [Google Scholar]; (e) Fang C, Shi B, Pei YY, Hong MH, Wu J, Chen HZ. Eur. J. Pharmaceut. Sci. 2006;27:27. doi: 10.1016/j.ejps.2005.08.002. [DOI] [PubMed] [Google Scholar]; (f) Xie J, Xu CJ, Kohler N, Hou YL, Sun SH. Adv. Mater. 2007;19:3163. [Google Scholar]
- 5(a).Okon E, Pouliquen D, Okon P, Kovaleva ZV, Stepanova TP, Lavit SG, Kudryavtsev BN, Jallet P. Lab. Invest. 1994;71:895. [PubMed] [Google Scholar]; (b) Bellin MF, Roy C, Kinkel K, Thoumas D, Zaim S, Vanel D, Tuchmann C, Richard F, Jacqmin D, Delcourt A, Challier E, Lebret T, Cluzel P. Radiology. 1998;207:799. doi: 10.1148/radiology.207.3.9609907. [DOI] [PubMed] [Google Scholar]
- 6(a).Cushing BL, Kolesnichenko VL, O’Connor CJ. Chem. Rev. 2004;104:3893. doi: 10.1021/cr030027b. [DOI] [PubMed] [Google Scholar]; (b) Lu AH, Salabas EL, Schuth F. Angew. Chem., Int. Ed. 2007;46:1222. doi: 10.1002/anie.200602866. [DOI] [PubMed] [Google Scholar]; (c) Latham AH, Williams ME. Acc. Chem. Res. 2008;41:411. doi: 10.1021/ar700183b. [DOI] [PubMed] [Google Scholar]
- 7(a).Sun SH, Zeng H, Robinson DB, Raoux S, Rice PM, Wang SX, Li G. J. Am. Chem. Soc. 2004;126:273. doi: 10.1021/ja0380852. [DOI] [PubMed] [Google Scholar]; (b) Zeng H, Rice PM, Wang SX, Sun SH. J. Am. Chem. Soc. 2004;126:11458. doi: 10.1021/ja045911d. [DOI] [PubMed] [Google Scholar]; (c) Song Q, Zhang ZJ. J. Am. Chem. Soc. 2004;126:6164. doi: 10.1021/ja049931r. [DOI] [PubMed] [Google Scholar]; (d) Lee JH, Huh YM, Jun YW, Seo JW, Jang JT, Song HT, Kim SJ, Cho EJ, Yoon HG, Suh JS, Cheon JW. Nat. Med. 2006;13:95. doi: 10.1038/nm1467. [DOI] [PubMed] [Google Scholar]
- 8.Peng S, Wang C, Xie J, Sun SH. J. Am. Chem. Soc. 2006;128:10676. doi: 10.1021/ja063969h. [DOI] [PubMed] [Google Scholar]
- 9.Desvaux C, Amiens C, Fejes P, Renaud P, Respaud M, Lecante P, Snoeck E, Chaudret B. Nat. Mater. 2005;4:750. doi: 10.1038/nmat1480. [DOI] [PubMed] [Google Scholar]
- 10(a).Gu H, Yang Z, Gao J, Chang CK, Xu B. J. Am. Chem. Soc. 2005;127:34. doi: 10.1021/ja045220h. [DOI] [PubMed] [Google Scholar]; (b) Yu H, Chen M, Rice PM, Wang SX, White RL, Sun SH. Nano Lett. 2005;5:379. doi: 10.1021/nl047955q. [DOI] [PubMed] [Google Scholar]
- 11(a).Kang YS, Risbud S, Rabolt JF, Stroeve P. Chem. Mater. 1996;8:2209. [Google Scholar]; (b) Hong CY, Jang IJ, Horng HE, Hsu CJ, Yao YD, Yang HC. J. Appl. Phys. 1997;81:4275. [Google Scholar]; (c) Fried T, Shemer G, Markovich G. Adv. Mater. 2001;13:1158. [Google Scholar]; (d) Zhang ZJ, Wang ZL, Chakoumakos BC, Yin JS. J. Am. Chem. Soc. 1998;120:1800. [Google Scholar]; (e) Neveu S, Bec A, Robineau M, Talbol D. J. Colloid Interface Sci. 2002;255:293. doi: 10.1006/jcis.2002.8679. [DOI] [PubMed] [Google Scholar]
- 12(a).Pileni MP, Moumen N. J. Phys. Chem. B. 1996;100:1867. [Google Scholar]; (b) Liu C, Zou B, Rondinone AJ, Zhang ZJ. J. Phys. Chem. B. 2000;104:1141. [Google Scholar]
- 13(a).Hyeon T, Lee SS, Park J, Chung Y, Na HB. J. Am. Chem. Soc. 2001;123:12798. doi: 10.1021/ja016812s. [DOI] [PubMed] [Google Scholar]; (b) Guo Q, Teng X, Rahman S, Yang H. J. Am. Chem. Soc. 2003;125:630. doi: 10.1021/ja0275764. [DOI] [PubMed] [Google Scholar]; (c) Casula MF, Jun YW, Zaziksi DJ, Chan EM, Corrias A, Alivisatos AP. J. Am. Chem. Soc. 2006;128:1675. doi: 10.1021/ja056139x. [DOI] [PubMed] [Google Scholar]
- 14.Sun SH, Zeng H. J. Am. Chem. Soc. 2002;124:8204. doi: 10.1021/ja026501x. [DOI] [PubMed] [Google Scholar]
- 15.Hou Y, Xu Z, Sun SH. Angew. Chem., Int. Ed. 2007;46:6329. doi: 10.1002/anie.200701694. [DOI] [PubMed] [Google Scholar]
- 16(a).Hyeon T. Chem. Commun. 2003;8:927. doi: 10.1039/b207789b. [DOI] [PubMed] [Google Scholar]; (b) Park J, An KJ, Hwang YS, Park JG, Noh HJ, Kim JY, Park JH, Hwang NM, Hyeon T. Nat. Mater. 2004;3:891. doi: 10.1038/nmat1251. [DOI] [PubMed] [Google Scholar]; (c) Park J, Lee E, Hwang NM, Kang MS, Kim SC, Hwang Y, Park JG, Noh HJ, Kini JY, Park JH, Hyeon T. Angew. Chem., Int. Ed. 2005;44:2872. doi: 10.1002/anie.200461665. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, Zhuang J, Peng Q, Li Y. Nature. 2005;437:121. doi: 10.1038/nature03968. [DOI] [PubMed] [Google Scholar]
- 18.Xie J, Chen K, Lee HY, Xu CJ, Hsu AR, Peng S, Chen X, Sun SH. J. Am. Chem. Soc. 2008;130:7542. doi: 10.1021/ja802003h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19(a).Dumestre F, Chaudret B, Amiens C, Renaud P, Fejes P. Science. 2004;303:821. doi: 10.1126/science.1092641. [DOI] [PubMed] [Google Scholar]; (b) Huber DL. Small. 2005;1:482. doi: 10.1002/smll.200500006. [DOI] [PubMed] [Google Scholar]
- 20.Chaubey GS, Barcena C, Poudyal N, Rong CB, Gao JM, Sun SH, Liu JP. J. Am. Chem. Soc. 2007;129:7214. doi: 10.1021/ja0708969. [DOI] [PubMed] [Google Scholar]
- 21.Seo WS, Lee JJ, Sun XM, Suzuki Y, Mann D, Liu Z, Terashima M, Yang PC, McConnell MV, Nishimura DG, Dai H. Nat. Mater. 2006;5:971. doi: 10.1038/nmat1775. [DOI] [PubMed] [Google Scholar]
- 22.Xu Z, Hou Y, Sun SH. J. Am. Chem. Soc. 2007;129:8698. doi: 10.1021/ja073057v. [DOI] [PubMed] [Google Scholar]
- 23(a).Yang J, Gunn J, Dave SR, Zhang MQ, Wang YA, Gao XH. Analyst. 2008;133:154. doi: 10.1039/b700091j. [DOI] [PubMed] [Google Scholar]; (b) Gupta AK, Naregalkar RR, Vaidya VD, Gupta M. Nanomedicine. 2007;2:23. doi: 10.2217/17435889.2.1.23. [DOI] [PubMed] [Google Scholar]
- 24(a).Xie J, Peng S, Brower N, Pourmand N, Wang SX, Sun SH. Pure Appl. Chem. 2006;78:1003. [Google Scholar]; (b) Robinson DB, Persson HHJ, Zeng H, Li G, Pourmand N, Sun SH, Wang SX. Langmuir. 2005;21:3096. doi: 10.1021/la047206o. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Grancharov SG, Zeng H, Sun SH, Wang SX, O’Brien S, Murray CB, Kirtley JR, Held GA. J. Phys. Chem. B. 2005;109:13030. doi: 10.1021/jp051098c. [DOI] [PubMed] [Google Scholar]; (d) Dubertret B, Skourides P, Norris DJ, Noireaux V, Brivanlou AH, Libchaber A. Science. 2002;298:1759. doi: 10.1126/science.1077194. [DOI] [PubMed] [Google Scholar]
- 25(a).Jun YW, Huh YM, Choi JS, Lee JH, Song HT, Kim S, Yoon S, Kim KS, Shin JS, Suh JS, Cheon J. J. Am. Chem. Soc. 2005;127:5732. doi: 10.1021/ja0422155. [DOI] [PubMed] [Google Scholar]; (b) Gu H, Xu K, Xu CJ, Xu B. Chem. Commun. 2006:941. doi: 10.1039/b514130c. [DOI] [PubMed] [Google Scholar]
- 26(a).Lacava LM, Lacava ZG, Da Silva MF, Silva O, Chaves SB, Azevedo RB, Pelegrini F, Gansau C, Buske N, Sabolovic D, Morais PC. Biophys. J. 2001;80:2483. doi: 10.1016/S0006-3495(01)76217-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Strable E, Bulte JWM, Moskowitz B, Vivekanandan K, Allen M, Douglas T. Chem. Mater. 2001;13:2201. [Google Scholar]; (c) Kohler N, Sun C, Fichtenholtz A, Gunn J, Fang C, Zhang MQ. Small. 2006;2:785. doi: 10.1002/smll.200600009. [DOI] [PubMed] [Google Scholar]; (d) Quaglia F, Ostacolo L, De Rosa G, La Rotonda MI, Ammendola M, Nese G, Maglio G, Palumbo R, Vauthier C. Int. J. Pharm. 2006;324:56. doi: 10.1016/j.ijpharm.2006.07.020. [DOI] [PubMed] [Google Scholar]; (e) Moghimi SM, Hunter AC, Murray JC. Pharm. Rev. 2001;53:283. [PubMed] [Google Scholar]
- 27.Brigger I, Dubernet C, Couvreur P. Adv. Drug Deliv. Rev. 2002;54:631. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]
- 28(a).Huh YM, Jun YW, Song HT, Kim S, Choi JS, Lee JH, Yoon S, Kim KS, Shin JS, Suh JS, Cheon J. J. Am. Chem. Soc. 2005;127:12387. doi: 10.1021/ja052337c. [DOI] [PubMed] [Google Scholar]; (b) Yarden Y, Sliwkowski MX. Nat. Rev. Mol. Cell Biol. 2001;2:127. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 29.Xu CJ, Xie J, Ho D, Wang C, Kohler N, Walsh EG, Morgan JR, Chin YE, Sun SH. Angew. Chem., Int. Ed. 2008;47:173. doi: 10.1002/anie.200704392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30(a).Haigler H, Ash JF, Singer SJ, Cohen S. Proc. Natl. Acad. Sci. U. S. A. 1978;75:3317. doi: 10.1073/pnas.75.7.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kawamoto T, Sato JD, Le A, Polikoff J, Sato GH, Mendelsohn J. Proc. Natl. Acad. Sci. U. S. A. 1983;80:1337. doi: 10.1073/pnas.80.5.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jain TK, Moralles MA, Sahoo SK, Lesllie-Pellecky DL, Labhasetwar V. Mol. Pharm. 2005;2:194. doi: 10.1021/mp0500014. [DOI] [PubMed] [Google Scholar]
- 32(a).Langer RS. Nature. 1998;392:5. [Google Scholar]; (b) Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Chem. Rev. 1999;99:3181. doi: 10.1021/cr940351u. [DOI] [PubMed] [Google Scholar]
- 33.Kohler N, Sun C, Wang J, Zhang MQ. Langmuir. 2005;21:8858. doi: 10.1021/la0503451. [DOI] [PubMed] [Google Scholar]
- 34(a).Barve V, Ahmed F, Adsule S, Banerjee S, Kulkarni S, Katiyar P, Anson CE, Powell AK, Padhye S, Sarkar FH. J. Med. Chem. 2006;49:3800–3808. doi: 10.1021/jm051068y. [DOI] [PubMed] [Google Scholar]; (b) Pisco L, Kordian M, Peseke K, Feist H, Michalik D, Estrada E, Carvalho J, Hamilton G, Rando D, Quincoces J. Eur. J. Med. Chem. 2006;41:401–407. doi: 10.1016/j.ejmech.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 35.Wang B, Xu CJ, Xie J, Yang Z, Sun SH. J. Am. Chem. Soc. 2008;130:14436. doi: 10.1021/ja806519m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36(a).Kratz F, Beyer U, Schutte MT. Crit. Rev. Ther. Drug Carrier Syst. 1999;16:245–288. doi: 10.1615/critrevtherdrugcarriersyst.v16.i3.10. [DOI] [PubMed] [Google Scholar]; (b) Saito H, Hoffman AS, Ogawa HI. J. Bioact. Compat. Polym. 2007;22:589–601. [Google Scholar]