

Abstract
Matrix metalloproteinase inhibitors (MMPi) possessing a glucose protecting group on the zinc-binding group (ZBG) show a dramatic increase in inhibitory activity upon cleavage by β-glucosidase.
Matrix metalloproteinases (MMPs) are a ubiquitous class of zinc(II)-dependent hydrolytic enzymes that have been associated with a wide range of pathologies including cancer, arthritis, heart disease, and stroke.1–3 Clinical trials of matrix metalloproteinase inhibitors (MMPi) have frequently been hampered by the onset of musculoskeletal syndrome (MSS), which manifests as severe joint pain, and has been attributed to non-specific, systemic inhibition of MMPs and other metalloenzymes.4,5 One approach to eliminate or minimize MSS and other complications would be to develop prodrugs or ‘proinhibitors’ that are triggered in a localized fashion, hence restricting inhibitory activity both spatially and temporally. The prodrug concept has been applied to a variety of targets, triggered by stimuli such as light, changes in pH, and enzymatic activity.6,7
A common strategy in the development of prodrugs and proinhibitors is through the use of carbohydrate protecting groups. The development of glycoconjugate prodrugs offers the ability to increase water solubility, enhance targeting, and minimize the side effects associated with systemic delivery of cytotoxic drugs.8–11 The use of glycoside protecting groups, such as glucoside moieties, have been reported for use with targeted treatments such as ADEPT (antibody directed enzyme prodrug therapy) and PMT (prodrug monotherapy).12–14 The ADEPT approach, developed over 20 years ago, has been widely investigated as a means to deliver drugs specifically to cancerous tissue and has shown promise in both animal studies and clinical trials.15–17 Glucose-derivatized compounds can also enhance uptake in cancer cells18–20 and the brain.9,10,21
Metalloenzyme inhibitors such as MMPi are particularly suitable to the proinhibitor approach because such compounds generally employ a metal-binding moiety, which if blocked with a protecting group, abolishes inhibitory activity. Specifically, using glucose as a protecting group, a proinhibitor MMPi can be developed to release the active MMPi in the presence of β-glucosidase to restore MMP inhibitory activity. Surprisingly, metalloenzyme proinhibitors have not been widely investigated and very few proinhibitors have been designed for MMPs.22–29 Only one example of a glycoside MMP proinhibitor has been prepared; however, efforts to release the active MMPi through enzymatic cleavage proved unsuccessful.27 In this report, we show that an MMPi can be formulated and inactivated as a proinhibitor, deprotected and released by an enzymatic signal, and hence exhibit triggered control over inhibitory activity. The compounds described here allow for spatial and temporal control of MMP inhibition, making them potentially valuable as therapeutics or as chemical tools for studying the roles of MMPs in disease models and developmental biology.30,31
Building on the strategy of glycosidic protecting groups, we first focused our attention on the development of glucoseprotected non-hydroxamate zinc-binding groups (ZBGs)32,33 that can be activated by enzymatic cleavage of the protecting group with β-glucosidase to release the ZBG and glucose (Fig. 1). The synthesis of the protected ZBGs (2, 4, and 6) was accomplished following a literature procedure used by Orvig and coworkers for enzyme-activated metal-binding chelators (Scheme S1).9,34 The ZBG was protected with acetobromo-α-D-glucose in a 1: 1 solution of 1.0 M NaOH and CH2Cl2 in the presence of (nBu)4NBr. The desired products were obtained by cleavage of the glucose acetate groups using NaOMe in MeOH.
Fig. 1.

Structures of inhibitors and proinhibitors tested in this study. Compounds include the ZBGs 1-hydroxy-pyridin-2(1H)-one (1), 3-hydroxy-2-methyl-4-pyrone (3) and 3-hydroxy-2-methyl-4-pyrothione (5) as well as the full-length MMPi 1,2-HOPO-2 (7).
To evaluate the ability of these compounds to be enzymatically activated, cleavage of the protected ZBGs in the presence of β-glucosidase (from almond extract, Fluka) was followed using electronic spectroscopy. To a solution of the protected ZBG in HEPES buffer was added β-glucosidase, and the change in absorbance was monitored over time. As can be seen in Fig. 2 for compound 2, the absorbance over time shows a decrease at 292 nm while a band at 312 nm emerges, indicative of the deprotected ZBG 1-hydroxy-2-pyridin- 2(1H)-one (1). Similar spectra were observed for the hydroxypyrone derivatives 4 and 6 (Fig. S1, S2). In addition, cleavage of the protected ZBGs was confirmed by HPLC analysis (Fig. S3–S5). These studies demonstrate that in aqueous buffer at room temperature, the glucose-protected ZBGs can be readily activated in the presence of β-glucosidase providing compelling evidence that hydroxypyridinone and hydroxypyrone ZBGs are well suited for the development of enzymeactivated MMP proinhibitors.
Fig. 2.

Absorption spectra of the glucose-protected ZBG 2 (0.05 mM, HEPES buffer, pH = 7.5) in the presence of β-glucosidase (16 U) monitored over time. The heavy lines are the initial (dashed) and final (solid) spectra and arrows indicate the change in spectra over time. The spectrum of an authentic sample of ZBG 1 (dotted) is also shown.
Having demonstrated the use of glucose as an effective protecting group for the aforementioned ZBGs, we aimed to incorporate a glucose protecting group into a full-length MMPi to develop an MMP proinhibitor. We selected the full-length inhibitor 1,2-HOPO-2 (7), a potent non-hydroxamate inhibitor of MMPs that uses a 1-hydroxy-2-pyridin-2(1H)-one (1) ZBG.35 Synthesis of the MMP proinhibitor was achieved by addition of acetobromo-α-D-glucose and Cs2CO3 to 7 in DMF at room temperature to give 8a in high yields (>80%, Scheme 1). These reaction conditions were a vast improvement in yield over the aqueous reaction conditions used to protect the ZBGs. Surprisingly, these high-yield reaction conditions did not produce the desired products with the ZBGs (1, 3, and 5). The final proinhibitor (8) was obtained by deprotection of the glucose acetate groups with NaOMe in MeOH at 0 °C for one hour.
Scheme 1.

Synthesis of the glucose-protected MMPi 8.
The MMP proinhibitor 8 was first evaluated for activation by β-glucosidase using electronic spectroscopy and HPLC analysis (Fig. S6, S7). Results from these studies indicate that while cleavage of 8 to produce the active MMPi 1,2-HOPO-2 (7) goes to completion, the kinetics of the reaction are noticeably slower than that observed for the protected ZBGs. Complete conversion of 8 to 7 required ~4 h at 37 °C; a Km value of 210 μM was determined (Fig. S8). Notably, compound 8 was not cleaved under acidic conditions (0.1 M HCl) over 24 h (Fig. S9). Overall, this is the first example of a glucosidase proinhibitor that can be enzymatically cleaved to yield the active MMPi.
The ability of the protected compounds to inhibit MMP-9 (gelatinase-B) in the presence of β-glucosidase was evaluated using a fluorescence-based assay.36 Compounds 1–8 were evaluated at a concentration close to their reported IC50 values in the presence and absence of β-glucosidase (Fig. 3). The percent inhibition of MMP-9 with the ZBGs (1, 3, and 5) is close to 50% when tested with and without β-glucosidase indicating that the presence of low concentrations of β-glucosidase in the assay has little effect on MMP inhibition. The protected ZBGs (2, 4, and 6) show attenuated inhibition when evaluated without the activating enzyme and complete restoration of inhibition when exposed to β-glucosidase. Compounds 2, 4, and 6 do show some inhibition of MMP-9 (Table 1), which is likely due to non-specific binding at the high concentrations of the ZBGs (0.125–4 mM) used in these experiments.
Fig. 3.
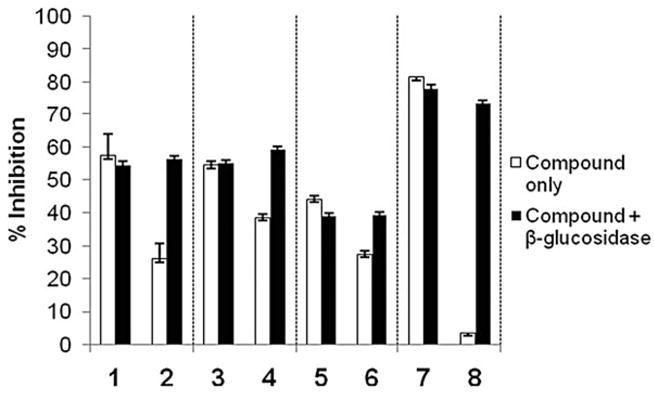
Percent inhibition of MMP-9 with compounds 1–8 tested at 1mMfor 1 and 2, 4mMfor 3 and 4, 125 μM for 5 and 6, and 16 μM for 7 and 8, in the absence (white) and presence (black) of β-glucosidase.
Table 1.
IC50 values of inhibitors and protected inhibitors against MMP-9 and MMP-8 as measured using fluorescence based assay
| Inhibitor | IC50 | Proinhibitor | IC50 | QIC50a | Enzyme |
|---|---|---|---|---|---|
| 1 | 1.02 mM (± 0.03) | 2 | 3.99 mM (± 0.04) | 3.9 | MMP-9 |
| 3 | 4.03 mM (± 0.05) | 4 | 5.7 mM (± 0.3) | 1.4 | MMP-9 |
| 5 | 138 μM (± 5) | 6 | 302 μM (± 5) | 2.2 | MMP-9 |
| 7 | 2.7 μM (± 0.2) | 8 | 587 μM (± 7) | 202 | MMP-9 |
| 7 | 0.075 μM (± 0.005) | 8 | 84 μM (± 1) | 1120 | MMP-8 |
QIC50 = quotient IC50 = IC50 proinhibitor/IC50 inhibitor.
In the absence of β-glucosidase, the complete MMPi 8 (16 μM) displays very little inhibition of MMP-9, but upon activation, MMP-9 activity is inhibited by 73%, which is essentially identical to an authentic sample of inhibitor 7. Even greater potency was observed against MMP-8, where MMPi 8 could be activated with β-glucosidase to obtain 33% inhibition at only 150 nM(Fig. S10), representing a > 1000-fold increase in activity upon enzymatic activation (Table 1). The factor of 1000 difference in the quotient IC50 (IC50 value of proinhibitor in the presence and absence of enzyme) has been reported as an optimal value for the ADEPT approach to targeted therapy,11 and hence MMPi 8 exceeds this threshold with MMP-8. The higher activity against MMP-8 is consistent with the potency of the cleavage product (7) against this metalloenzyme.35 The results for 8 show that incorporation of a ZBG protection strategy into an MMPi gives near complete abolition and recovery of inhibitory activity with these enzyme inhibitors.
In summary, we have demonstrated that it is possible to passivate an MMPi as a proinhibitor, activate it through an enzymatic reaction, and inhibit MMPs in a controlled manner. We have shown that protection of the metal-binding moiety of MMPi can be achieved and enzymatically removed to release an active, intact MMPi. This general strategy of protecting the ZBG with a cleavable group is expected to permit the development of a variety of MMP proinhibitors that respond to a range of stimuli. The ability to selectively utilize MMPi with different chemical and biological triggers will provide a new level of spatial and temporal control over MMP inhibition, which can be useful for both biological studies and therapeutic approaches.
Supplementary Material
Acknowledgments
We thank Dr Y. Su for help with HPLC analysis. This work was supported by the National Institutes of Health (R01 HL00049-01) and the American Heart Association (0970028N). J.L.M.J. is supported by a National Institutes of Health Training Grant (5 T32 HL007444-27).
Footnotes
Electronic supplementary information (ESI) available: Synthetic details, characterization of all compounds, and details of cleavage assays. Scheme S1, Fig. S1–S10. see DOI: 10.1039/b923302d
Notes and references
- 1.Coussens LM, Fingleton B, Matrisian LM. Science. 2002;295:2387–2392. doi: 10.1126/science.1067100. [DOI] [PubMed] [Google Scholar]
- 2.Whittaker M, Floyd CD, Brown P, Gearing AJH. Chem Rev. 1999;99:2735–2776. doi: 10.1021/cr0100345. [DOI] [PubMed] [Google Scholar]
- 3.Overall CM, Lüpez-Otín C. Nat Rev Cancer. 2002;2:657–672. doi: 10.1038/nrc884. [DOI] [PubMed] [Google Scholar]
- 4.Renkiewicz R, Qiu L, Lesch C, Sun X, Devalaraja R, Cody T, Kaldjian E, Welgus H, Baragi V. Arthritis Rheum. 2003;48:1742–1749. doi: 10.1002/art.11030. [DOI] [PubMed] [Google Scholar]
- 5.Fingleton B. Semin Cell Dev Biol. 2008;19:61–68. doi: 10.1016/j.semcdb.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denny WA. Cancer Invest. 2004;22:604–619. doi: 10.1081/cnv-200027148. [DOI] [PubMed] [Google Scholar]
- 7.Fry FH, Jacob C. Curr Pharm Des. 2006;12:4479–4499. doi: 10.2174/138161206779010512. [DOI] [PubMed] [Google Scholar]
- 8.Houston TA. Curr Drug Delivery. 2007;4:264–268. doi: 10.2174/156720107782151278. [DOI] [PubMed] [Google Scholar]
- 9.Schugar H, Green DE, Bowen ML, Scott LE, Storr T, Bohmerle K, Thomas F, Allen DD, Lockman PR, Merkel M, Thompson KH, Orvig C. Angew Chem, Int Ed. 2007;46:1716–1718. doi: 10.1002/anie.200603866. [DOI] [PubMed] [Google Scholar]
- 10.Storr T, Merkel M, Song-Zhao GX, Scott LE, Green DE, Bowen ML, Thompson KH, Patrick BO, Schugar HJ, Orvig C. J Am Chem Soc. 2007;129:7453–7463. doi: 10.1021/ja068965r. [DOI] [PubMed] [Google Scholar]
- 11.Tietze LF, Feuerstein T. Aust J Chem. 2003;56:841–854. [Google Scholar]
- 12.Wang S, Liu D, Zhang X, Li S, Sun Y, Li J, Zhou Y, Zhang L. Carbohydr Res. 2007;342:1254–1260. doi: 10.1016/j.carres.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 13.Syrigos KN, Rowlinson-Busza G, Epenetos AA. Int J Cancer. 1998;78:712–719. doi: 10.1002/(sici)1097-0215(19981209)78:6<712::aid-ijc8>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 14.Tietze LF, Schuster HJ, Krewer B, Schuberth I. J Med Chem. 2009;52:537–543. doi: 10.1021/jm8009102. [DOI] [PubMed] [Google Scholar]
- 15.Niculescu-Duvaz I, Springer CJ. Expert Opin Invest Drugs. 1996;3:289–308. doi: 10.1517/13543784.6.6.685. [DOI] [PubMed] [Google Scholar]
- 16.Bagshawe KD. Curr Drug Targets. 2009;10:152–157. doi: 10.2174/138945009787354520. [DOI] [PubMed] [Google Scholar]
- 17.Bagshawe KD, Begent RHJ. Adv Drug Delivery Rev. 1996;22:365–367. [Google Scholar]
- 18.Tian YS, Lee HY, Lim CS, Park J, Kim HM, Shin YN, Kim ES, Jeon HJ, Park SB, Cho BR. Angew Chem, Int Ed. 2009;48:8027–8031. doi: 10.1002/anie.200901175. [DOI] [PubMed] [Google Scholar]
- 19.Gillies RJ, Robey I, Gatenby RA. J Nucl Med. 2008;49:24S–42S. doi: 10.2967/jnumed.107.047258. [DOI] [PubMed] [Google Scholar]
- 20.Lin YS, Tungpradit R, Sinchaikul S, An FM, Liu DZ, Phutrakul S, Chen ST. J Med Chem. 2008;51:7428–7441. doi: 10.1021/jm8006257. [DOI] [PubMed] [Google Scholar]
- 21.Fernández C, Nieto O, Rivas E, Montenegro G, Fontenla JA, Fernandez-Mayoralas A. Carbohydr Res. 2000;327:353–365. doi: 10.1016/s0008-6215(00)00073-2. [DOI] [PubMed] [Google Scholar]
- 22.De Simone G, Vitale RM, Di Fiore A, Pedone C, Scozzafava A, Montero JL, Winum JY, Supuran CT. J Med Chem. 2006;49:5544–5551. doi: 10.1021/jm060531j. [DOI] [PubMed] [Google Scholar]
- 23.Failes TW, Cullinane C, Diakos CI, Yamamoto N, Lyons JG, Hambley TW. Chem–Eur J. 2007;13:2974–2982. doi: 10.1002/chem.200601137. [DOI] [PubMed] [Google Scholar]
- 24.Failes TW, Hambley TW. J Inorg Biochem. 2007;101:396–403. doi: 10.1016/j.jinorgbio.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 25.Suzuki T, Hisakawa S, Itoh Y, Suzuki N, Takahashi K, Kawahata M, Yamaguchi K, Nakagawa H, Miyata N. Bioorg Med Chem Lett. 2007;17:4208–4212. doi: 10.1016/j.bmcl.2007.05.040. [DOI] [PubMed] [Google Scholar]
- 26.Bowers A, West N, Taunton J, Schreiber SL, Bradner JE, Williams RM. J Am Chem Soc. 2008;130:11219–11222. doi: 10.1021/ja8033763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitchell MB, Whitcombe IWA. Tetrahedron Lett. 2000;41:8829–8834. [Google Scholar]
- 28.Thomas M, Rivault F, Tranoy-Opalinski I, Roche J, Gesson JP, Papot S. Bioorg Med Chem Lett. 2007;17:983–986. doi: 10.1016/j.bmcl.2006.11.042. [DOI] [PubMed] [Google Scholar]
- 29.Thomas M, Clarhaut J, Tranoy-Opalinski I, Gesson JP, Roche J, Papot S. Bioorg Med Chem. 2008;16:8109–8116. doi: 10.1016/j.bmc.2008.07.048. [DOI] [PubMed] [Google Scholar]
- 30.Liu K, Wahlberg P, Leonardsson G, Hägglund AC, Ny A, Bodén I, Wibom C, Lund LR, Ny T. Dev Biol. 2006;295:615–622. doi: 10.1016/j.ydbio.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 31.Gill SE, Pape C, Leco KJ. Dev Biol. 2006;298:540–554. doi: 10.1016/j.ydbio.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 32.Puerta DT, Lewis JA, Cohen SM. J Am Chem Soc. 2004;126:8388–8389. doi: 10.1021/ja0485513. [DOI] [PubMed] [Google Scholar]
- 33.Puerta DT, Griffn MO, Lewis JA, Romero-Perez D, Garcia R, Villarreal FJ, Cohen SM. J Biol Inorg Chem. 2006;11:131–138. doi: 10.1007/s00775-005-0053-x. [DOI] [PubMed] [Google Scholar]
- 34.Scott LE, Page BDG, Patrick BO, Orvig C. Dalton Trans. 2008:6364–6367. doi: 10.1039/b815404j. [DOI] [PubMed] [Google Scholar]
- 35.Agrawal A, Romero-Perez D, Jacobsen JA, Villarreal FJ, Cohen SM. ChemMedChem. 2008;3:812–820. doi: 10.1002/cmdc.200700290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knight CG, Willenbrock F, Murphy G. FEBS Lett. 1992;296:263–266. doi: 10.1016/0014-5793(92)80300-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.