

Abstract
The synthesis and characterization of TiO2-based core-shell nanoparticles as surface-enhanced Raman Scattering (SERS) tags are reported. A hydrolysis approach is first used to generate colloidal TiO2 nanoparticles, which are subsequently tagged with Raman probe molecules and encapsulated within a gold nanoshell. The resulting core-shell nanoparticles are characterized by using a number of techniques including UV-visible spectroscopy, transmission electron microscopy, and energy-dispersive X-ray spectroscopy (EDX) to confirm the successful coating of the Au shells. These core-shell nanoparticles exhibit very strong and reproducible SERS signals of the Raman probe molecules. Three different types of Raman probe molecules are used to prepare different SERS-active nanoparticles (SERS-tags), which demonstrates the versatility of the design. Such TiO2-based metal-coated core-shell nanoparticles will be useful as SERS-tags in biological assay and imaging applications. They may also provide a platform for fundamental studies in the ongoing investigations on the mechanisms of SERS.
1. Introduction
Surface-enhanced Raman scattering (SERS) has been known, intensely studied and applied for over three decades.1–5 In recent years, SERS has been transformed into a powerful microanalytical technique with several significant advantages for ultrasensitive chemical analysis and interfacial studies,6–10 due to the advances in nanofabrication combined with an increasing understanding of the plasmonic properties of nanomaterials.11–21 The highly enhanced Raman signals have allowed SERS to be used as a tool for single-molecule spectroscopy.22–27
From the application point of view, SERS is usually being exploited in two ways. Based on the molecular fingerprint information associated with the Raman spectroscopy, there have been ongoing efforts to develop highly efficient SERS substrates, which can be used for detections of targets of interest, such as, biological molecules, pollutants, explosives and bacteria, among others.28–31 Alternatively, SERS-active nanoparticles (SERS-tags) are developed for detection in bioassays and labeling in bioimaging.32,33 Typically, Raman probe molecules are placed on the surface of an Ag or Au nanoparticle, which together are subsequently encapsulating within a silica or polymer shell. Such SERS-tags can be further functionalized with targeting elements, such as antibodies or oligonucleotides, which would selectively bind to the targets of interest. Due to the inherently narrow and fingerprint-specific spectra, SERS-tags could potentially offer a greater degree of multiplexing capability, as compared to the more widely used fluorescence-based tags.
The strongest SERS effects are usually observed in metal substrates, typically Ag and Au, that are very rough on a microscopic scale, such as electrode surfaces roughened by redox cycle(s), films deposited by evaporation or sputtering in vacuum onto cold supports, island films deposited on glass surfaces, aggregated colloids, single ellipsoidal nanoparticles, arrays of metal nanoparticles prepared by lithographic techniques, and metal nanoshells.34–38
Metal nanoshells, especially those with SiO2-cores, have been widely studied as SERS substrates for a decade.39,40 Most of the experimental studies on metal nanoshells consider the SERS effect in the proximal distance outside the shells. Very recently, we reported the observation of SERS signals of probe molecules residing inside a metal nanoshell.5 The probe molecules, typically some dyes, were first encapsulated inside the SiO2 nanoparticles through doping during the nanoparticle formation. The dye-doped SiO2 nanoparticles were subsequently coated with a thin layer of Au or Ag shell. The resulting core-shell nanostructures displayed highly enhanced Raman signals, with great potential to be used as SERS-tags for Raman-based assay and imaging. In practice, however, there appears to be not as many Raman probe molecules as desired that can be readily doped inside the SiO2 nanoparticles. This is probably due to the fact that the sol-gel method used to synthesize SiO2 nanoparticles in the reverse microemulsion tends to exclude the probe molecules which are either negative charged or oil-soluble from being encapsulated inside the SiO2 matrix. Thus it is worthwhile to explore other types of material as the core, which ideally can encapsulate a wider variety of Raman probes.
Molecules adsorbed chemically on the metal oxides have been intensively investigated over the years, especially in the field of dye-sensitized nanocrystalline TiO2 solar cells.41–44 It is well recognized that the surface of TiO2 particles in an aqueous environment may have different electric charges, depending on the pH value of the solution. The positive charge occurs at acidic conditions (pH < 5) due to the presence of the ≡TiOH2+ groups, where the zeta potentials are sufficiently high to form stable colloids. Nearly neutral charge at close to zero zeta potential takes place at around pH=5–7. At pH > 7, the TiO2 surface becomes negatively charged, owing to the ≡TiO- groups.45–47
In colloidal solution, the development of a net charge at the TiO2 nanoparticle surface affects the distribution of ions in the neighboring interfacial region, resulting in an increased concentration of counter ions close to the surface. By adjusting the pH of the colloidal solution, the surface charge of the colloidal TiO2 nanoparticles can be controlled, so that different Raman probe molecules can adsorb to the TiO2 nanoparticles depending on the net charge of these molecules.
Taking advantage of this surface property of the TiO2 nanoparticles, we adopt them as the core material to prepare the TiO2-core/metal-shell nanoparticles with Raman probes embedded inside. To demonstrate the versatility of the synthetic strategy and the diversity of the produced core-shell nanoparticles, we use three Raman probe molecules as examples, namely, Meso-tetra(4-carboxylphenyl) porphine (TCPP), tris(2,2′-bipyridyl) ruthenium(II) chloride (Ru(bpy)) and Rhodamine 6G (R6G). Results show that the resulting core-shell nanoparticles all exhibit very strong and reproducible SERS signals.
2. Experimental Section
Materials
Meso-tetra(4-carboxylphenyl) porphine (TCPP) was purchased from Frontier Scientific. Tris(2,2′-bipyridyl) ruthenium(II) chloride (Ru(bpy)), Rhodamine 6G (R6G), sodium citrate, sodium borohydride (NaBH4) and hydrogen tetrachloroaurate (HAuCl4, 99.99%) were obtained from Sigma-Aldrich. TiCl4 was from Fisher. Reagents and solvents were obtained commercially and used without further purification. Other chemicals, unless specified, were reagent grade. Deionized water, with a resistivity greater than 18.0 MΩ·cm (Millipore Milli-Q system), was used in preparing the aqueous solutions. All glassware used in the following procedures was cleaned with freshly prepared aqua regia and rinsed thoroughly in water prior to use.
Preparation of colloidal TiO2 nanoparticles
Colloidal stock solutions of TiO2 were prepared by the hydrolysis of TiCl4 as previously described.48,49 Briefly, 5 mL of 99.8% TiCl4 at 0 °C was introduced under a stream of nitrogen and vigorous stirring into 70 mL water/ice solution of 0.1 M HCl. After 30 min stirring at 0 °C, the solution was dialyzed against aqueous HCl of pH 2.3 using Spectrapor membrane tubing (Spectrum Medical Industries) with MW 6000–8000 cutoff pores. A transparent solution containing ca. 20 g/L TiO2 at pH 2.3 was obtained. The solution was kept in the refrigerator at 4 °C and used within 3 months.
Preparation of colloidal TCPP-TiO2 nanoparticles
TCPP-TiO2 nanoparticles were prepared by mixing 3.0 mg TCPP with 20.00 mL of 0.20 g/L colloidal TiO2 solution (pH=2.3) for 7–8 days under vigorous stirring. The mixture was kept in dark during the preparation in order to avoid photocatalytic decomposition of TCPP. The excessive TCPP molecules were then separated from aqueous phase by centrifugation. The prepared colloidal TCPP-TiO2 solutions are extremely stable at pH 1.0–3.3.
Preparation of colloidal Ru(bpy)-TiO2
20.00 mL of 0.20 g/L colloidal TiO2 solution (pH=2.3) was mixed with 2.00 mL of 1.0 mM Ru(bpy) solution under vigorous stirring. Then 0.10 M of NaOH was added into the mixture to adjust the pH value to 8. The excessive Ru(bpy) molecules were separated from aqueous phase after centrifugation.
Preparation of colloidal R6G-TiO2
20.00 mL of 0.20 g/L colloidal TiO2 solution (pH=2.3) was mixed with 2.00 mL of 1.0 mM R6G solution under vigorous stirring. Then 0.10 M of NaOH was used to adjust the pH value to 8. The excessive R6G molecules were removed from aqueous phase after centrifugation.
Preparation of Au nanoshells onto the surface of dye-embedded TiO2 nanostructures
To prepare Au-coated, TiO2-based core-shell nanoparticles, 0.20 mL of 25 mM sodium citrate solution was added to 19.8 ml of aqueous solution containing 2.5 mM HAuCl4 and 0.02 g/L dye-TiO2 under vigorous stirring for 5 minutes. Then 0.60 ml of freshly prepared 0.10 M NaBH4 in 25 mM sodium citrate solution was added to the above mixture under vigorous stirring at room temperature for another 10 minutes. The mixture turned to orange red color immediately. Lastly, after washing with DI water a few times, the mixture was kept in refrigerator at 4 °C until use.
Extinction spectra measurements
The extinction spectra were recorded by a Shimadzu 2550 Spectrophotometer (Shimadzu Corporation Japan) using a 1-cm path length quartz cell at room temperature.
TEM and EDX measurements
A drop of suspension containing dye-embedded TiO2-core Au-shell nanoparticles was deposited on a Formvar-covered carbon-coated copper grid (Electron Microscopy Sciences, PA) and let dried at room temperature. A JEOL 2010 high-resolution transmission electron microscope (HRTEM) was used to obtain the TEM images and EDX spectra at 200 kV.
Raman spectra measurements
The dye-embedded TiO2-core Au-shell nanoparticles were dispersed thoroughly in DI water by sonication. A drop of the diluted solution was cast onto a tilted, clear cover slip, and let dried. Raman measurements were carried out on a LabRAM Raman microscope (HORIBA Jobin Yvon Inc., NJ). Laser intensities at the samples were set at approximately 130 μW for the 632.8-nm HeNe laser for all measurements. Between different Raman sessions, the 520.7 cm−1 peak of a silicon wafer was used to calibrate the spectrograph and to normalize the measured intensity to compensate for possible fluctuation of the Raman system. Exposure time for all measurements was 1 sec. Each spectrum was the average of 10 scans.
3. Results and discussion
General strategy of nanoparticle synthesis
The molecular structures of the three dye molecules used as Raman probes are shown in Figure 1. TCPP is negatively charged, while the parts of Ru(bpy) and R6G containing the fluorophores are positively charged. The scheme of preparing the dye-embedded TiO2-core Au-shell is illustrated in Figure 2. Colloidal TiO2 nanoparticles are first formed by the hydrolysis of TiCl4. Then, depending on the type of Raman probe molecules to be adsorbed, appropriate pH values are set so that the surface charge of the TiO2 nanoparticles is opposite to the probes, either positively or negatively charged. Lastly, thin layer of Au shells are deposited on the nanoparticles. The final products are dye-embedded TiO2-core Au-shell nanoparticles. The scheme is shown to be versatile toward dyes with different charge properties. Note that, although the nanoparticles shown in this report are coated with Au shells, Ag shells also work out well, with highly enhanced and reproducible SERS signals under proper excitation.
Figure 1.
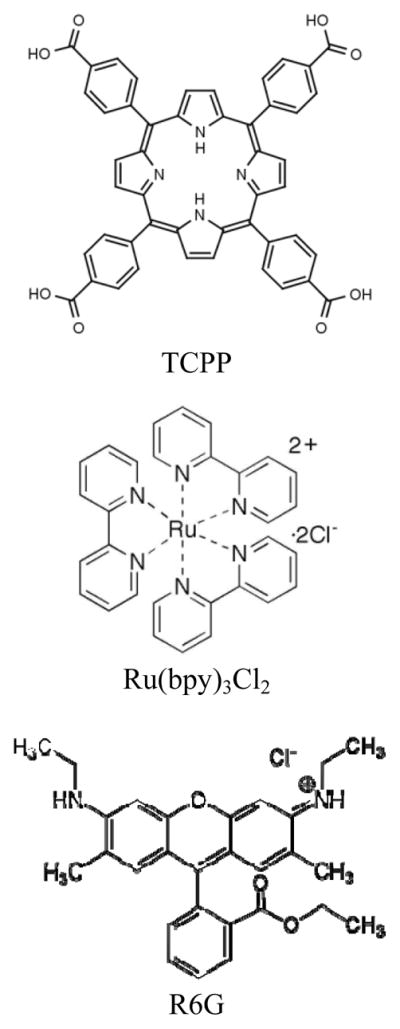
Molecular structure of TCPP, Ru(bpy)3Cl2 and R6G.
Figure 2.
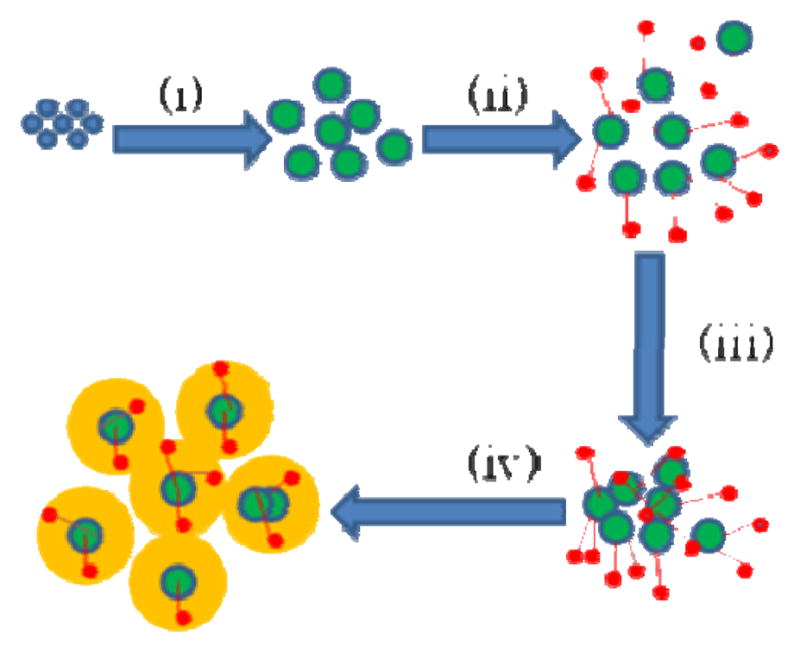
Preparation of Au-coated, TiO2-based core-shell nanoparticles: (i) hydrolysis of TiCl4 in 0.1 M HCl to form colloidal TiO2 solution, (ii) addition of Raman probes, (iii) controlled aggregation using NaOH, (iv) growth of Au shell using HAuCl4.
Determination of the concentration of colloidal TiO2 nanoparticles in solution
Colloidal TiO2 nanoparticles prepared by our method usually result in an average diameter of less than 3 nm.49 Molecular TiO2 concentration was determined by spectraphotometric measurements at 215 nm using molar extinction coefficient 6050 M−1cm−1.48 Using a TiO2 density of 4 g/cm3 and an average diameter of 3 nm, we can estimate the number of TiO2 nanoparticles in the reaction mixture.
Extinction spectra of dye-embedded TiO2-core Au-shell nanoparticles
Figure 3 shows the extinction spectra of the TiO2, TCPP, Ru(bpy), R6G, Au and various combinations. The spectra indicate the adsorption of dyes to the colloidal TiO2 nanoparticles. For TCPP-TiO2, it is reasonable to assume that TCPP molecules bind to the surface of TiO2 nanoparticles through the carboxylic acid anchoring groups. The absorption peak of free TCPP at 405 nm is red-shifted to 418 nm for TCPP-TiO2. For Ru(bpy)-TiO2, the absorption peak of free Ru(bpy) at 452 nm is also slightly red-shifted to 456 nm. Interestingly, the absorption peak of R6G-TiO2 is blue-shifted to 512 nm, as compared to 526 nm for the free R6G. This observation is consistent with previous reports that the absorption peaks were shifted in other dye-sensitized semiconductor nanoparticles, with the direction and magnitude being highly system-dependent.50,51 The different shifts of absorption peak are probably due to the different extents of charge transfer between the respective dye and TiO2.
Figure 3.

Extinction spectra of TiO2, TCPP, Ru(bpy), R6G, AuNP and their combinations. (a) TCPP as dye; (b) Ru(bpy) as dye; and (c) R6G as dye.
The coating of Au nanoshell on the surface of dye-embedded TiO2 nanoparticles can also be observed from the extinction spectra, where the Au-coated dye-embedded TiO2 nanoparticles distinctively display the combined absorption features of both Au nanoparticle and the respective dyes. Thus, the extinction spectra support the notion that the final products contain the three components: Au-shell, TiO2-core and dyes.
TEM and EDX of dye-embedded TiO2-core Au-shell nanoparticles
TEM and EDX results are the other evidences to support the formation of dye-embedded TiO2-core Au-shell nanostructures. Figure 4 shows the typical TEM images of the (a) TiO2, (b) TiO2-TCPP, (c) Au-TiO2-TCPP, (d) TiO2-Ru(bpy), (e) Au-TiO2-Ru(bpy), (f) TiO2-R6G and (g) Au-TiO2-R6G. In general, the particle size distribution is small. The colloidal TiO2 nanoparticles are rather uniform, with particle size less than 3 nm. However, after dyes adsorption of and the coating of Au shells, the particle sizes increase to more than 5 nm. The final sizes and shapes of the TiO2-core Au-shell nanoparticles appear to be dependent on the type of dye molecules used. Among the three dyes used in this work, Ru(bpy) seems to enlarge the TiO2 core more than the other two dyes. This could be due to the fact that Ru(bpy) is more positively charged than R6G while TCPP is negatively charged. EDX data collected on the nanoparticles in respective samples (see Figure S1 in supporting material) clearly indicate the presence of the major elements expected in the final dye-embedded TiO2-core Au-shell nanoparticles. Compared to the dye-embedded SiO2-core metal-shell nanoparticles reported previously, the TiO2-core Au-shell nanoparticles are much smaller, which should be beneficial to imaging applications.
Figure 4.

TEM image of (a) TiO2, (b) TiO2-TCPP, (c) Au-TiO2-TCPP, (d) TiO2-Ru(bpy), (e) Au-TiO2-Ru(bpy), (f) TiO2-R6G and (g) Au-TiO2-R6G. Scale bars in all images: 20 nm.
SERS of dye-embedded TiO2-core Au-shell nanoparticles
Results of Raman measurements of the dye-embedded TiO2-core Au-shell nanoparticles are shown in Figure 5. It can be seen that all the dye-embedded TiO2-core Au-shell nanoparticles display very strong SERS, similar to those SiO2-core Au-shell nanoparticles reported previously.5 The Raman spectra also shed some light to the charge transfer between the respective dye and TiO2 nanoparticles. Compared to the SERS spectra of free TCPP on Ag colloid,52 several peaks in the 1000–1650 cm−1 region for the TCPP-embedded TiO2-core Au-shell nanoparticles display a downshift of a few wavenumbers, suggesting that the carbonyl groups of TCPP coordinate to the surface Ti4+ ions. Similarly, the several peaks in the 1000–1650 cm−1 region for the Ru(bpy)-embedded TiO2-core Au-shell nanoparticles are also downshifted by a few wavenumbers in comparison with the SERS spectra of Ru(bpy)-embedded SiO2-core Au-shell nanoparticles previously reported.5 It appears that both these two dyes have appreciable interaction with TiO2 nanoparticles after the adsorption. In contrast, the peak positions in the spectra of the R6G-embedded TiO2-core Au-shell nanoparticles are very close to free R6G adsorbed on Ag colloid,53 suggesting less interaction between R6G and the TiO2 nanoparticles.
Figure 5.

SERS spectra of dye-embedded TiO2-core Au-shell nanoparticles under 632.8 nm laser excitation: (a) Au-TiO2-TCPP, (b) Au-TiO2- Ru(bpy) and (c) Au- TiO2-R6G.
4. Conclusion
In summary, we report a general strategy to synthesize dye-embedded TiO2-core Au-shell nanoparticles, which display highly enhanced SERS signals of the encapsulated dyes. The same strategy can also be applied to prepare Ag-shell nanoparticles with very strong SERS signals. The versatility of the scheme is demonstrated by successfully incorporating dyes with different charge properties, which may be extended to include many more commonly-used Raman probe molecules. The increased variety of the embedded dyes in these TiO2-based metal-coated core-shell nanoparticles should generate more optically unique SERS-tags used as identifiers in multiplexed biological assays and imaging applications. They can serve as a platform for fundamental studies in the ongoing investigations on the mechanisms of SERS. Such nanostructures may also have profound implication in the design of other plasmonic devices.54
Supplementary Material
Acknowledgments
Support from the Natural Science Foundation (CHE-0632071) and the National Center for Research Resources (NCRR) of the National Institutes of Health (RR-016480) are gratefully acknowledged.
Footnotes
Supporting Information Available: EDX spectra of TiO2, TiO2-Ru(bpy) and Au-TiO2-Ru(bpy). These materials are available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Fleischmann M, Hendra PJ, McQuillan A. J Chem Phys Lett. 1974;26:163. [Google Scholar]
- 2.Jeanmaire DL, Van Duyne RP. J Electroanal Chem. 1977;84:1. [Google Scholar]
- 3.Moskovits M. Rev Mod Phys. 1985;57:783. [Google Scholar]
- 4.Kneipp K, Kneipp H, Itzkan I, Dasari RR, Feld MS. Chem Rev. 1999;99:2957. doi: 10.1021/cr980133r. [DOI] [PubMed] [Google Scholar]
- 5.Zhang P, Guo Y. J Am Chem Soc. 2009;131:3808. doi: 10.1021/ja8086642. [DOI] [PubMed] [Google Scholar]
- 6.Yonzon CR, Haynes CL, Zhang XY, Walsh JT, Jr, Van Duyne RP. Anal Chem. 2004;76:78. doi: 10.1021/ac035134k. [DOI] [PubMed] [Google Scholar]
- 7.Schultz DA. Curr Opin Biotechnol. 2003;14:13. doi: 10.1016/s0958-1669(02)00015-0. [DOI] [PubMed] [Google Scholar]
- 8.Cao YW, Jin R, Mirkin CA. Science. 2002;297:1536. doi: 10.1126/science.297.5586.1536. [DOI] [PubMed] [Google Scholar]
- 9.Brus L. Acc Chem Res. 2008;41(12):1742. doi: 10.1021/ar800121r. [DOI] [PubMed] [Google Scholar]
- 10.Anker JN, Hall WP, Lyandres O, Shah NC, Zhao J, Van Duyne RP. Nat Mater. 2008;7:442. doi: 10.1038/nmat2162. [DOI] [PubMed] [Google Scholar]
- 11.Gates BD, Xu Q, Stewart M, Ryan D, Willson CG, Whitesides GM. Chem Rev. 2005;105:1171. doi: 10.1021/cr030076o. [DOI] [PubMed] [Google Scholar]
- 12.Xia YN, Halas NJ. MRS Bull. 2005;30:338. [Google Scholar]
- 13.Henzie J, Barton JE, Stender CL, Odom TW. Acc Chem Res. 2006;39:249. doi: 10.1021/ar050013n. [DOI] [PubMed] [Google Scholar]
- 14.Murphy CJ, Sau TK, Gole AM, Orendorff CJ, Gao J, Gou L, Hunyadi SE, Li T. J Phys Chem B. 2005;109:13857. doi: 10.1021/jp0516846. [DOI] [PubMed] [Google Scholar]
- 15.Dieringer JA, Lettan RB, Scheidt KA, Van Duyne RP. J Am Chem Soc. 2007;129:16249. doi: 10.1021/ja077243c. [DOI] [PubMed] [Google Scholar]
- 16.Jadzinsky PD, Calero G, Ackerson CJ, Bushnell DA, Kornberg RD. Science. 2007;318:430. doi: 10.1126/science.1148624. [DOI] [PubMed] [Google Scholar]
- 17.Zhang X, Young MA, Lyandres O, Van Duyne RP. J Am Chem Soc. 2005;127:4484. doi: 10.1021/ja043623b. [DOI] [PubMed] [Google Scholar]
- 18.Yang WH, Schatz GC, Van Duyne RP. J Chem Phys. 1995;103:869. [Google Scholar]
- 19.Prodan E, Radloff C, Halas NJ, Nordlander P. Science. 2003;302:419. doi: 10.1126/science.1089171. [DOI] [PubMed] [Google Scholar]
- 20.Kelly KL, Coronado EA, Zhao LL, Schatz GC. J Phys Chem B. 2003;107:668. [Google Scholar]
- 21.Wang H, Brandl DW, Nordlander P, Halas NJ. Acc Chem Res. 2007;40:53. doi: 10.1021/ar0401045. [DOI] [PubMed] [Google Scholar]
- 22.Nie SM, Emory SR. Science. 1997;275:1102. doi: 10.1126/science.275.5303.1102. [DOI] [PubMed] [Google Scholar]
- 23.Kneipp K, Wang Y, Kneipp H, Perelman LT, Itzkan I, Dasari R, Feld MS. Phys Rev Lett. 1997;78:1667. doi: 10.1103/PhysRevLett.76.2444. [DOI] [PubMed] [Google Scholar]
- 24.Otto A, Mrozek I, Grabhorn H, Akemann W. J Phys: Condens Matter. 1992;4:1143. [Google Scholar]
- 25.Otto A. Phys Status Solidi A. 2001;188:1455. [Google Scholar]
- 26.Michaels AM, Nirmal M, Brus LE. J Am Chem Soc. 1999;121:9932. [Google Scholar]
- 27.Jiang J, Bosnick K, Maillard M, Brus L. J Phys Chem B. 2003;107(37):9964. [Google Scholar]
- 28.Pavel I, McCarney E, Elkhaled A, Morrill A, Plaxco K, Moskovits M. J Phys Chem C. 2008;112(13):4880. doi: 10.1021/jp710261y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alvarez-Puebla RA, dos Santos DS, Aroca RF. Analyst. 2007;132:1210. doi: 10.1039/b711361g. [DOI] [PubMed] [Google Scholar]
- 30.Sengupta A, Mujacic M, Davis EJ. Anal Bioanal Chem. 2006;386:1379. doi: 10.1007/s00216-006-0711-z. [DOI] [PubMed] [Google Scholar]
- 31.Premasiri WR, Moir DT, Klempner MS, Krieger N, Jones G, Ziegler LD. J Phys Chem B. 2005;109(1):312. doi: 10.1021/jp040442n. [DOI] [PubMed] [Google Scholar]
- 32.Penn SG, He L, Natan MJ. Curr Opin Chem Biol. 2003;7:609. doi: 10.1016/j.cbpa.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 33.Qian X, Peng XH, Ansari DO, Yin-Goen Q, Chen GZ, Shin DM, Yang L, Young AN, Wang MD, Nie S. Nature Biotech. 2008;26(1):83. doi: 10.1038/nbt1377. [DOI] [PubMed] [Google Scholar]
- 34.Constantino CJL, Lemma T, Antunes PA, Aroca R. Anal Chem. 2001;73:3674. doi: 10.1021/ac0101961. [DOI] [PubMed] [Google Scholar]
- 35.Ren B, Lin XF, Yang ZL, Liu GK, Aroca RF, Mao BW, Tian ZQ. J Am Chem Soc. 2003;125:9598. doi: 10.1021/ja035541d. [DOI] [PubMed] [Google Scholar]
- 36.Dou XM, Ozaki Y. Rev Anal Chem. 1999;18(4):285. [Google Scholar]
- 37.Vo-Dinh T. Trends Anal Chem. 1998;17:557. [Google Scholar]
- 38.Moskovits M. J Raman Spectrosc. 2005;36:485. [Google Scholar]
- 39.Oldenburg SJ, Averitt RD, Westcott SL, Halas NJ. Chem Phys Lett. 1998;288:243. [Google Scholar]
- 40.Jackson JB, Halas NJ. J Phys Chem B. 2001;105:2743. [Google Scholar]
- 41.Campbell WM, Burrell AK, Officer DL, Jolley KW. Coord Chem Rev. 2004;248:1363. [Google Scholar]
- 42.A small sample of references on TCPP or metallo-TCPP sensitized TiO2 solar cells are: Kroeze JE, Savenije TJ, Warman JM. J Am Chem Soc. 2004;126:7608. doi: 10.1021/ja039303u.Clifford JN, Palomares E, Nazeeruddin MK, Gratzel M, Nelson J, Li X, Long NJ, Durrant JR. J Am Chem Soc. 2004;126:5225. doi: 10.1021/ja039924n.Walter MG, Wamser CC, Ruwitch J, Zhao Y, Braden D, Stevens M, Denman A, Pi R, Rudine A, Pessiki PJ. J Porphyrins and Phthalocyanines. 2007;11:601.Jasieniak J, Johnston M, Waclawik ER. J Phys Chem B. 2004;108:12962.Ma T, Inoue K, Yao K, Noma H, Shuji T, Abe E, Yu J, Wang X, Zhang B. J Electroanal Chem. 2002;537:31.Ma T, Inoue K, Noma H, Yao K, Abe E. J Mat Sci Lett. 2002;21:1013.
- 43.Ingo Z, Lars P, Zhan G, Lars KA, Ogilby PR. Langmuir. 2003;19:8927. [Google Scholar]
- 44.Zheng P, Zhang W. J Cat. 2007;250:324. [Google Scholar]
- 45.Wendell WD, Aikawa Y, Bard AJ. J Am Chem Soc. 1981;103:3456. [Google Scholar]
- 46.Hoffmann MR, Martin ST, Choi W, Bahnemann DW. Chem Rev. 1995;95:69. [Google Scholar]
- 47.Cohen-Tannoudji C. Quantum Mechanics. Wiley; New York: 1977. [Google Scholar]
- 48.Gao R, Safrany A, Joseph R. Radiat Phys Chem. 2002;65:599. [Google Scholar]
- 49.Gao R, Safrany A, Joseph R. Radiat Phys Chem. 2003;67:2599. [Google Scholar]
- 50.Huber R, Sporlein S, Moser JE, Gratzel M, Wachtveitl J. J Phys Chem B. 2000;104:8995. [Google Scholar]
- 51.Shoute LCT, Loppnow GR. J Chem Phys. 2002;117:842. [Google Scholar]
- 52.Vlckova B, Matejka P, Simonova J, Cermakova K, Pancoska P, Baumruk V. J Phys Chem. 1993;97:9719. [Google Scholar]
- 53.Zhang P, Smith S, Rumble G, Himmel M. Langmuir. 2005;21:520. doi: 10.1021/la048037a. [DOI] [PubMed] [Google Scholar]
- 54.Jin Y, Gao X. Nature Nanotech. 2009;4:571. doi: 10.1038/nnano.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.