

Abstract
We analyze how lethal mutagenesis operates in a compartmentalized host. We assume that different compartments receive different amounts of mutagen and that virions can migrate among compartments. We address two main questions: 1. To what extent can refugia, i.e., compartments that receive little mutagen, prevent extinction? 2. Does migration among compartments limit the effectiveness of refugia? We find that if there is little migration, extinction has to be achieved separately in all compartments. In this case, the total dose of mutagen administered to the host needs to be so high that the mutagen is effective even in the refugia. By contrast, if migration is extensive, then lethal mutagenesis is effective as long as the average growth in all compartments is reduced to below replacement levels. The effective-ness of migration is governed by the ratio of virion replication and death rates, R0. The smaller R0, the less migration is necessary to neutralize refugia and the less mutagen is necessary to achieve extinction at high migration rates.
Keywords: lethal mutagenesis, mutagen, drug refugia, structured host
1 Introduction
Lethal mutagenesis is a promising new antiviral therapy in which viruses are driven to extinction by chemically increasing their mutation rate (Freistadt et al., 2004). Lethal mutagenesis is appealing because it has the potential to lead to a broad-spectrum antiviral drug. Unlike other antiviral drugs, which generally have to be tailored to a specific virus, chemical mutagens such as nucleoside analogs tend to work on large classes of viruses. In the laboratory setting, lethal mutagenesis has been achieved with a variety of different viruses (Loeb et al., 1999; Sierra et al., 2000; Grande-Pérez et al., 2002; Pariente et al., 2001, 2003; Ruiz-Jarabo et al., 2003; Grande-Perez et al., 2005; Ojosnegros et al., 2008). In the clinical setting, an effective broad-spectrum antiviral drug is ribavirin (Fernandez et al., 1986; Pawlotsky, 2003), and one of its modes of action is mutagenesis (Crotty et al., 2000; Graci and Cameron, 2006).
The idea of killing a virus by mutagenesis is not new; experimental investigations date back at least twenty years (Holland et al., 1990). Yet a systematic theoretical investigation of lethal mutagenesis has begun only recently (Bull et al., 2007; Zeldovich et al., 2007; Bull and Wilke, 2008). The key insight of the recent works is that a theoretical description of lethal mutagenesis requires a model that keeps track of absolute virion frequencies (Bull et al., 2005; Wilke, 2005; Bull et al., 2007). Models that work with relative virion frequencies, such as the ones commonly studied in the quasispecies literature (Swetina and Schuster, 1982; Eigen et al., 1989; Campos and Fontanari, 1998; Woodcock and Higgs, 1996; Tannenbaum et al., 2004; Tannenbaum and Shakhnovich, 2004; Wilke, 2005; Saakian and Hu, 2006; Saakian et al., 2006; Stich et al., 2007; Takeuchi and Hogeweg, 2007), provide limited insight into the conditions under which viral extinction takes place.
Here, we extend the recent modeling work to the question of how lethal mutagenesis acts in a structured environment. We envision the situation of a patient suffering from a viral infection that has spread to multiple tissues, and ask how effective lethal mutagenesis will be if the mutagen penetrates certain tissues more readily than others and if there is migration of virus among the tissues. Specifically, we are asking to what extent refugia, i.e., tissues that admit only very little mutagen, interfere with successful lethal mutagenesis.
2 Model
2.1 Model assumptions
We model an infected host consisting of k compartments. The compartments reflect different tissues in the host. We assume that viruses replicate with rate r and die with rate d. Virus death may be caused by the immune system, by spontaneous decay of virus particles, or by other mechanisms. We do not make any specific assumptions about the causes of virus death. Upon replication, the virus can mutate. We assume that all mutations are either neutral or lethal. We keep track of only the lethal mutations.
We further assume that the viral infection in the host is being treated with a mutagen, which however does not penetrate all compartments equally well. We denote the efficiency with which a tissue absorbs mutagen by the mutagen absorption coefficient αi; αi is a number between 0 and 1. The overall amount of mutagen administered to the host is proportional to U. We choose the units of U such that the mean number of lethal mutations per virion per round of replication in compartment i is αiU. We assume that the total amount of mutagen administered to the host is proportional to U, and we neglect all mutations that arise as a consequence of the virus’s baseline mutation rate. Therefore, in compartment i, the mutagen attenuates virus growth by a factor of gi = e−αiU (Bull et al., 2007).
We denote the absolute number of virions in compartment i by ni, and the total virus population size by . We assume that virions migrate among compartments with rate m. The change in the number of virions in compartment i is given by
| (1) |
The matrix (cji) describes the connectivity graph among compartments. The elements of (cji) are between 0 and 1, indicating what fraction of virions that migrate from compartment j will end up in compartment i. Note that . Further, we assume that there is one compartment whose αi is smaller than all other αi values. Without loss of generality, we number compartments such that g1 > g2 ≥ g3 ≥ … ≥ gk.
Equation (1) describes a system in which virus can grow without bound. Clearly, unbounded growth is not realistic. A more realistic model would include tissue-specific saturation terms −ni2(t)/Ki, where Ki is the carrying capacity of the ith compartment. In this case, we would replace Eq. (1) with
| (2) |
The saturation terms dramatically alter the model dynamics for large viral population sizes, when ni(t) ~ Ki. Near extinction, however, we have ni(t) ≪ Ki and the saturation terms are negligible. In the present work, we are primarily interested in lethal mutagenesis and the mutation rate necessary to achieve it. Therefore, we neglect the saturation terms in what follows and focus exclusively on Eq. (1). Because of this modeling decision, all results we report are only valid far from saturation.
2.2 Solution
We can rewrite Eq. (1) in vector notation as dn(t)/dt = Gn(t), where n = (n1, … , nk) and G = (Gij) is given by
| (3) |
Here, δij is the Kronecker Delta; it is equal to 1 if i = j and 0 otherwise.
Given the initial state of the virus population at time t = 0, n(0), we can write n(t) as
| (4) |
ϕi and λi represent the ith right-eigenvector and eigenvalue of G, respectively, and βi is chosen to satisfy the initial conditions of n(0).
Without loss of generality, we order eigenvalues by declining real part. In other words, we assume that ℜ(λi) ≥ ℜ(λj) for all i < j. The asymptotic behavior of n(t) for t → ∞ is determined by ℜ(λ1). The total virus population size N(t) will decrease over time if ℜ(λ1) < 0. Thus, lethal mutagenesis will be successful for all mutation rates U > Ucrit, where Ucrit satisfies ℜ[λ1(Ucrit)] = 0. For simplicity, in the following we will only consider cases in which λ1 is real and non-degenerate. The Frobenius-Perron theorem guarantees that these two conditions are satisfied if (cji) is primitive (Varga, 2000).
3 Results
3.1 Two compartments
We first analyze the case of two compartments, k = 2. For this special case, we can calculate λ1 exactly. Because migration from one compartment must lead to the other, we have cji = 1 − δij. The matrix G reads
| (5) |
The principal eigenvalue of G is given by
| (6) |
If there is no migration, m = 0, the principal eigenvalue is λ1 = rg1 − d. This result makes intuitive sense. In the absence of migration, we expect the asymptotic growth of the virus population to be determined by the growth in the compartment that receives less mutagen. (Recall that g1 > g2 by earlier assumption.) Consequently, the population will go extinct if and only if it goes extinct in the compartment that absorbs less mutagen.
By contrast, for large amounts of migration, m → ∞, the principal eigenvalue approaches r(g1 + g2)/ 2 − d. Thus, for a sufficiently large migration rate, the viral population behaves like a homogeneous population growing in a single tissue whose effective mutagen absorption coefficient is αeff = (−1/U) ln[(g1 + g2)/2].
3.2 Multiple compartments
We now consider the case of an arbitrary number of compartments. For this case, a general analytic expression for λ1 does not exist, but we can use perturbation theory (Mathews and Walker, 1970) to investigate the limits of either very little migration (m ≈ 0) or extensive migration (m → ∞). To keep the analytic expressions manageable, we only treat the special case of uniform migration among all compartments, cij = (1 − δij)/(k − 1).
We begin with the case of small m. We write G as the sum of two matrices G0 and H, G = G0 + H. We choose the matrices G0 and H such that we know the spectrum of G0 and that we can consider H a small perturbation to G0. We use
| (7) |
and
| (8) |
Clearly, for sufficiently small m, H is negligible compared to G0. Using standard non-degenerate perturbation theory (Mathews and Walker, 1970), we obtain to second order in m:
| (9) |
For large m, we follow the same procedure. We write G = m(G∞ + H′) − dI, where I is the k-byk identity matrix,
| (10) |
and
| (11) |
We apply perturbation theory to the matrix G∞ + H′. Clearly, H′ is negligible compared to G∞ if r ≪ m.
For any k, the principal eigenvalue of G∞ is 0, and the corresponding normalized eigenvector is . We obtain the leading correction to the principal eigenvalue of G from the expression ϕ1 · H′ · ϕ1T. We find:
| (12) |
These approximations recapitulate the results for two compartments: If migration is weak, then the overall growth of the virus population is determined by the growth in the compartment that provides the best environment for replicating viruses, i.e., αi ≪ 1. If migration is strong, then the overall growth is determined by the average growth over all compartments. In general, migration tends to reduce the growth rate of the viral population, i.e., the larger m, the smaller λ1. Since our approximation to the principal eigenvalue of G for large m is only valid if r is small compared to m, migration will be more efficient in reducing the average growth rate of the viral population for smaller r.
The maximal amount by which migration can reduce the average growth rate depends on the mutagen absorption coefficients αi. We see from eqs. (9) and (12) that a system with large differences in absorption coefficients αi (hence large differences in growth factors gi) will experience an overall larger effect from migration, because the average Σi rgi/k will be smaller compared to the maximum value rg1.
Next, we determine the level of migration at which the overall viral growth becomes comparable to the average growth over all compartments. We calculate the point m* at which the two approximations Eqs. (9) and (12) intersect (Fig. 1), and find
| (13) |
Fig. 1.
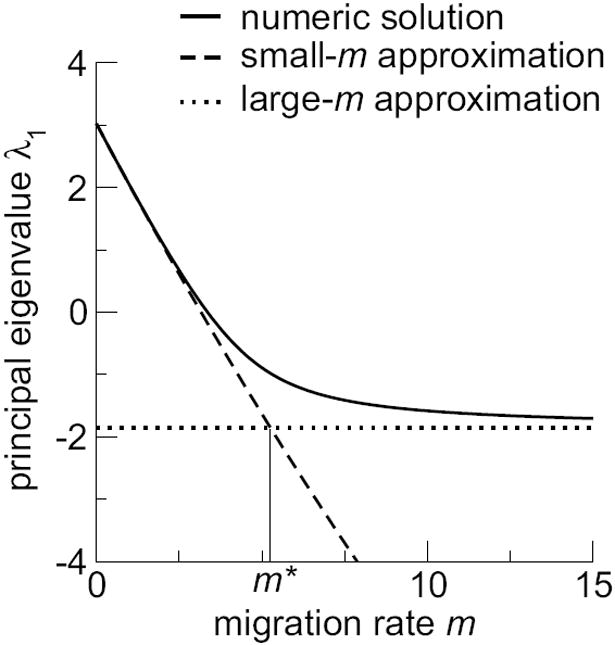
Analytic approximations to the principal eigenvalue λ1. The dashed line corresponds to Eq. (9), and the dotted line corresponds to Eq. (12). Paramter values are k = 15, α1 = 0.3, α2 = ⋯ = α8 = 0.7, α9 = ⋯ = α15 = 1.0, r = 20, d = 3, and U = 4.
For values of m ≪ m*, the small-m approximation is appropriate, whereas for values of m ≫ m*, the large-m approximation is appropriate.
From Eq. (13), we see that m* is proportional both to r and to the difference between the largest gi and the mean gi. The proportionality to r simply reflects that m and r need to be measured in the same units for meaningful comparison. Migration will generally be small if little migration happens on the time scale on which viruses replicate, and it will be large if little replication happens on the time scale on which viruses migrate. The proportionality to g1 − Σigi/k means that the larger the difference in effective growth rates among compartments, the more migration will be required to reach optimal conditions for lethal mutagenesis. Thus, the condition under which migration reduces overall virus growth the most, large g1 − Σigi/k, is also the condition under which comparatively more migration is needed to achieve the maximum reduction in virus growth.
3.3 Sectors
In the previous subsection, we found simple expressions for the overall virus growth in the limiting cases of small or large m. The advantage of these expressions is that they are asymptotically exact. Their drawback, however, is that they were derived under the assumption of homogeneous migration among all compartments. We will now develop an alternative approximation method that does not require homogeneous migration. The alternative method does not employ an asymptotically correct expansion, but instead relies on a phenomenological description of the system.
We make use of the observation that a system with a large difference in growth rates between compartments will experience a larger effect from migration. We assume that we can subdivide all compartments into two groups, one group in which the growth rate is comparatively high and one group in which it is comparatively low. We assume that there are p compartments in the first group (small αi) and q compartments in the second (large αi), with p + q = k. We refer to these two groups as sectors.
We denote by the fraction of virions that travel out of sector j and into sector i, and we assume that any migration from a sector to itself can be ignored. We then consider a population vector such that the ith position corresponds to the total number of virions in sector i. Our growth matrix G takes on the form
| (14) |
We determine by assuming that we can take the sum of all fractions of virions going from a compartment in sector i to a compartment in sector j and then average each of these sums over the total number of compartments in sector i. In other words,
| (15) |
| (16) |
Under the sector approximation, G is a 2 × 2 matrix, and we can calculate the principal eigenvalue λ1 exactly. We obtain
| (17) |
Strictly speaking, Eqs. (14) and (17) assume that all compartments within each sector have the same αi. But the sector approximation works also if there is variation in absorption coefficients αi within sectors. To model this variation, we use an appropriate effective absorption coefficient α̃i for each sector. We calculate our effective absorption coefficients as follows:
| (18) |
| (19) |
A priori, it is not necessarily clear into which sector a specific compartment should be grouped. Through extensive numerical simulation, we determined the following rule of thumb. If , compartment i can be put into the high growth rate sector and vice versa for the low growth rate sector, where αmax and αmin are the maximum and minimum absorption coefficients, respectively, of our system.
We find that the sector approximation loses accuracy for very low levels of migration and a large spread in absorption rates αi within sectors. Nonetheless, by and large, the sector approximation provides a useful and reliable description of the viral growth rate under mutagenesis and migration. See Fig. 2.
Fig. 2.
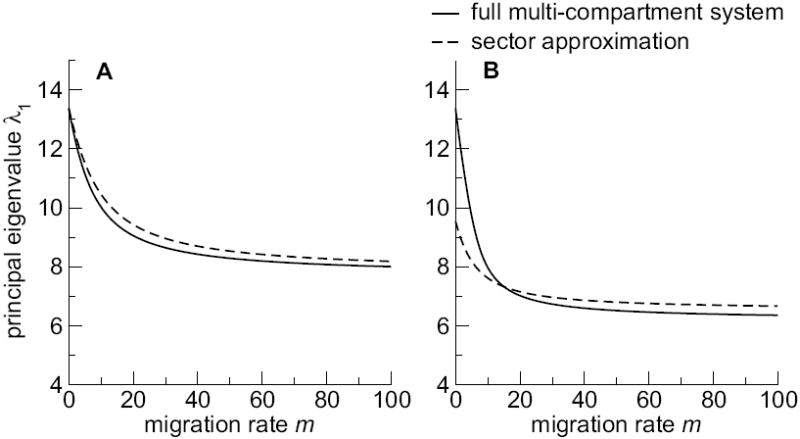
Comparison of sector approximation to the full multi-compartment system. Parameters are p = 12, q = 8, r = 20, u = 2, and d = 3. We selected the cji’s at random from a uniform distribution on the unit interval, conditional on Σi,i≠j cji = 1, and set cjj = 0. In (A), absorption and replication coefficients were uniform within the low- and highabsorption sector (α1 = ⋯ = α12 = 0.1, and α13 = ⋯ = α20 = 1.0). In (B), we used three absorption coefficients in both the low- and the high-absorption sector (α1 = ⋯ = α5 = 0.1, α6 = α7 = 0.4, α8 = ⋯ = α12 = 0.3, α13 = α14 = α15 = 0.6, α16 = α17 = 0.7, and α18 = α19 = α20 = 1.0).
3.4 The critical mutation rate
We now turn our attention to the critical mutation rate Ucrit, i.e., the minimum mutation rate sufficiently large to guarantee viral extinction. The critical mutation rate is simply the root of the equation λ1(Ucrit) = 0. Since this equation is transcendental in the present context, we cannot derive an exact analytical expression for Ucrit. Instead, we employ further approximations.
We will again take advantage of the fact that migration has a more pronounced effect when the difference between absorption rates is large for different compartments, and will assume that either α1 ≪ α2 (for k = 2) or α̃1 ≪ α̃2 (for the sector approximation). We first consider the case k = 2.
In Eq. (6), we omit g2, set λ1(Ucrit) = 0, and find that
| (20) |
When m = 0, this expression simplifies to Ucrit = (1/α1) ln R0, where R0 = r/d is the basic reproduction number. As m approaches ∞, this expression simplifies to Ucrit = (1/α1)[ln R0−ln 2]. Eq. (20) loses validity as R0 approaches 2, but serves as a faithful approximation as long as R0 ≫ 2. See Fig. 3.
Fig. 3.
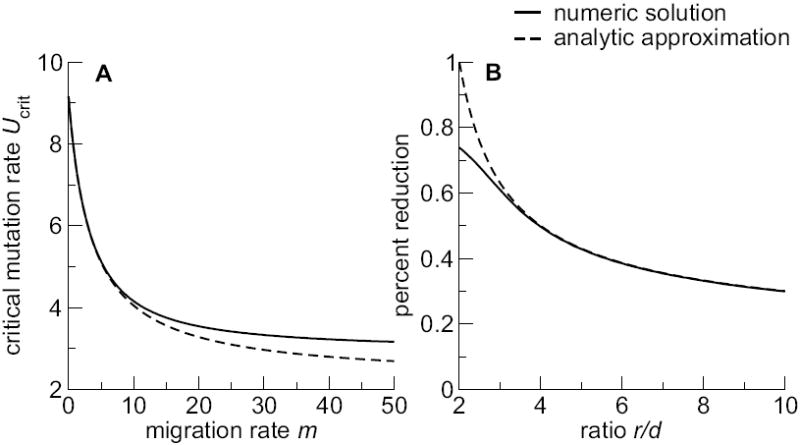
Effect of migration on the critical mutation rate. (A) Critical mutation rate vs. migration rate m, for fixed ratio R0 = 5/2. The dashed line shows Eq. (20). (B) Percent reduction in the critical mutation rate between no migration and maximum migration, as a function of the ratio R0. The dashed line shows Eq. (21). For both (A) and (B), parameters are α1 = 0.1, α2 = 1.0, and d = 5.
Using these two limiting cases, we can calculate the maximum percent reduction in Ucrit caused by migration:
| (21) |
The percent reduction depends only on the ratio R0. The larger R0, the smaller the percent reduction. In other words, if a virus population has a very large R0, then migration does not substantially alter the value of the critical mutation rate. However, the ratio R0 enters Eq. (21) only logarithmically. For realistic values of R0, which will probably not exceed 103 and will usually be smaller, Eq. (21) predicts at least a 10% reduction in the critical mutation rate.
We now consider the sector approximation. Once again, we omit g2 and set Eq. (17) equal to 0. We find
| (22) |
This approximation works well if the mutagen absorption coefficients within each sector are homogeneous (Fig. 4A), but it loses accuracy if the absorption or replication coefficients vary substantially (Fig. 4B).
Fig. 4.
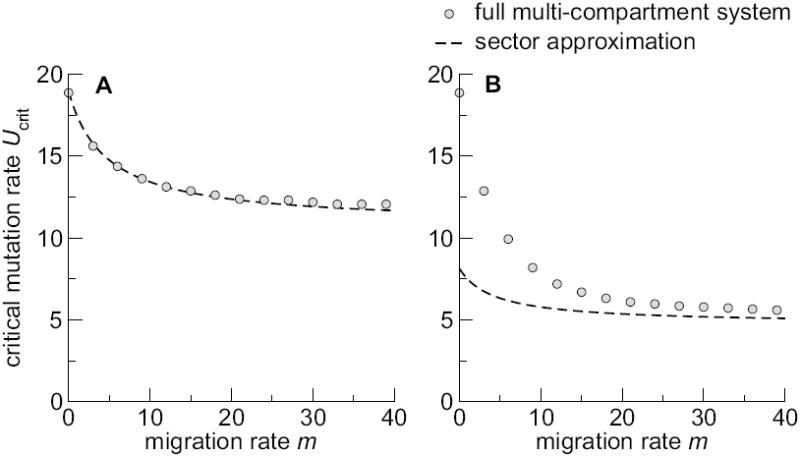
Comparison of sector approximation to the full multi-compartment system. Parameters are identical to Fig. 2A and B. The dashed line corresponds to Eq. (22).
When m = 0, we have Ucrit = (1/α̃1) ln R0. As m approaches ∞, we have . Therefore, the maximum percent reduction in this case is
| (23) |
Eq. (23) reduces to Eq. (21) if , i.e., if the two sectors have equal sizes, so that the amount of virions flowing into a sector is exactly balanced by the amount of virions flowing out. If , more virions flow out of sector 1 than into sector 1. As a consequence, the maximum percent reduction caused by migration is larger than for even-sized sectors. If , on the other hand, the maximum percent reduction caused by migration is less than for even-sized sectors. As with our approximation for k = 2, we have an analogous restriction that for the approximation to be valid.
3.5 The effect of replication and absorption abilities on the system
In the previous analysis, we have assumed that viruses have constant replicating and infecting ability in every compartment. In a real infection, this is not necessarily the case, and we will now explore the consequences of variations in viral replication rate among compartments. Suppose we take r to be a base replication rate and denote the deviation of replication or infecting ability in compartment i from this base rate as ρi, so that the growth rate in compartment i becomes rρi. We expect ρi to be of order one, but in principle it could be much larger or much smaller than this value. (We assume ρi > 0.) The system of differential equations describing viral growth then becomes
| (24) |
All our previous results carry through if we replace all occurences of gi by ρigi. In our expressions for the critical mutation rate, Eqs. 20 and 22, we have to use rρ1 in place of r. Thus, in principle, the inclusion of varied replication and infecting ability can substantially alter the phenomenology of the system. For example, a compartment could absorb only a small amount of mutagen (i.e., gi ≈ 1) yet hinder replication dramatically (i.e., ρi ≪ 1). In this case, the effective viral growth rate in the compartment, ρigi, would be small. Similarly, a compartment could absorb a large amount of mutagen (i.e., gi ≈ 1) but facilitate replication substantially (i.e., ρi ≫ 1). In this case, the effective viral growth rate in the compartment would be large. We will briefly discuss these effects in a two-compartment system.
Suppose we have a two-compartment system with R0 = 20, m = 10, α1 = 0.1, and α2 = 1. We assume that ρ2 is fixed at ρ2 = 1, and investigate the effect of ρ1 on the system behavior. In this case, the value of ρ1 directly determines whether or not compartment 1 can serve as a refugium for the virus. Alternatively, if we keep ρ1 fixed at ρ1 = 1 and vary α1, then the value of α1 directly determines whether or not compartment 1 can serve as a refugium for the virus. Fig. 5 visualizes both scenarios. It shows that the critical mutation rate does not respond to absorption and replication in the same way. In the regime in which the first compartment can act as a refugium, for α1 ≪ 1, we find that even a small reduction in α1 results in a substantial increase in Ucrit. By contrast, ρ1 needs to increase by orders of magnitude to have a similar effect on the critical mutation rate. We can explain this result by calculating an approximate expression for the critical mutation rate in a two-compartment system which contains a well protected compartment (i.e. ρ1g1 ≪ ρ2g2). We obtain this expression by writing rρ1 in place of r in Eq. (20):
| (25) |
Fig. 5.
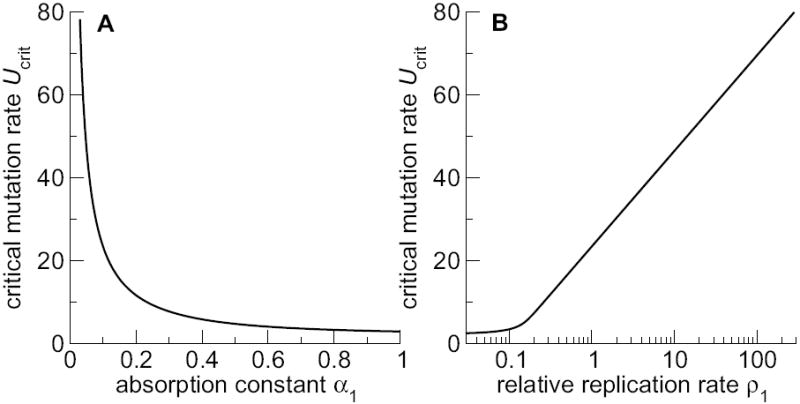
Critical mutation rate as a function of mutagen absorption and viral replication rate, as determined by exact numerical solution. Parameters are k = 2, R0 = 20, m = 10, α2 = 1, and ρ2 = 1. (A) α1 varies and ρ1 = 1. (B) ρ1 varies and α1 = 0.1.
We see that the critical mutation rate is proportional to the logarithm of ρ1 and inversely proportional to α1. Thus, a small reduction in α1 has a large effect on Ucrit, but a large increase in ρ1 has only a small effect on Ucrit. Since it is unlikely that a specific tissue will speed up viral replication by many orders of magnitude, we conclude that differences in replication rate among different compartments have only a minor effect and can be neglected to first order.
4 Discussion
We have developed a model of lethal mutagenesis in a structured host with limited migration among compartments and with uneven mutagen penetration of compartments. We have found, not surprisingly, that in the absence of migration virus extinction is determined by the compartment that is the least susceptible to mutagen. The virus population will go extinct if and only if it goes extinct in this specific compartment.
Migration generally increases the efficiency of mutagenesis. The more migration, the lower the critical mutation rate at which the virus population goes extinct. But there is a limit to the extent to which migration can reduce the critical mutation rate. In general, in the limit of infinitely fast migration, the total virus population grows with the average growth rate of all compartments, and the critical mutation rate in this limit is the mutation rate at which this average growth rate turns negative. Consequently, migration will affect the extinction threshold considerably if the average growth rate is substantially lower than the growth rate in the compartment least affected by the mutagen.
The effect of migration on the extinction threshold is largely determined by the ratio R0. This ratio interacts with migration in two ways. First, the large-m approximation is valid for m ≫ R0. Thus, the smaller R0, the less migration is needed to achieve the maximum reduction in Ucrit. Second, the size of the maximum reduction also depends on R0. The smaller R0, the larger the reduction in Ucrit as we move from m = 0 to m = ∞. In this context, we have to emphasize that R0 is not the epidemiological basic reproductive ratio, which describes the average number of newly infected cases arising from each infected individual in an epidemic. Instead, here R0 is the within-host basic reproductive ratio and describes the average number of newly infected host cells arising from every infected host cell.
In practice, we will be interested in the question under what circumstances a tissue that is not well penetrated by mutagen can serve as a refugium for the virus and can prevent successful lethal mutagenesis. Our results suggest that if the tissue is small and there is significant migration into and out of the tissue, it will not hinder lethal mutagenesis much. However, if the tissue is large and/or well isolated, then the amount of mutagen required to clear the virus may be substantial.
The method of lethal mutagenesis we studied here was using a mutagen to drive the viral replication rate below replacement level. Another method of lethal mutagenesis is lethal defection, whereby defective interfering particles accumulate and lead to the collapse of the virus population (Iranzo and Manrubia, 2009). Lethal defection is reliant on coinfection of viable and non-viable virions and has to date only been observed in systems that show persistent infections (Grande-Perez et al., 2005) or that have been artificially enriched with defective sequences (González-López et al., 2004). Whether lethal defection could occur in lytic viruses, in which we expect the multiplicity of infection to drop below one near the extinction threshold, remains an open question.
We made several simplifying assumptions in our model, to keep the mathematics tractable. First, we used a deterministic model. Since lethal mutagenesis is to first order a deterministic process that will work in arbitrarily large populations (Bull et al., 2007), this assumption seems reasonable. However, with a deterministic model, we do not take into account fluctuations in the viral population size. When viral population numbers are low, i.e. near extinction, the rate of deleterious mutation fixation increases (Lynch et al., 1993). In addition, stochastic fluctuations in a small population can drive a population to extinction even when the mutation rate is below the deterministic threshold (Demetrius et al., 1985). Thus, we would expect that the critical mutation rate of a finite population is lower than our deterministic calculation predicts.
Second, we assumed that all mutations are either neutral or lethal, and we kept track of only the lethal mutations. The resulting growth rate within a compartment, R0e−αiU, is identical to the equilibrium growth rate in a model that takes into account arbitrary deleterious mutations (Kimura and Maruyama, 1966; Bull et al., 2007). Therefore, our predictions will be approximately correct for any virus that sits at the top of a local fitness optimum. Some deviations from our predictions will arise because the virus population is not at equilibrium at the beginning of the mutagenic treatment. However, as long as beneficial mutations can be neglected, as will be the case for a virus at the top of a fitness optimum, the out-of-equilibrium mean fitness of the viral population will exceed R0e−αiU. Consequently, for any U smaller than the Ucrit predicted based on the equilibrium theory the viral population will survive at all times, and for any U larger than Ucrit the population fitness will decline until the population goes extinct. Thus, Ucrit remains the same even if the population is initially out of equilibrium. Additional deviations from our predictions will arise if beneficial mutations increase in frequency and effect as the mutation rate increases and fitness decreases, as we would expect near extinction via lethal mutagenesis (Silander et al., 2007). The Kimura-Maruyama formula R0e−αiU holds only approximately in this case. By contrast, if the virus initiating the infection is far from a fitness optimum or if new fitness optima become available under mutagenesis, then our predictions will likely not match experimental outcomes. A comprehensive treatment of beneficial mutations is beyond the scope of this study and we leave this topic for future work.
Third, our model does not include any delay in migration. Thus, we assume that migration from one tissue to another is immediate. We believe that neglecting delays is a reasonable assumption for vertebrates with a cardiovascular system because virus dispersal through the blood will happen much faster than viral replication. In an average-sized human body, the blood turns over about once per minute (Jacob et al., 1982). By contrast, viral replication time is on the order of hours to days. For example, HIV-1 in-vivo generation time is estimated to be 2 days (Markowitz et al., 2003).
Fourth, we assumed that the multiplicity of infection (m.o.i.) remains low at all times, so that viral coinfection can be ignored. Low m.o.i. is a reasonable assumption for a viral population close to extinction.
In this study, we introduced the sector approximation as a way of analytically analyzing lethal mutagenesis in structured systems with multiple compartments. The sector approximation subsumes all compartments into two classes (sectors), compartments that allow a lot of viral replication and compartments that do not. While the sector approximation did not necessarily achieve high numerical accuracy in all cases, it captured the qualitative behavior of all multi-compartment systems to which we applied it. Therefore, the sector approximation will likely be a powerful tool for future studies that aim to remove some of the other simplifying assumptions we made here.
Acknowledgments
This work was supported in part by NIH grant AI 065960 and by NSF grant EF-0742373.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bull JJ, Meyers LA, Lachmann M. Quasispecies made simple. PLoS Comp Biol. 2005;1:e61. doi: 10.1371/journal.pcbi.0010061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull JJ, Sanjuán R, Wilke CO. Theory of lethal mutagenesis for viruses. J Virol. 2007;81:2930–2939. doi: 10.1128/JVI.01624-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull JJ, Wilke CO. Lethal mutagenesis of bacteria. Genetics. 2008;180:1061–1070. doi: 10.1534/genetics.108.091413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos PRA, Fontanari JF. Finite-size scaling of the quasispecies model. Phys Rev E. 1998;58:2664–2667. [Google Scholar]
- Crotty S, Maag D, Arnold JJ, Zhong W, Lau JYN, Hong Z, Andino R, Cameron CE. The broad-spectrum antiviral ribonucleoside ribavirin is an rna virus mutagen. Nature Medicine. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- Demetrius L, Schuster P, Sigmund K. Polynucleotide evolution and branching processes. Bull of Math Biol. 1985;47:239–262. doi: 10.1007/BF02460034. [DOI] [PubMed] [Google Scholar]
- Eigen M, McCaskill J, Schuster P. The molecular quasi-species. Adv Chem Phys. 1989;75:149–263. [Google Scholar]
- Fernandez H, Banks G, Smith R. Ribavirin: A clinical overview. European J Epidemiology. 1986;2:1–14. doi: 10.1007/BF00152711. [DOI] [PubMed] [Google Scholar]
- Freistadt MS, Meades GD, Cameron CE. Lethal mutagens: broad-spectrum antivirals with limited potential for development of resistance? Drug Resistance Updates. 2004;7:19–24. doi: 10.1016/j.drup.2003.12.003. [DOI] [PubMed] [Google Scholar]
- González-López C, Arias A, Pariente N, Gómez-Mariano G, Domingo E. Preextinction viral RNA can interfere with infectivity. J Virol. 2004;78:3319–3324. doi: 10.1128/JVI.78.7.3319-3324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graci JD, Cameron CE. Mechanisms of action of ribavirin against distinct viruses. Rev Med Virol. 2006;16:37–48. doi: 10.1002/rmv.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande-Perez A, Lazaro E, Lowenstein P, Domingo E, Manrubia SC. Suppression of viral infectivity through lethal defection. Proc Natl Acad Sci USA. 2005;102:4448–4452. doi: 10.1073/pnas.0408871102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grande-Pérez A, Sierra S, Castro M, Domingo E, Lowenstein P. Molecular indetermination in the transition to error catastrophe: System elimination of lymphocytic choriomeningitis virus through mutagenesis does not correlate linearly with large increases in mutant spectrum complexity. Proc Natl Acad Sci USA. 2002;99:12938–12943. doi: 10.1073/pnas.182426999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland JJ, Domingo E, de la Torre JC, Steinhauer DA. Mutation frequencies at defined single codon sites in vesicular stomatitis virus and poliovirus can be increased only slightly by chemical mutagenesis. J Virol. 1990;64:3960–3962. doi: 10.1128/jvi.64.8.3960-3962.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iranzo J, Manrubia SC. Stochastic extinction of viral infectivity through the action of defectors. Europhys Lett. 2009;85:18001. [Google Scholar]
- Jacob SW, Ashworth Francone C, Lossow WJ. Structure and Function in Man. 5 W B Saunders; Philadelphia: 1982. [Google Scholar]
- Kimura M, Maruyama T. The mutational load with epistatic gene interactions in fitness. Genetics. 1966;54:1337–1351. doi: 10.1093/genetics/54.6.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb LA, Essigmann JM, Kazazi F, Zhang J, Rose KD, Mullins JI. Lethal mutagenesis of hiv with mutagenic nucleoside analogs. Proc Natl Acad Sci USA. 1999;96:1492–1497. doi: 10.1073/pnas.96.4.1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Bürger R, Butcher D, Gabriel W. The mutational meltdown in asexual populations. J Heredity. 1993;84:339–344. doi: 10.1093/oxfordjournals.jhered.a111354. [DOI] [PubMed] [Google Scholar]
- Markowitz M, Louie M, Hurley A, Sun E, Di Mascio M, Perelson AS, Ho DD. A novel antiviral intervention results in more accurate assessment of human immunodeficiency virus type 1 replication dynamics and T-Cell decay in vivo. J Virol. 2003;77:5037–5038. doi: 10.1128/JVI.77.8.5037-5038.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews J, Walker RL. Mathematical Methods of Physics. 2 Addison-Wesley; 1970. [Google Scholar]
- Ojosnegros S, Agudo R, Sierra M, Briones C, Sierra S, Gonzalez-Lopez C, Domingo E, Cristina J. Topology of evolving, mutagenized viral populations: quasispecies expansion, compression, and operation of negative selection. BMC Evol Biol. 2008;8:207. doi: 10.1186/1471-2148-8-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariente N, Sierra S, Lowenstein PR, Domingo E. Efficient virus extinction by combinations of a mutagen and antiviral inhibitors. J Virol. 2001;75:9723–9730. doi: 10.1128/JVI.75.20.9723-9730.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariente N, Sierra S, Lowenstein PR, Domingo E. Mutagenesis versus inhibition in the efficiency of extinction of foot-and-mouth disease virus. J Virol. 2003;77:7131–7138. doi: 10.1128/JVI.77.12.7131-7138.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlotsky JM. Mechanisms of antiviral treatment efficacy and failure in chronic hepatitis C. Antiviral Res. 2003;59:1–11. doi: 10.1016/s0166-3542(03)00088-3. [DOI] [PubMed] [Google Scholar]
- Ruiz-Jarabo CM, Ly C, Domingo E, de la Torre JC. Lethal mutagenesis of the prototypic arenavirus lymphocytic choriomeningitis virus (LCMV) Virology. 2003;308:37–47. doi: 10.1016/s0042-6822(02)00046-6. [DOI] [PubMed] [Google Scholar]
- Saakian DB, Hu C-K. Exact solution of the Eigen model with general fitness functions and degradation rates. Proc Natl Acad Sci USA. 2006;103:4935–4939. doi: 10.1073/pnas.0504924103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saakian DB, noz EM, Hu C-K, Deem MW. Quasispecies theory for multiple-peak fitness landscapes. Phys Rev E. 2006;73:041913. doi: 10.1103/PhysRevE.73.041913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra S, Dávila M, Lowenstein PR, Domingo E. Response of foot-and-mouth disease virus to increased mutagenesis: Influence of viral load and fitness in loss of infectivity. J Virol. 2000;74:8316–8323. doi: 10.1128/jvi.74.18.8316-8323.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silander OK, Tenaillon O, Chao L. Understanding the evolutionary fate of finite populations: The dynamics of mutational effects. PLoS Comp Biol. 2007;5:e94. doi: 10.1371/journal.pbio.0050094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stich M, Briones C, Manrubia SC. Collective properties of evolving molecular quasispecies. BMC Evol Biol. 2007;7:110. doi: 10.1186/1471-2148-7-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swetina J, Schuster P. Self-replication with errors: a model for polynucleotide replication. Biophys Chem. 1982;16:329–345. doi: 10.1016/0301-4622(82)87037-3. [DOI] [PubMed] [Google Scholar]
- Takeuchi N, Hogeweg P. Error-threshold exists in fitness landscapes with lethal mutants. BMC Evol Biol. 2007 doi: 10.1186/1471-2148-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannenbaum E, Deeds EJ, Shakhnovich EI. Semiconservative replication in the quasispecies model. Phys Rev E. 2004;69:061916. doi: 10.1103/PhysRevE.69.061916. [DOI] [PubMed] [Google Scholar]
- Tannenbaum E, Shakhnovich EI. Solution of the quasispecies model for an arbitrary gene network. Phys Rev E. 2004;70:021903. doi: 10.1103/PhysRevE.70.021903. [DOI] [PubMed] [Google Scholar]
- Varga RS. Matrix Iterative Analysis. 2 Springer-Verlag; New York: 2000. [Google Scholar]
- Wilke CO. Quasispecies theory in the context of population genetics. BMC Evol Biol. 2005;5:44. doi: 10.1186/1471-2148-5-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock G, Higgs PG. Population evolution on a multiplicative single-peak fitness landscape. J Theor Biol. 1996;179:61–73. doi: 10.1006/jtbi.1996.0049. [DOI] [PubMed] [Google Scholar]
- Zeldovich KB, Chen P, Shakhnovich EI. Protein stability imposes limits on organism complexity and speed of molecular evolution. Proc Natl Acad Sci USA. 2007;104:16152–16157. doi: 10.1073/pnas.0705366104. [DOI] [PMC free article] [PubMed] [Google Scholar]