

Abstract
Recovery of fine motor skills after traumatic brain injury (TBI) is variable, with some patients showing progressive improvements over time while others show poor recovery. We therefore studied possible cellular mechanisms accompanying the recovery process in a non-human primate model system, in which the lateral frontal motor cortex areas controlling the preferred upper limb were unilaterally lesioned, and the animals eventually regained fine hand motor function. Immunohistochemical staining of the cervical spinal cord, the site of compensatory sprouting and degeneration of corticospinal axons, showed profound increases in immunoreactivities for major histocompatibility complex class II molecule (MHC-II) and extracellular signal-regulated kinases (ERK1/2) up to 12 months post lesion, particularly within the lateral corticospinal tract (LCST). Double immunostaining demonstrated that phosphorylated ERK1/2 colocalized within the MCH-II + microglia, suggesting a trophic role of long-term microglia activation after TBI at the site of compensatory sprouting. Active sprouting was observed in the LCST as well as in the spinal gray matter of the lesioned animals, as illustrated by increases in growth associated protein 43. Upregulation of Nogo receptor and glutamate transporter expression was also observed in this region after TBI, suggesting possible mechanisms for controlling aberrant sprouting and/or synaptic formation en route and interstitial glutamate concentration changes at the site of axon degeneration, respectively. Taken together, these changes in the non-human primate spinal cord support a long-term trophic/tropic role for reactive microglia, in particular, during functional and structural recovery after TBI.
Key words: injury, microglia, motor recovery, spinal cord, synapse
Introduction
In the United States alone, an estimated 1.4 million people are afflicted by traumatic brain injury (TBI) annually, and, as a consequence, many suffer from motor and cognitive disabilities (Langlois et al., 2004). TBI is also a major consequence of modern-day battlefield interactions and has become the signature injury of the Afghanistan/Iraq conflicts, with approximately 25% of soldiers in action reporting TBI (Hodge et al., 2008; Okie, 2005; Xydakis et al., 2005). Considering the prevalent and devastating nature of this neurological disorder, there is a critical need to develop new and more advanced treatments than is currently available in clinical practice. Furthermore, effective post-injury therapies developed for TBI may be beneficial to other groups, including stroke patients, since ischemic damage elicits similar post-traumatic events (Leker and Shohami, 2002). Indeed, fundamental processes of brain degeneration and regeneration after injury likely apply to recovery from even more controlled injury conditions, such as that occurring in some neurosurgical patients. In order to determine possible therapeutic intervention and treatment strategies to facilitate functional recovery after TBI or related brain injury, understanding the degenerative/regenerative processes that occur in the central nervous system (CNS) is crucial. Specific post-traumatic events after TBI have been well documented in both human TBI patients and experimental animal models. These events include but are not limited to: (a) inflammatory responses by glial cells in the perilesion site and along pathways of wallerian degeneration (Streit, 2000); (b) elevation of glutamate concentration at the site of injury and at distant areas innervated by injured neurons (Ashwal et al., 2004; Faden et al., 1989; Palmer et al., 1993; Rose et al., 2002; Yamamoto et al., 1999); (c) acute and delayed apoptotic and/or necrotic cell death at the site of injury and in the penumbra region (Conti et al., 1998; Dietrich et al., 1994; Newcomb et al., 1999; Rink et al., 1995); and (d) adaptive structural changes such as axonal sprouting by surviving neurons (Bütefisch, 2006; Carmichael, 2003a; Dancause et al., 2005; Nudo, 1999).
TBI-mediated inflammatory responses are rapidly initiated by disruption of the blood–brain barrier, allowing accumulation of leukocytes and cytokines (Morganti-Kossman et al., 1997, 2007) and activation of microglia/macrophage (Czigner et al., 2007; Streit, 2000). Using a non-human primate TBI model that selectively induced upper extremity paresis, we previously conducted an immunohistological study to investigate microglia activation and their phenotype changes after TBI (Nagamoto-Combs et al., 2007). In this model, the primary motor cortex and lateral premotor cortex controlling the preferred arm of the animal were specifically lesioned by aspiration/coagulation, as performed in human neurosurgery for the selective removal of tumors, epileptogenic foci, and arteriovenous malformations. Following resection, the animals recovered most of their limb functions during the recovery period (Darling et al., 2006; Pizzimenti et al., 2007). Our results demonstrated that microglia activation, as indicated by positive immunoreactivity to a phagocytic marker, CD68, was observed not only at the site of injury but also in the cervical spinal cord for up to 12 months after the injury (Nagamoto-Combs et al., 2007). Furthermore, we found that the activated microglia co-expressed brain-derived neurotrophic factor (BDNF) and its receptors, TrkB and TrkB[TK−], suggesting a long-term trophic/tropic function of microglia after TBI.
These results led us to hypothesize further that microglia may play an important role in assisting the survival of neurons undergoing apoptosis/necrosis after TBI, especially at distal sites from the injury. They may also be involved in modulating other post-traumatic processes, such as regulating extracellular glutamate concentrations. It is reasonable to assume the presence of a mechanism to control local glutamate concentration during recovery, since both ionotropic and metabotropic glutamate receptors are expressed in periaxonal astrocytes (Agrawal and Fehlings, 1997; Agrawal et al., 1998) and oligodendrocytes (Matute et al., 1997) within the central white matter, and have been postulated to play a role in myelin and axonal degradation after injury (Ouardouz et al., 2009; Tekkok and Goldberg, 2001). In support of this hypothesis, earlier studies have reported increased expression of glutamate transporter-1 (GLT-1) in microglia activated by facial-nerve axotomy in vivo (López-Redondo et al., 2000) or by TBI in humans (Beschorner et al., 2007). Furthermore, microglia may be involved in structural reorganization during recovery. A finding that microglia and sprouting neurites are closely associated after needle injury in the sensorimotor cortex of the rat (King et al., 2001) supports this notion, suggesting that microglia may regulate axonal growth and/or synaptic formation.
In our present exploratory study, we again utilized the non-human primate TBI model system, and examined one sham-operated animal and six animals following localized lateral motor cortex lesion. Molecular changes that might be responsible for regulating post-TBI events at the site of axonal degeneration and/or regeneration in the cervical spinal cord were examined using immunohistochemical techniques. To identify reactive microglia, we employed an antibody against major histocompatibility complex class II molecule (MHC-II), a well-described injury-elicited phenotype of reactive microglia (Popovich et al., 1997; Streit et al., 1989). Possible involvement of microglia in cell survival was examined by evaluating the activation of the extracellular signal-regulated kinases (ERK1/2), which are downstream kinases that can mediate BDNF signaling and have been shown to relay cell survival cascades (Bonni et al., 1999). Changes in immunoreactivity to two different glutamate transporters, GLT-1 and glutamate aspartate transporter (GLAST), were also investigated for the potential role of activated microglia and/or other glial cells in controlling local glutamate concentration. To identify possible sites of axonal growth and synapse formation, we used antibodies against growth associated protein-43 (GAP-43) and synaptophysin, a growth cone protein and presynaptic vesicle protein respectively. Furthermore, we investigated the expression of the Nogo-66 receptor (NgR) and one of its ligands, Nogo A, since the receptor is thought to control directions of axonal growth and sites of synaptic formation by interacting with myelin-associated growth proteins (Fournier et al., 2001; Schwab, 2004). Lastly, we present behavioral data obtained from the experimental animals before lesion and during recovery to show their varied levels of motor-function recovery following injury.
Methods
Materials
The mouse monoclonal anti-human MHC-II (HLA-DR) antibody (LN3) (Lab Vision Products, Fremont, CA) was diluted to 1:1000 for immunostaining. The rabbit polyclonal antibody against human phospho-p44/42 ERK1/2 (Thr202/Tyr204; Cell Signaling Technology, Danvers, MA) for detection of phospho-ERKs (pERKs) was used at a 1:1000 dilution. The rabbit polyclonal antibodies against GAP-43, Nogo A (H-300) and NgR (Nogo-R, H-120; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) were used at 1:500, 1:500, and 1:300 dilutions respectively. The mouse monoclonal anti-synaptophysin antibody (Chemicon International, Inc., Temecula, CA) was used at a 1:1000 dilution. The rabbit polyclonal antibodies against GLT-1 and GLAST (Alpha Diagnostic International, Inc., San Antonio, TX) were both used at a concentration of 5 μg/mL. The VECTASTAIN Elite ABC kit, Vector VIP, Vector SG, Vector NovaRED, biotinylated secondary antibodies, VectaMount mounting medium and other immunodetection reagents were obtained from Vector Laboratories (Burlingame, CA). ProLong Gold anti-fade reagent with DAPI was purchased from Molecular Probes, Inc. (Eugene, OR). The mouse monoclonal anti-GFAP antibody, used at a 1:500 dilution, and all the other reagents were purchased from Sigma Chemical Co. (St. Louis, MO).
Animals
Six adult rhesus monkeys (Macaca mulatta) with ages ranging from 3.8 to 14 years (Table 1) were maintained in a primate facility approved by the US Department of Agriculture. All experimental and surgical procedures employed in this study were conducted in accordance with US Department of Agriculture, National Institutes of Health and Society for Neurosciences guidelines for the ethical treatment of experimental animals, and were approved by the Institutional Animal Care and Use Committee at the University of South Dakota.
Table 1.
A Summary of the Experimental Animals Used in this Study
| Monkey | Total survival time | Age (years) | Gender | Weight (kg) | Lesion side |
|---|---|---|---|---|---|
| SDM49 | 1 month post lesion (33 days) | 3.8 | Male | 6.2 | Left |
| SDM45 | 6 months post lesion (33 days) | 5.4 | Male | 7.2 | Left |
| SDM64 | 6 months post lesion (33 days) | 14 | Female | 6.9 | Right |
| SDM24 | 12 months post lesion (31 days) | 13.5 | Male | 11 | Right |
| SDM48 | 12 months post lesion (33 days) | 7.7 | Female | 5.3 | Left |
| SDM54 | 1 month post sham operation (33 days) | 9 | Male | 9.2 | N/A |
Total survival time indicates the recovery periods after surgery. The numbers in parentheses to the right of the survival time period show the survival days after neural tract tracer injection.
Surgery
The preparation and procedure of animal surgery were performed as previously described (Darling et al., 2006; Morecraft et al., 2001, 2002, 2004, 2007; Nagamoto-Combs et al., 2007; Pizzimenti et al., 2007). Briefly, each animal was prepared for surgery by an initial administration of ketamine hydrochloride (10 mg/kg) and kept anesthetized during cortical exposure with a continuous administration of isofluorane (1–1.5% with surgical grade air/oxygen mixture) with the assistance of a mechanical respirator. The frontal motor cortices in the hemisphere contralateral to the preferred hand were mapped electrophysiologically under intravenous ketamine anesthesia. The primary motor cortex (M1) and dorsolateral premotor cortex (LPMCd) in the hemisphere contralateral to the animals preferred arm were identified using intracortical microstimulation. A detailed description of the intracortical microstimulation procedure has been published previously (Darling et al., 2009; McNeal et al., 2010; Morecraft et al., 2001, 2002), and cortical gray matter at these sites was removed by aspiration and coagulation (Fig. 1). Specifically, large and medium-sized arteries within the cortical field to be resected were cauterized using a Bovie Specialist Electrosurgical Unit (Bovie Aaron Medical, St. Petersburg, FL), disposable electrophysiological foot-control pencil, and needle electrode. Subpial aspiration was performed using an angled glass pipette or angled Frazier surgical suction tube. The pipette or suction tube was attached to a Schuco vacuum (Model 130; Schuco Inc., Toledo, OH). The tip was carefully placed on the cortical surface using microscopic guidance, and the gray matter was gently removed until subcortical white matter was identified. Surgery was completed by suturing the dural opening, replacing and anchoring the bone flap, and suturing the temporalis muscle. The skin was closed with sterile staples. Pre-surgical and post-surgical antibiotics (bicillin L-A or amoxicillin) were administered to all animals.
FIG. 1.

Schematic diagrams of aspiration/coagulation lesion made in the lateral motor cortex of adult rhesus monkeys. The size and location of lesion in each case was confirmed using histological techniques and is indicated as the blackened area on the lateral cortical surface of the lesioned cases. The pullout shows the electrophysiological map that was generated following cortical exposure to localize the arm area of the primary motor cortex (M1) and dorsal lateral premotor cortex (LPMCd). A = arm, cs = central sulcus, D = digit, ecs = ectocalcarine sulcus, El = elbow, Hp = hip, ilas = inferior limb of the arcuate sulcus, ios = inferior occipital sulcus, ips = intraparietal sulcus, L = leg, lf = lateral fissure, LL = lower lip, LPMCd = dorsolateral premotor cortex, ls = lunate sulcus, M1 = primary motor cortex, N = neck, NR = no response, ps = principle sulcus, Sh = shoulder, slas = superior limb of the arcuate sulcus, sts = superior temporal sulcus, T = toe, Th = thumb, To = tongue, Tr = trunk, UL = upper lip, Wr = wrist.
The total recovery periods after surgery were 1 month (SDM49), 6 months (SDM45, SDM64), or 12 months (SDM24, SDM48). For animals SDM45, SDM64, SDM24, and SDM48, a second surgery was performed 31–33 days prior to sacrifice in order to inject neural tract tracers into various regions of the spared motor cortices. The neural tracers were utilized for our separate neuroplasticity studies quantifying sprouting from spared supplemental motor cortex, and the tract-tracing results are not discussed in the present report (McNeal et al., 2010). One animal, SDM54, was sham-operated without lesion, received neural tract tracer injections following intracortical stimulation, and survived for 33 days (sham with 1-month survival). Specifically, the 1-month experiment was terminated at 33 days, the 6-month experiment at 24 weeks (168 days), and the 12-month experiment after 48 weeks (336 days) of survival (see Table 1).
Behavioral assessment
Prior to surgery, all animals were tested to determine hand preference using a standard dexterity board. We then trained the monkeys on two fine hand motor tasks before and after the motor cortex lesion using two techniques that allowed control over which hand the monkey could use for successful task performance without constraining a limb and allowing the monkey to be free to roam the cage. These tasks thus permitted testing recovery of the contralesional hand fine motor skill after the lesion. Briefly, in one task (modified movement assessment panel – mMAP), the forces applied while the monkey removed a carrot chip with a central hole from a flat surface and over straight and curved rods (Darling et al., 2006). Time to remove the carrot chip and total absolute impulse (equation 1) were used to assess performance quantitatively using a performance score normalized to each monkey's best single-trial pre-lesion performance in terms of both duration and impulse. The equation used to compute the performance score is given in equation 2. In the second task (modified dexterity board – mDB), the monkey removed small food pellets from a Plexiglas plate (with a small dimple to hold the pellet in place) and from 10-mm deep tapered wells that ranged from 10–25 mm in diameter routed into the plate (Pizzimenti et al., 2007). Performance in calibrated three-dimensional space was recorded on four video cameras, permitting measurements of reach duration and accuracy (fingertip position relative to pellet position), grip aperture (distance between tips of index and thumb), manipulation duration, and number of lost digit contacts with the pellet during manipulation. These were used to compute an overall performance score using an equation similar to the one used to quantify mMAP performance on each trial (Pizzimenti et al., 2007). Performances were also qualitatively assessed to determine whether monkeys used a similar strategy to perform the tasks during post-lesion trials to strategies used prior to the lesion. Pre-lesion tests were administered approximately weekly until performance was stable and then at weekly intervals for the first 2 months post lesion, followed by tests every 2 weeks in the following months. Skill was measured by taking the mean performance score over five consecutive tests sessions (last five pre-lesion test sessions, best five consecutive post-lesion test sessions) and dividing by the standard deviation of performance scores over that period. Pre- and post-lesion skills were computed for the well with highest pre-lesion skill on the mDB task (Pizzimenti et al., 2007) and the curved rod mMAP task. The ratio of post- to pre-lesion skill was used as measure of recovery of fine motor skill (i.e., this ratio is zero if there is no recovery and equals 1 if skill returns to pre-lesion levels). It should be noted that the animal designated as SDM24 was tested only on the mMAP task without a load cell to record forces, so that only duration to retrieve the food could be measured to assess recovery of performance, and there was no measure of recovery of skill. Also for SDM49, the animal with 1-month post-recovery period, post-lesion skill was not measured because of its brief recovery period limited to having only four consecutive testing sessions.
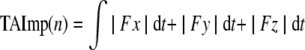 |
(1) |
where: TAImp (n) is the total absolute impulse of trial n; ∫ is the integral over duration of trial t with respect to time (dt); Fx is the force applied in a left/right direction; Fy is the force applied in an anterior/posterior direction; Fz is the force applied in a vertical direction.
If outcome ≥2 (i.e., successful grasp and lift/manipulation of the carrot chip) then:
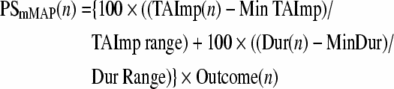 |
(2) |
if PSmMAP(n) < 200 then PSmMAP(n) = 200,
else:
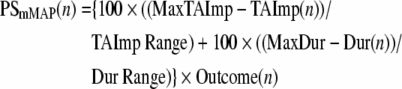 |
if PSmMAP(n) > 200 then PSmMAP(n) = 200,
where: PSmMAP(n) is the performance score on mMAP trial n; Outcome(n) is the success on trial n (0 for no attempt with the correct hand, 1 for unsuccessful attempt with the correct hand, 2 if the carrot chip is successfully grasped and lifted over the rod but then dropped and not removed from the food chamber, 3 if the carrot chip is successfully grasped and lifted over the rod but then dropped and removed from the food chamber, 4 for successful acquisition without dropping the carrot chip); MinTAImp is the minimum single trial pre-lesion total absolute impulse within a difficulty level for either hand; TAImp Range is the maximum single trial pre-lesion total absolute impulse–MinTAImp; Dur(n) is the duration of trial n; MinDur is the minimum single trial duration during pre-lesion tests; and Dur Range is the maximum single trial duration during pre-lesion tests–MinDur.
Preparation of tissue sections
At the end of the recovery period, each animal was given an overdose of pentobarbital (50 mg/kg or higher) and terminated via transcardial perfusion with solutions of 0.9% saline followed in sequence by 4% paraformaldehyde in 0.1M phosphate buffer (PB, pH 7.2–7.4), 10% sucrose in PB (sucrose/PB), and 30% sucrose/PB. The brain, brainstem, and spinal cord were removed and documented before being stored in 30% sucrose/PB for 2–4 days at 4°C. The spinal cord was frozen sectioned at 50 μm in the transverse (horizontal) plane on a sliding microtome (American Optical AO 860; Buffalo, NY) and collected in 0.05M PB. The spinal-cord sections within each series represented every 300 or 400 μm intervals through each tissue block. One series was immediately mounted on subbed slides and stained for Nissl substance as a reference for cytoarchitectural analysis along with the sections from the brain and brainstem (Morecraft et al., 1992, 2004). For the purpose of this study, two to three sections per animal per antibody from the lower cervical spinal cords at C5–C8 level were examined. Unprocessed tissue sections were stored in a cryoprotective solution (30% glycerol, 30% ethylene glycol in 0.1M PB) at −80°C until use.
Immunohistological staining
Tissue sections stored in the cryoprotective solution were first rinsed in phosphate-buffered saline (PBS) with gentle agitation for 1 h with several changes of PBS for removal of ethylene glycol. To reduce endogenous peroxidase activity, the washed tissue sections were then treated for 5 min at room temperature with 0.01% hydrogen peroxide, prepared in PBS/TritonX-100 solution (PBS-T; 0.1% v/v TritonX-100), supplemented with 0.5% (w/v) bovine serum albumin and 10% goat serum. An additional 1-h incubation in the serum-supplemented PBS-T followed to block non-specific tissue interaction of the primary antibodies. Since biotinylated neuronal track tracers were utilized in the animals to be analyzed for separate neuroplasticity studies, Avidin/Biotin Blocking Solutions (Vector Laboratories) was used according to the manufacturer's protocol for the purpose of this current study to prevent potential staining of the tracers during visualization of antibodies.
Incubations of the prepared tissue sections with appropriate primary antibodies were carried out at 4°C overnight, with gentle agitation in the serum-containing PBS-T at the antibody concentrations indicated in the Materials section. Some sections were incubated in the buffer without any primary antibody to determine background staining by each secondary antibody. After the primary antibody incubation and subsequent rinses (10 min × 3) at room temperature, tissue sections were subjected to further 1-h incubation with a species-specific biotinylated secondary antibody diluted to 1:2000. The tissue sections were rinsed again then treated with the avidin-biotin complex solution provided in the Vector ABC Elite Kit (Vector Laboratories) for a further 1 h. For visualization of the antigens, Vector VIP was used as a chromogen (Vector Laboratories), according to the manufacturer's protocol. Color development was terminated by rinsing the tissues extensively with PBS. Alternatively, when fluorescent double staining was utilized, the tissue sections were not treated with 0.01% hydrogen peroxide in PBS-T prior to the blocking procedure. Furthermore, tissues were incubated with two primary antibodies of different animal origins simultaneously overnight at 4°C. The subsequent procedures were performed as described above, except Texas Red- or FITC-conjugated species-specific secondary antibodies were used (Santa Cruz Biotechnology, Inc.).
The sections were mounted onto gelatin-coated glass slides after being rinsed briefly with water. The slides mounted with VIP-stained tissue sections were air-dried at least overnight, and dehydrated through a series of ethanol solutions (50%, 70%, 90%, and 100%) and Histo-Clear (National Diagnostics, Atlanta, GA), and coverslipped using VectaMount (Vector Laboratories). The slides with fluorescent stained sections were mounted and coverslipped using ProLong Gold antifade reagent with DAPI (Invitrogen, Carlsbad, CA) without the air-drying and dehydration procedures.
Microscopy and photography
In all animal subjects, the location and general size of the lesion to M1 and LPMCd were verified as described in Figure 1. The tissue sections processed for immunohistochemical staining were examined using a Leica DM4000B microscope (Leica Microsystems, Nussloch, Germany) with 1.25 ×, 10 ×, 20 ×, 40 ×, and 63 × objectives. Each light photograph was taken using a Leica DFC300FX digital camera supported by Leica FW4000 Software (Leica Microsystems). Confocal images of fluorescent labeled tissue sections were obtained using the Zeiss LSM 510 Meta confocal microscope (Zeiss, Thornwood, NY), as previously described (Jara et al., 2007). Images were viewed and processed using a Zeiss LSM Image Browser. Captured photographic images were combined into each figure and labeled using Adobe Photoshop 7.0 software. The immunohistochemical images in the figures were adjusted using the software so that the left panels show ipsilateral side and the right panels show contralateral side to the lesion for the purpose of easier comparison.
Results
Microglia/macrophage exhibited MHC-II immunoreactivity over a prolonged period in the lateral corticospinal tract of the cervical spinal cord after traumatic brain injury of the lateral motor cortex
Phenotypes of reactive microglia can vary across different experimental paradigms. Although our prior study demonstrated increased immunoreactivity (IR) to a trophic factor (BDNF) and phagocytic marker (CD68), we sought to define the phenotype of microglia further in our TBI long-term recovery paradigm. In particular, expression of MHC-II is one of the well-described characteristics of reactive microglia in pathological conditions (Stoll et al., 2006; Streit et al., 1989), and its increased expression signifies acquisition of antigen presentation ability (Redwine et al., 2001) and immune-responsiveness by microglia (Gehrmann et al., 1995). Therefore, we investigated whether the microglia in the cervical spinal cord increase MHC-II-IR as a general marker of reactive phenotype in lesioned and sham-operated animals. MHC-II-IR was observed in all the animals examined, including the sham-operated animal (Fig. 2). The staining was primarily found perivascularly and within the white-matter parenchyma, but was more widely distributed throughout the spinal-cord sections than that of CD68-IR demonstrated in our prior report (Nagamoto-Combs et al., 2007). More importantly, sections from lesioned animals at 1 (Fig. 2E–H), 6 (Fig. 2I–L) and 12 (Fig. 2M–P) months post lesion (m.p.l.) showed intensely stained groups of cells in the LCST contralateral to the lesion side, some displaying a dense clustered appearance with stout processes (Fig. 2G,K,O, inset). This morphology was consistent with reactive microglia, and was especially profound in the animals sacrificed at 1 and 6 m.p.l. (Fig. 2G,K respectively). MHC-II-IR was also found in the sham-operated animal in the same region (Fig. 2B–D), but the immunoreactive cells showed distinct, more ramified morphology, and staining intensity was markedly less compared to that observed in the lesioned animals. Some densely stained cells were apparent in the spinal gray matter of the 1-m.p.l. animal (Fig. 2F, arrowheads). Though moderate staining was observed in the gray matter of other lesioned (Fig. 2I–P) and sham-operated animals (Fig. 2B–D), it was less intense, and the staining was found mostly around the central canal and vasculature. These observations indicated that MHC-II is basally expressed, as seen in the sham-operated animal, and TBI induces its expression within the spinal cord white matter for up to 12 months. Furthermore, the level of MHC-II might be differentially regulated in the spinal white matter compared with the gray matter.
FIG. 2.

MHC-II immunoreactivity increased in the LCST of the lower cervical spinal cord from adult rhesus monkeys after injury of the lateral motor cortex. (A) A schematic diagram of a transverse cervical spinal cord section. Contra/Ipsi indicates contralateral or ipsilateral side to each animal's lesion respectively. The rectangular boxes labeled LCST and VH indicate approximate areas where photomicrographs were taken for the lateral corticospinal tract and spinal gray matter in the ventral horn respectively. (B–P) MHC-II positive cells were visualized using the ABC/Vector VIP immunodetection method in the transverse spinal-cord sections from the sham-operated control (B–D), 1- (E–H), 6- (I–L), and 12-m.p.l. (M–P) animals. The sections have been arranged so that the left-most panels, E, I, and M show low-magnification photographs of the ipsilateral LCST region, while F, J, and N show the contralateral side of each animal. The boxes within B, F, J, and N indicate the areas where high-power photomicrographs, C, G, K, and O, were taken. The inset in each of these photomicrographs shows a higher-power photomicrograph of the MHC-II-IR cell(s) designated by the arrow. Note the significant increases in MHC-II-IR and different morphology of the stained cells within the contralateral LCST regions of the lesioned animals (G, K, and O) compared to the sham-operated (C) animal. Panels D, H, L, and P depict MHC-II-IR within the ventral spinal gray matter (VH) of the sham-operated, 1-, 6-, or 12-m.p.l. animals respectively. Scale bar sizes are as indicated.
Phosphorylation of ERK1/2 occurred within MHC-II + , activated microglia within the cervical spinal cord after traumatic brain injury of the lateral motor cortex
In our previous study, we demonstrated that immunoreactivities to BDNF and its receptors, TrkB[gp145] and TrkB[TK−], were increased and partially colocalized to CD68-IR microglia in the spinal cords of lesioned animals (Nagamoto-Combs et al., 2007). Therefore, in the current study, we investigated whether TBI also led to activation of ERK1/2, the major protein kinases downstream of BDNF-TrkB activation. To assess ERK activity indirectly, cervical spinal-cord sections from the sham-operated and lesioned animals were immunostained to detect active, phosphorylated forms of ERK1/2 (pERK). Profound, apparently chronic, pERK-IR was detected in the LCST of the lesioned animals at 1 (Fig. 3D–F), 6 (Fig. 3G–I), and 12 (Fig. 3J–L) months recovery. The morphology of the labeled cells was similar to that of MHC-II immunoreactive microglia (Fig. 3F,I,L), supporting the notion that microglia had maintained their increased ERK activity for up to 12 months after the lesion. Some staining was also noted in the LCST region of the sham-operated animal but without the dense, cluster-like appearance observed in the lesioned animals (Fig. 3C). With the exception of the area around the central canal, no significant staining in the spinal gray matter was observed at these time points.
FIG. 3.

pERK immunoreactivity was elevated in the LCST of the lower cervical spinal cord from adult rhesus monkeys after injury of the lateral motor cortex. (A) A schematic diagram of a transverse cervical spinal-cord section. Contra/Ipsi indicates contralateral or ipsilateral side to each animal's lesion respectively. The rectangular box labeled LCST indicates approximate area of lateral corticospinal tract where photomicrographs were taken. (B–L): The immunoreactivity to pERK was visualized using the ABC/Vector VIP method in the transverse spinal cord sections from the sham-operated control (B and C), 1- (D–F), 6- (G–I), and 12-m.p.l. (J–L) animals. The sections have been arranged so that the left-most panels, D, G, and J, show low-magnification photographs of the ipsilateral LCST region, while E, H, and K show the contralateral side of each animal. The boxes within B, E, H, and K indicate the areas where high-power photomicrographs, C, F, I, and L, were taken. The inset in each of these photomicrographs shows a higher-power photomicrograph of the pERK-IR cell(s) designated by the arrow. Robust labeling for pERK-IR was observed in the contralateral LSCT region of each lesioned animal (F, I, and L) compared to that of the sham-operated animal (C). No significant labeling was found in the spinal gray matter. Scale bar sizes are as indicated.
In order to verify that the immunostaining for pERK1/2 colocalized with activated microglia following TBI, we double labeled sections from the 1-m.p.l. animal using pERK and MHC-II antibodies (Fig. 5A–C). The pERK-IR (shown as green signals in Fig. 5A) and MHC-II-IR (shown as red signals in Fig. 5B) were most intensely localized in the LCST, as shown previously, and showed clear overlap (Fig. 5C). This observation was particularly evident in clusters of cells as depicted in a group of the blue DAPI-stained nuclei in Figure 5C.
FIG. 5.

MHC-II immunoreactivity overlapped that of pERK and was in proximity to that of GAP-43 within the LCST of the lower cervical spinal cord from the 1-m.p.l. adult rhesus monkey after injury of the lateral motor cortex. Transverse cervical spinal-cord sections from the 1-m.p.l. animal (SDM49) were processed for double immunofluorescent labeling against pERK (A, green) or GAP-43 (D, green) in conjunction with MHC-II (red, B, E). The merged images of pERK/MHC-II and GAP-43/MHC-II staining are shown in panels C and F respectively (MERGE). The blue fluorescence in panel C and F depicts nuclear labeling by DAPI. Photomicrographs were taken from the LCST region using a standard or confocal fluorescent microscope for pERK/MHC-II staining (A–C) and for GAP-43/MHCII (D–F) respectively. Note that pERK and MHC-II labels greatly overlap (C) while GAP-43 and MHC-II labels show little colocalization (F). Scale bar in panel C = 25 μm; scale bar in panel F = 20 μm.
Immunoreactivity to GAP-43 increased in the lateral corticospinal tract and the ventral horn of the cervical spinal cord after traumatic brain injury of the lateral motor cortex
Since all of our lesioned animals recovered the use of the TBI-afflicted contralesional hand (to varying levels in relation to pre-lesion skill) and our prior data demonstrated increased BDNF-IR within microglia up to 12 months post-lesion, we next determined whether the reactive microglia in the spinal cord after TBI were associated with axonal regrowth and/or sprouting. To achieve this objective, actively growing nerve endings in the sham-operated, 1-, 6-, and 12-m.p.l. animals were visualized using an antibody against GAP-43, a protein found at high levels in developing and regenerating axons and growth cones (Karns et al., 1987). We found that GAP-43-IR was notably higher in the LSCT of all the lesioned animals, except one of the 12-m.p.l. animals (SDM48), compared with the sham control (Fig. 4). Particularly, in SDM49 (1 m.p.l.; Fig. 4E–H), SDM45 (6 m.p.l.; Fig. 4I–L), and SDM64 (6 m.p.l.; Fig. 4M–P), the staining appeared as a bushy, cluster-like formation in the LSCT. Lesion-associated increases in GAP-43-IR were also found in the gray matter, although there seemed to be some individual differences. For example, the GAP-43-IR in the gray matter of SDM49 (1 m.p.l.; Fig. 4H) and SDM24 (12 m.p.l.; Fig. 4T) appeared compatible, while that of SDM45 (6 m.p.l.; Fig. 4L), SDM64 (6 m.p.l.; Fig. 4P) showed less intense labeling, which was still above the sham level (Fig. 4D). In some locations within the cervical spinal gray matter, the positive staining was especially intense around the large oval structures that morphologically appeared to be spinal motor neurons.
FIG. 4.

GAP-43 immunoreactivity increased in the LCST of the lower cervical spinal cord of adult rhesus monkeys after injury of the lateral motor cortex. (A) A schematic diagram of a transverse cervical spinal-cord section. Contra/Ipsi indicates contralateral or ipsilateral side to each animal's lesion respectively. The rectangular boxes labeled LCST and VH indicate approximate areas where photomicrographs were taken for the lateral corticospinal tract and spinal gray matter in the ventral horn respectively. (B–X) Transverse sections of the lower cervical spinal cord from the sham-operated (SDM54; B–D), 1- (SDM49; E–H), 6- (SDM45; I–L and SDM64; M–P), and 12-m.p.l. (SDM24; Q–T and SDM48; U–X) animals were immunostained for GAP-43 using the ABC/Vector VIP method. The sections have been arranged so that left-most panels E, I, M, Q, and U show low-magnification photographs of the ipsilateral LCST region, while F, J, N, R, and V show the contralateral side of each animal. The boxes within B, F, J, N, R, and V indicate the areas where high-power photomicrographs, C, G, K, O, S, and W, were taken. The inset in each of these photomicrographs shows a higher-power photomicrograph of the GAP-43-IR cell(s) designated by the arrow. The panels D, H, L, P, T, and X show GAP-43-IR in the spinal gray matter within the VH region. Specific recovery-time-dependent changes were not consistently observed. Scale bar sizes are as indicated.
To confirm that these growth processes were associated with the microglia in the LCST region, sections were double labeled for MHC-II and GAP-43 (Fig. 5D–F). Confocal microscopy was utilized to obtain high-resolution images of the immunofluorescence to localize the two labels. The GAP-43-IR (shown in green in Fig. 5D, GAP-43) and MHC-II-IR (shown in red in Fig. 5E, MHC-II) were closely associated, and the merged image of the two signals showed that they were indeed adjacent to one another (Fig. 5F, MERGE), showing little overlap (shown in yellow). This result indicated that MHC-II + , reactive microglia were in close proximity to GAP-43-expressing processes and/or growth cones.
Synaptophysin immunoreactivity modestly increased in the lateral corticospinal tract and the ventral spinal gray only in some lesioned animals
Because GAP-43-IR suggested changes in neurite growth was concurrent with the reactive gliosis, we next examined a more functional assessment of the sprouting response, synaptic formation, at the 1-, 6-, and 12-month recovery periods. Cervical spinal cords of lesioned versus sham-operated animals were compared for changes in immunoreactivity to the pre-synaptic vesicle associated protein, synaptophysin. Synaptophysin-immunoreactive neurites seemed slightly more apparent in the LCST of the 1-m.p.l. animal (Fig. 6G), one of the 6-m.p.l. animals (SDM45; Fig. 6K), and also to a lesser extent, one of the 12-m.p.l. animals (SDM48; Fig. 6W). In contrast, other lesioned animals (SDM64 & 24; Fig. 6O,S respectively) showed more compatible levels of synaptophysin-IR when compared to the control animal (SDM54; Fig. 6C). In the former group of animals, the immunopositive neurites were associated with or were at least in close proximity to a labeled cell or what appeared to be a cluster(s) of cells (Fig. 6G,K,W, inset). While there was evidence of clustered synaptophysin-IR in the LCST region, note the difference in the extent of immunostaining from that of GAP-43-IR (Fig. 4). This relatively modest synaptophysin-IR with regard to GAP-43-IR may indicate that sprouting neurites do not form aberrant functional synapses in the LCST, or such synapses get pruned as the animals recover their motor function. Synaptophysin-IR was also found in varying degrees throughout the spinal gray matter of both lesioned (Fig. 6H,L,P,T,X) and sham-operated (Fig. 6D) animals, appearing particularly intense around motor neuron cell bodies. Such immunoreactivity was more abundant in 1- (SDM49; Fig. 6H) and 12-m.p.l. (SDM24 & 48; Fig. 6T,X respectively) animals, but not in the 6-m.p.l. (SDM45 & 64; Fig. 6L,P respectively) animals.
FIG. 6.

Synaptophysin immunoreactivity showed modest increases in the LCST and ventral spinal gray matter of lower cervical spinal cords from only some of the lesioned animals after lateral motor cortex injury. (A) A schematic diagram of a transverse cervical spinal cord section. Contra/Ipsi indicate contralateral or ipsilateral side to each animal's lesion respectively. The rectangular boxes labeled LCST and VH indicate approximate areas where photomicrographs were taken for the lateral corticospinal tract and spinal gray matter in the ventral horn respectively. (B–X) Transverse sections of the lower cervical spinal cord from the sham-operated (SDM54; B–D), 1- (SDM49; E–H), 6- (SDM45; I–L and SDM64; M–P), and 12-m.p.l. (SDM24; Q–T and SDM48; U–X) animals were immunostained for synaptophysin using the ABC/Vector VIP method. The sections have been arranged so that left-most panels E, I, M, Q, and U show low-magnification photographs of the ipsilateral LCST region, while F, J, N, R, and V show the contralateral side of each animal. The boxes within B, F, J, N, R, and V indicate the areas where high-power photomicrographs, C, G, K, O, S, and W, were taken. Synaptophysin-IR neurites were found most prominently in the spinal gray matter in all animals examined (D, H, L, P, T, and X). In the LCST region, no striking differences were observed between the lesioned and sham-operated animals, though slightly more synaptophysin-labeled neurites were found in SDM49 (G) and SDM45 (K), and also to a lesser extent in SDM48 (W). As depicted in panels G, K, and W, the labeled neurites (arrowheads) appeared to be in close proximity to a lightly stained cell or cluster-like structure(s) (arrows), which is shown at a higher power in the inset. Scale bar sizes are as indicated.
The level of NgR expression was elevated in the lateral corticospinal tract, while the level of NgR ligand, Nogo A, showed no apparent changes in the cervical spinal cord after traumatic brain injury
The lesion-associated increases, and gradual-recovery-period-associated decreases, in synaptophysin-IR and GAP-43-IR in the spinal white matter after TBI suggested possible pruning mechanisms of sprouting corticospinal axons. Since NgR and one of its ligands, Nogo A, are now generally accepted as inhibitors of neurite outgrowth (Fournier et al., 2001; Schwab, 2004), we next investigated changes in these proteins as potential candidates for dysregulation causing aberrant sprouting and/or synaptic formation. Immunohistochemical staining using an antibody against NgR in the sham-operated animal showed that there were basal levels of NgR-IR in the LCST (Fig. 7C) and spinal gray matter (Fig. 7D), particularly in the ventral horns (VH). From the morphology of the stained cells in the VH, NgR appeared to localize in the spinal motor neurons. Such NgR-IR in the ventral gray matter was also present in the lesioned animals (Fig. 7H,L,P), but additional increases in the staining in the VH parenchyma were observed. This change was especially evident at 1 and 12 m.p.l. (Fig. 7H,P respectively), perhaps due to individual differences across the animals, as seen in other staining.
FIG. 7.

NgR immunoreactivity increased in the LCST of lower cervical spinal cords of adult rhesus monkeys after injury of the lateral motor cortex. (A) A schematic diagram of a transverse cervical spinal cord section. Contra/Ipsi indicate contralateral or ipsilateral side to each animal's lesion respectively. The rectangular boxes labeled LCST and VH indicate approximate areas where photomicrographs were taken for the lateral corticospinal tract and spinal gray matter in the ventral horn respectively. (B–P) Transverse sections from the lower cervical spinal cords from the sham-operated (B–D), 1- (E–H), 6- (I–L), and 12-m.p.l. (M–P) animals were immunostained for NgR using the ABC/Vector VIP method. The sections have been arranged so that left-most panels E, I, and M show low-magnification photographs of the ipsilateral LCST region, while F, J, and N show the contralateral side of each animal. When compared to the sham control (C), profound NgR-IR was detected in this region of the 1- (G) and 6-m.p.l. (K) animals, and of the 12-m.p.l. animal (O) to a lesser extent. The boxes within B, F, J, and N indicate the areas where high-power photomicrographs, C, G, K, and O, were taken. The inset in each of these panels shows a higher-power photograph of the NgR-immunoreactive structure indicated by the arrow. Panels D, H, L, and P show NgR labeling in the spinal gray matter within the VH region. Scale bar sizes are as indicated.
NgR-IR notably increased in the LCST of the lesioned animals, especially at 6 m.p.l., with the presence of intensely labeled, cluster-like formations strikingly similar to those already described for MHC-II-IR microglia (Fig. 7J,K). The increased level of NgR-IR in the LCST was observed as early as 1 m.p.l. (Fig. 7F,G), and returned to near basal levels after 12 months (Fig. 7N,O). Importantly, this temporal pattern of NgR expression suggests that aberrant neurite sprouting and growth is attenuated or normalized by 12 months of recovery.
Despite the robust changes in NgR-IR in the LCST after TBI, the immunoreactivity of its ligand, Nogo A, in this region showed very little recovery time-dependent changes and did not differ appreciably from the sham-operated animal at any recovery period (i.e., 1, 6, and 12 m.p.l.; Fig. 8E,H,K respectively). Intense labeling in the VH of the spinal cords was observed in all animals examined (Fig. 8C,F,I,L), with no apparent differences between ipsilateral and contralateral sides (Fig. 8A,D,G,J). The Nogo A-IR localized within the soma of the lower motor neurons and their dendritic processes in the VH.
FIG. 8.

Nogo A immunoreactivity did not appreciably change in the LCST or VH in the lower cervical spinal cords of adult rhesus monkeys after injury of the lateral motor cortex. Transverse sections from the lower cervical spinal cords of the sham-operated (A–C), 1- (D–F), 6- (G–I), and 12-m.p.l. (J–L) animals were immunostained for Nogo A using the ABC/Vector VIP method. The sections are arranged so that left side of each photograph is the ipsilateral side to each animal's lesion. (A, D, G, and J) Low-power photographs. The rectangles and arrows in these panels respectively indicate where the high-power photographs of the LCST (B, E, H, and K) and ventral spinal gray matter (C, F, I, and L) from the corresponding animals were taken. Scale bar sizes are as indicated.
Traumatic brain injury in the lateral motor cortex did not markedly alter astrocyte proliferation in the cervical spinal cord
The immunohistochemical results thus far demonstrated that microglial activation was maintained until at least 12 months post-lesion recovery, and their activation state correlated with increased markers of axon growth and synapse formation not only in the VH but also the associated white matter in the LCST. Because microglia are only one component of a typical reactive glial response to injury, we next assessed changes in astrocyte phenotype in our TBI paradigm. Reactive astrocytes were assessed via changes in glial fibrillary acidic protein (GFAP)-IR (Fig. 9). Cervical spinal-cord sections from 1- (SDM49; Fig. 9B,F), 6- (SDM45; Fig. 9C,G), or 12-m.p.l. (SDM24; Fig. 9D,H) animals, as well as the sham-operated animals (SDM54; Fig. 9A,E), showed GFAP-IR astrocytes in both white matter (LCST; Fig. 9A–D) and ventral gray matter (VH; Fig. 9E–H). It is noteworthy that the staining pattern within the VH formed a “basket-like” structure, possibly surrounding the spinal motor neurons in the anterior horn. We did not observe, however, obvious differences in GFAP-IR intensity or patterns in either the white or gray matter between the lesioned and sham-operated animals.
FIG. 9.

GFAP immunoreactivity did not change in the LCST or VH in the lower cervical spinal cords of adult rhesus monkeys at different recovery time points after injury of the lateral motor cortex. Transverse sections of the lower cervical spinal cords from the sham-operated (A, E), 1- (B, F), 6- (C, G), and 12-m.p.l. (D, H) animals were immunostained for GFAP using the ABC/Vector VIP method. The schematic drawing of the spinal cord section indicates the orientation of the sections and the approximate areas of lateral corticospinal tract (LCST) and ventral spinal gray matter (VH) where the photographs A–D and E–H were taken respectively. Only the contralateral (Contra) sides of the sections are shown. No significant temporal change in GFAP-IR was detected at the examined recovery time points. Scale bar = 50 μm.
The glutamate transporters, GLT-1 and GLAST, were differentially regulated in the spinal gray matter of the spinal cord after traumatic brain injury
Although GFAP-IR showed little change in astrocytic proliferation at the post-lesion recovery periods examined, there can be multiple changes in astrocyte protein expression that indicate a plethora of variations in reactive phenotype. For example, astrocytes, as well as microglia, have been reported to increase glutamate transporter protein expression after injury (Beschorner et al., 2007; Van Landeghem et al., 2001). Therefore, we assessed whether alteration in glial expression of GLT-1 and GLAST would occur after TBI. Immunodetection of GLT-1 (Fig. 10) and GLAST (Fig. 11) in the cervical spinal cords from the sham-operated or 1-, 6-, or 12-m.p.l. animals demonstrated higher levels of staining for both glutamate transporters in the LCST in the lesioned animals than the sham-operated animal (Figs. 10 & 11G,K,O). The expression of these glutamate transporters were also found in the VH region, especially around the large motor neurons (Figs. 10 & 11D,H,L,P). While basal levels of GLT-1 (Fig. 10D) and GLAST (Fig. 11D) were observed in the sham-operated animals, immunoreactivity for these glutamate transporters seemed to be elevated in the lesioned animals in the environs of motor neurons, giving rather diffused staining patterns in the VH region (Figs. 10 & 11H,L,P). This result suggested that glial expression of GLT-1 and GLAST increase both in the white and gray matters of the cervical spinal cord after TBI.
FIG. 10.

GLT-1 immunoreactivity increased in the LCST and VH in the lower cervical spinal cord of adult rhesus monkeys after injury in the lateral motor cortex. (A) A schematic diagram of a transverse cervical spinal cord section. Contra/Ipsi indicate contralateral or ipsilateral side to each animal's lesion respectively. The rectangular boxes labeled LCST and VH indicate approximate areas where photomicrographs were taken for the lateral corticospinal tract and spinal gray matter in the ventral horn respectively. (B–P) GLT-1 positive cells were visualized using the ABC/Vector VIP immunodetection method in the transverse spinal-cord sections from the sham-operated control (B–D), 1- (E–H), 6- (I–L), and 12-m.p.l. (M–P) animals. The sections have been arranged so that left-most panels, E, I, and M, show low-magnification photographs of the ipsilateral LCST region, while F, J, and N show the contralateral side of each animal. The boxes within B, F, J, and N indicate the areas of LCST where the high power photomicrographs, C, G, K, and O, were taken. The inset in each of these photomicrographs shows a higher-power photomicrograph of the GLT-1 immunoreactive area designated by the arrow. Note the presence of different morphology in the LCST of the lesioned animals, especially in the 1- and 6-m.p.l. animals (G and K respectively) compared to the sham-operated animal (C). Panels D, H, L, and P depict GLT-1-IR within the ventral spinal gray matter (VH) of the sham-operated, 1-, 6-, or 12-m.p.l. animals respectively. The inset photographs in these panels also show higher-power images of the areas designated by the arrows, illustrating immunolabeling around motor neurons. Scale bar sizes are as indicated.
FIG. 11.

GLAST immunoreactivity increased in LCST and VH in the lower cervical spinal cord of adult rhesus monkeys after injury in the lateral motor cortex. (A) A schematic diagram of a transverse cervical spinal cord section. Contra/Ipsi indicate contralateral or ipsilateral side to each animal's lesion respectively. The rectangular boxes labeled LCST and VH indicate approximate areas where photomicrographs were taken for the lateral corticospinal tract and spinal gray matter in the ventral horn, respectively. (B–P) GLAST-IR was visualized using the ABC/Vector VIP immunodetection method in the transverse spinal cord sections from the sham-operated control (B–D), 1- (E–H), 6- (I–L), and 12-m.p.l. (M–P) animals. The sections have been arranged so that left-most panels, E, I, and M, show low-magnification photographs of the ipsilateral LCST region, while F, J, and N show the contralateral side of each animal. The boxes within B, F, J, and N indicate the areas of LCST where the high power photomicrographs, C, G, K, and O, were taken. Overall increases in the staining in this region were observed in the lesioned animals (G, K, and O) compared to the sham control (C). GLAST-IR was also found in the ventral spinal gray matter in all the animals examined (D, H, L, and P). There seem to be diffuse increases in the GLAST-IR surrounding strongly immunopositive motor neurons, although any obvious temporal pattern in the staining intensity was not determined. Scale bar sizes are as indicated.
Lesioned animals showed varied degrees of recovery on the traumatic brain injury afflicted fine motor skill following traumatic brain injury of the lateral motor cortex
Functional recovery in the lesioned animals for specific fine motor skills were quantified both pre- and post-lesion in order to record physiological outcomes that accompanied the observed immunohistochemical changes during the recovery periods. Performance on the fine motor tasks showed a clear decrement after the lesion with no attempts (performance score equal to zero) or successful acquisitions on the first post-lesion test in most animals on one or both tasks, followed by increases in performance scores over the next 3–5 weeks (e.g., Fig. 12A,B). However, one monkey (SDM45) was successful on all attempts in both tasks at 1 week post-lesion (Fig. 12C,D; white bars). Performance usually improved on the second test at 2 weeks after the lesion, as most animals had at least one success on one or both tasks, with the exception of SDM24 (only tested on the mMAP task – Fig. 12C; gray bars) and SDM64 (on the mDB task – no successful acquisitions until the 4th post-lesion week – Fig. 12D; gray bar). Recovery of fine motor skill on the two tasks varied from about 50% of pre-lesion skill (i.e., post/pre-lesion skill ratio of 0.5) to better than pre-lesion skill (i.e., post to pre-lesion skill ratio greater than 1.0) on the two tasks (Fig. 12C,D). Notably, SDM45 recovered to better than pre-lesion skill on both tasks, and SDM48 recovered to better than pre-lesion skill on the mMAP, but to only 50% of pre-lesion skill on the mDB task. SDM64 recovered to about 50% of pre-lesion skill on both tasks.
FIG. 12.

Recovery of fine motor function after lesion to M1 and LPMC. Average performance scores from individual testing sessions on the mMAP (A) and mDB (B) tasks for the final five pre-lesion testing sessions and for all post-lesion testing sessions for one monkey (SDM48). Each plotted symbol shows the mean performance score for five trials in a single test session in A and B (error bars are 1 standard deviation). C and D show the post-lesion week of first successful acquisition of the food targets (white bar) and consistent successful acquisition (i.e., on all trials – black bar) for the mMAP curved rod (C) and mDB best well (D) tasks for each monkey. The ratio of post-lesion skill to pre-lesion skill in the mMAP curved rod (white bar) and mDB best well (black bar) tasks is shown in E. Note that SDM24 only performed the mMAP task without the load cell. Thus, there are no data presented for this animal on the mDB task in D and recovery of skill is not presented for the mMAP task in E.
Discussion
Although TBI and stroke often result in long-term physical and cognitive disabilities, many patients regain part of their functional losses over time. It is without question that a rapid and significant response occurs at the cellular/molecular level immediately following brain trauma, which continues to manifest throughout the first month of recovery following brain injury (Carmichael, 2003b; Kuhtz-Buschbeck et al., 2003; Sbordone et al., 1995; Wade et al., 1985). As a result, it is not surprising that the majority of studies tracking neuroplastic responses to brain injury have focused on this critical post-injury time period. Yet, the functional recovery process can continue for 2 or more years after TBI (Lotze et al., 2006; Sbordone et al., 1995) or stroke (Mark et al., 2006), indicating that long-term studies investigating the underlying neuroplastic responses of prolonged recovery are needed. Such information could assist in the development of long-term treatment strategies aimed to facilitate and enhance the recovery process in patients with brain injury.
To study this process as it relates to the primate CNS, the appropriate experimental model is also an important consideration (Fukuda and Del Zoppo, 2003). For example, in order to assess post-TBI recovery of fine motor skills, such as digit coordination, non-human primates provide an excellent and unique experimental model because they use the digits similarly to humans to manipulate hand-held objects, and the underlying anatomical infrastructure supporting this function is featured only in higher-order primates (Courtine et al., 2007; Lemon and Griffiths, 2005). In addition, we employed the aspiration/coagulation method, rather than contusion model, to generate a more isolated injury to the upper extremity region of the motor cortex. Thus this experimental model not only simulates certain neurosurgical procedures performed in human patients, it also allows investigators to control the locus and extent of injury and to observe and quantitatively record the functional recovery process of the animals in great detail (Darling et al., 2006, 2009; Pizzimenti et al., 2007). Importantly, such observations can be conducted to study the motor-recovery process following motor cortex injury in the absence of confounding variables resulting from the added affect of injury to subcortical white matter pathways and subcortical gray matter structures. Furthermore, histological analyses can also be used to examine structural and molecular alterations following injury that accompany favorable recovery of dexterous movements.
Indeed, changes in the molecular phenotype of CNS cells after TBI may provide an insight into the underlying mechanisms of pathology and recovery, and offer novel therapeutic targets. Based on our previous finding that TBI causes prolonged microglia activation at a distal location from the injury site with a persistent increase in the expression of BDNF, we continued to characterize phenotypic changes in this vital region of the neuraxis. Specifically, in the present study, we investigated the possible role of reactive microglia in promoting trophic (cell survival) and tropic (axonal growth) support in the white and gray matters of the spinal cord, where compensatory axon sprouting may occur in monkeys who recovered motor function, by immunohistochemically assessing the levels of select proteins thought to be involved in such events.
We previously used an antibody against CD68 to identify the population of reactive microglia/macrophage (Nagamoto-Combs et al., 2007). Here, we chose to use an antibody against MHC-II that is also a common marker of reactive phenotype changes in microglia. It has been shown that MHC-II is basally expressed, and the level of its expression increases in activated microglia in vivo (Redwine et al., 2001; Stoll et al., 2006; Streit et al., 1989) and in vitro (Hamo et al., 2007) during diseased or other inflammatory conditions. In addition, immunostaining against this molecule provided a more defined profile of the cell morphology. As we expected from the prior CD68 staining, MHC-II expression was significantly elevated in the cervical spinal cords of the lesioned animals (Fig. 2). Increases in the number of MHC-II + cells displaying characteristic reactive microglia morphology were especially striking at 1 and 6 m.p.l. in the LCST, indicating a large population of microglia remains activated up to 12 months at a distal location from the injury, as we had observed with CD68-IR (Nagamoto-Combs et al., 2007).
Since TBI-activated microglia express BDNF and its receptor, TrkB (Nagamoto-Combs et al., 2007), it is reasonable to hypothesize that the activated microglia provide neurotrophic support to local cells, including microglia themselves, by secreting BDNF. The presence of pERK1/2 within the MCH-II positive reactive microglia found in the LCST supports this hypothesis (Figs. 3&5). At the time points we examined (i.e., 1, 6, and 12 m.p.l.), significant pERK1/2-IR was not observed in the spinal gray matter or cortical neurons near the injury site (Nagamoto-Combs, unpublished observation). A similar result from a short-term study has been reported in rats, describing increased ERK phosphorylation in non-neuronal cells within the perilesional subcortical white matter, but not in neurons forming the cortical gray matter, for up to 3 days after fluid percussion injury (Raghupathi et al., 2003). Other studies that examined acute ERK activation in a rat TBI model, however, have described that the neuronal ERK activation occurs rapidly and transiently, especially in the damaged neurons around the injury site (Clausen et al., 2004; Otani et al., 2002a, 2002b). If this phenomenon occurs in the non-human primate CNS, it is likely that any phosphorylation of neuronal ERK1/2 in our paradigm might have occurred immediately after TBI in the cortical region perilesionally and/or subsided by the time of our examinations at 1, 6, and 12 m.p.l. Although ERK activation has been shown to support cell survival and differentiation (Bonni et al., 1999), the observations that pharmacological inhibition of ERKs leads to decreased secondary cell damage and better functional outcome after TBI (Clausen et al., 2004; Enomoto et al., 2005; Otani et al., 2007) make the role of ERKs rather complex, requiring further investigation to identify the precise function of these kinases in TBI, particularly within the chronically activated microglia in our paradigm.
In order to define a potential relationship between activated microglia and axonal growth/sprouting and synaptic formation, we examined the expression of GAP-43 and synaptophysin in the cervical spinal cord after TBI. Prior work has demonstrated that close association of reactive microglia and sprouting neurites occurs after striatal wound in mice (Batchelor et al., 2002) and needle injury in the sensorimotor cortex of the rat (King et al., 2001), suggesting that activated microglia can serve a role in promoting axonal growth and/or synaptic formation likely by supplying trophic and/or tropic factors, such as BDNF (Batchelor et al., 2008). In order to assess whether microglia in our paradigm had a similar tropic role, we first examined the state of axonal growth and sprouting by determining the levels of GAP-43, which has been well described for its association with presynaptic terminal membranes and its ability to promote process outgrowth and synaptic connections (see review by Benowitz and Routtenberg, 1997). Our results demonstrated that GAP-43-IR was more prominent in the lesioned animals than the sham-operated animal in both the white and gray matter (Fig. 4). Since activation of GAP-43 occurs upon phosphorylation by protein kinase C, which can be activated by a variety of extracellular signals such as growth factors and cytokines, it is possible that BDNF secreted from the local reactive microglia stimulates axonal processes to maintain the growth-promoting function of GAP-43 at the nerve endings of sprouting axons. The close association between the GAP-43-IR and MHC-II-IR we observed in our fluorescent double staining confocal microscopy supports this possibility (Fig. 5D–F).
We also examined post-lesion changes in another presynaptic protein, synaptophysin, to continue correlating the apparent sprouting response with maintained microgliosis. Although there seem to be modest increases in the LCST of some lesioned animals, such increases were of magnitudes that were less than what we observed with GAP-43. This observation may suggest that the GAP-43 immunoreactive sprouting associated with microglia was not being translated into aberrant functional synapses consistent with the recovery of motor function in these animals. It is feasible that the microglia offer a guidance system for the sprouting axons from undamaged motor cortex areas. Indeed, recent data derived from a rodent spinal cord injury model demonstrated that increasing microglial activation and BDNF expression via administration of interleukin-12 facilitated axon remyelination and functional recovery (Yaguchi et al., 2008). Batchelor and colleagues (2000) have also demonstrated in a rodent striatal injury paradigm that decreased amount of BDNF expressed in the striatum leads to decreased amount of dopaminergic fiber sprouting after injury. In their subsequent studies, they showed that the sprouting fibers were closely and intricately associated with activated microglia and macrophages in the periwound area where these cells expressed high concentrations of BDNF and GDNF, respectively (Batchelor et al., 2002). In addition, introduction of additional BDNF and GDNF promoted sprouting within the wound care where sprouting did not normally occur (Batchelor et al., 2008), further suggesting that activated microglia play a supportive role for sprouting axons.
On the other hand, the presence of reactive microglia may also produce neuronal dysfunction by specifically impairing axonal transport of synaptic vesicles. For example, Stagi and co-workers (2005) demonstrated that nitric oxide derived from activated microglia is capable of inhibiting axonal transport of synaptophysin. This may suggest that the reactive microglia might instead be impairing functional recovery post-lesion. Alternatively, this inhibitory effect of reactive microglia could be preventing sprouting axons from making inappropriate synaptic contact en route. Since synaptic interaction between neurons and immune cells such as macrophages have been previously reported (Felten et al., 1987; Straub et al., 1998), it is possible that the trophic factor-secreting microglia may communicate reciprocally with the growing axonal terminals, forming aberrant synaptic contact with microglia before they reach their appropriate target during recovery. Nonetheless, determining whether the prolonged gliosis is primarily beneficial or detrimental to the recovery process will ultimately have to be defined by manipulating microglial phenotype.
Regardless of the impact of the gliosis, the notable attenuation of MCH-II-IR along with GAP-43 in the LCST after 12 months of recovery suggests that the process of axonal regrowth/sprouting is actively regulated in this region. To begin determining what factors may be involved, we next examined the tissue for changes in proteins known to regulate process outgrowth in other paradigms. In particular, we examined expression of NgR, which has been shown to interact with myelin-associated growth proteins, such as Nogo A, myelin-associated glycoprotein (MAG), and oligodendrocyte myelin glycoprotein (OMgp) to inhibit axonal growth in the CNS (see review by Schwab, 2004). Activation of NgR has been shown to inhibit axonal growth, resulting in growth-cone collapse (Liu et al., 2002). On the other hand, inhibition of its activity by NgR antagonist peptides (GrandPré et al., 2002; Li and Strittmatter, 2003) and an antibody against its ligand, Nogo A (Bregman et al., 1995; Schnell and Schwab, 1990), is associated with promotion of axonal growth. Although NgR is generally observed in neurons, a recent study has found increased levels of NgR expression within astrocytes and microglia in the brains of multiple sclerosis patients (Satoh et al., 2005), and has been suggested to play a role in inflammatory and disease conditions (David et al., 2008). Our staining showed elevated NgR, but not Nogo A, in the LCST in the lesioned animals, especially in the 1- and 6-m.p.l. animals (Fig. 7). This result seems contradictory with our observations for GAP-43 and synaptophysin, but it may suggest a regulatory role of this receptor in restricting aberrant sprouting, counteracting the increased expression and/or phosphorylation of GAP-43 and synaptophysin in the LCST. Alternatively, the increased NgR in the LCST may contribute to the regulation of microglia population. Another study using a peripheral nerve-crush injury paradigm reported that NgR was expressed in migrating, phagocytic macrophages, and plays an important role in the efflux of macrophages from the injury site and thus resolution of inflammation (Fry et al., 2007). Because microglia share their monocytic origin with macrophage, they likely exploit a similar mechanism for down-regulating activation suggesting a novel function for NgR during microglial-mediated inflammatory changes.
Yet another important function of glial cells, particularly post injury, is the regulation of extracellular glutamate concentration via glutamate transporter proteins. Excitotoxic cell death caused by increases in the local concentration of glutamate is thought to be a major cause of delayed, secondary damage in the perilesional area after injury. Increased levels of glutamate and other excitatory amino acids have been found in human TBI patients (Ashwal et al., 2004; Yamamoto et al., 1999) and in fluid percussion (Faden et al., 1989), cortical contusion (Rose et al., 2002), and impact (Palmer et al., 1993) injury models in rats. At a distal site from the lesion, increases in excitatory amino acid concentration can lead to degeneration of oligodendrocytes, leading to degradation of myelin and dysfunction of myelinated axons (Matute et al., 2007). Some studies also indicated that the degree of glutamate increase after TBI is related to the severity of the injury (Faden et al., 1989) and the patient's clinical outcome (Koura et al., 1998). It is therefore crucial that glial cells maintain their capability to uptake glutamate after injury in both proximal and distal areas from the lesion site. In perilesional sites of cortical injuries, decreases in the glial levels of GLT-1 and GLAST have been reported within 72 h after injury (Rao et al., 1998; Van Landeghem et al., 2001). This change in the levels of glutamate transporters, however, is transient, perhaps due to decreases in the number of GLT-1 and GLAST expressing astrocytes (Van Landeghem et al., 2001). The levels of GLT-1 and GLAST increase and remain elevated thereafter, especially in ramified cortical microglia (Van Landeghem et al., 2001). Indeed, recent studies have found increased expression of GLT-1 in microglia activated by facial-nerve axotomy in vivo (López-Redondo et al., 2000), or by lipopolysaccharide in vitro (Persson et al., 2005), or by TBI in humans (Beschorner et al., 2007), suggesting a role for microglia as a regulator of local glutamate concentrations. In our GLT-1/GLAST immunodetection study, it was therefore our intention to substantiate this particular role of microglia, or possibly other glial cells, in our non-human primate TBI model at a site distant from the injury site where degeneration of axons from the damaged cortical neurons and sprouting from neighboring cortical area cells took place. Since oligodendrocytes, the component of myelin, are also susceptible to glutamate excitotoxicity (Matute et al., 1997), controlling glutamate concentration at a distal site could be imperative in the maintenance of favorable conditions for axonal regeneration. The elevated GLT-1/GLAST-IR we observed in the LCST and VH regions after TBI indeed supports this idea. However, the specific cell types elevating glutamate transporter levels in response to injury still need to be identified.
Although activation and proliferation of astrocytes are major events often observed after CNS injury, we did not detect significant changes in GFAP-IR in the lesioned animals we examined. This lack of indication for astrocyte activation/proliferation in the lesioned animals may be due to their relatively long-term recovery periods. It is possible that astrocytic activation/proliferation occurs more transiently in the spinal cord after cortical damage.
Our present immunohistochemical study was conducted as an exploratory investigation and was reported qualitatively. The animals utilized in this study were also part of previous and ongoing studies (Darling et al., 2009; McNeal et al., 2010), and the limitation of tissue availability, as well as animal number, did not allow quantitation of specific immunoreactivities. Therefore, representative images are necessarily presented in each Figure. Nevertheless, robust immunoreactivity profiles for microglial activation, active ERK levels, GAP-43, glutamate transporters, and Nogo R present a provocative, albeit preliminary, set of observations describing ongoing changes that occur in the distal spinal cord for up to 1 year following cortical lesion. The fact that these observations were also performed in a primate model of injury with a specific controlled lesion lends importance to the possibility of future extrapolation to human recovery-associated changes. Moreover, at the very least, these observations define a starting point for subsequent, mechanistic studies. For example, although the limitation of animal number in the current study prevented a comparison of recovery of performance scores with the immunohistochemical changes, this model provides a powerful means of fine motor recovery assessment compared to microglial phenotype manipulation in future study.
Taken together, our present results qualitatively support the hypothesis that prolonged gliosis, in particular reactive microgliosis, after TBI is supportive of functional and structural recovery. Continued quantitative collection of data detailing the gliosis and synaptic responses in this paradigm will offer clearer evidence of a specific trophic/tropic role for microglia up to at least 1 year post lesion. Prolonged microglia activation may facilitate recovery by continuously supplying trophic/tropic factor(s), including BDNF, to guide sprouting axons, while also preventing aberrant synaptic formations and/or increasing the efficacy of local glutamate removal. A schematic drawing illustrating this hypothesized function of long-term microgliosis after injury is presented in Figure 13. Future efforts to manipulate microglial phenotype by either promoting or inhibiting their activation state at specific times post lesion will determine whether their function impairs or improves regeneration and recovery. This knowledge could translate into specific temporal strategies of anti-inflammatory therapy following TBI.
FIG. 13.

Hypothesized contribution of prolonged microglial activation to surviving neuron sprouting and functional recovery. Potential events following injury to the upper motor neuron in the primary motor cortex or lateral premotor cortex (M1/LPMC) are schematically presented. Injury-mediated loss of the cortical motor neuron (shown as the neuron with dotted outline) causes degeneration of its descending axon and activation of microglia in the spinal cord. These reactive microglia may promote neurite sprouting from spared neurons of the supplemental motor cortex (Suppl. Ctx) by supplying trophic factors such as BDNF as well as by preventing aberrant synapse formation. They may also participate in maintenance of local environment by facilitating glutamate uptake and phagocytosis.
Acknowledgments
This study was supported by National Institutes of Health grants, NS046367 and 5P20RR017699-08, and The South Dakota Spinal Cord and Traumatic Brain Injury Research Council.
Author Disclosure Statement
No competing financial interests exist.
References
- Agrawal S.K. Fehlings M.G. Role of NMDA and non-NMDA ionotropic glutamate receptors in traumatic spinal cord axonal injury. J. Neurosci. 1997;17:1055–1063. doi: 10.1523/JNEUROSCI.17-03-01055.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal S.K. Theriault E. Fehlings M. Role of group I metabotropic glutamate receptors in traumatic spinal cord white matter injury. J. Neurotrauma. 1998;15:929–941. doi: 10.1089/neu.1998.15.929. [DOI] [PubMed] [Google Scholar]
- Ashwal S. Holshouser B. Tong K. Serna T. Osterdock R. Gross M. Kido D. Proton MR spectroscopy detected glutamate/glutamine is increased in children with traumatic brain injury. J. Neurotrauma. 2004;21:1539–1552. doi: 10.1089/neu.2004.21.1539. [DOI] [PubMed] [Google Scholar]
- Batchelor P.E. Liberatore G.T. Porritt M.J. Donnan G.A. Howells D.W. Inhibition of brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor expression reduces dopaminergic sprouting in the injured striatum. Eur. J. Neurosci. 2000;12:3462–3468. doi: 10.1046/j.1460-9568.2000.00239.x. [DOI] [PubMed] [Google Scholar]
- Batchelor P.E. Porritt M.J. Martinello P. Parish C.L. Liberatore G.T. Donnan G.A. Howells D.W. Macrophages and microglia produce local trophic gradients that stimulate axonal sprouting toward but not beyond the wound edge. Mol. Cell Neurosci. 2002;21:436–453. doi: 10.1006/mcne.2002.1185. [DOI] [PubMed] [Google Scholar]
- Batchelor P.E. Wills T.E. Hewa A.P. Porritt M.J. Howells D.W. Stimulation of axonal sprouting by trophic factors immobilized within the wound core. Brain Res. 2008;1209:49–56. doi: 10.1016/j.brainres.2008.02.098. [DOI] [PubMed] [Google Scholar]
- Benowitz L.I. Routtenberg A. GAP-43: An intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 1997;20:84–91. doi: 10.1016/s0166-2236(96)10072-2. [DOI] [PubMed] [Google Scholar]
- Beschorner R. Dietz K. Schauer N. Mittelbronn M. Schluesener H.J. Trautmann K. Meyermann R. Simon P. Expression of EAAT1 reflects a possible neuroprotective function of reactive astrocytes and activated microglia following human traumatic brain injury. Histol. Histopathol. 2007;22:515–526. doi: 10.14670/HH-22.515. [DOI] [PubMed] [Google Scholar]
- Bonni A. Brunet A. West A.E. Datta S.R. Takasu M.A. Greenberg M.E. Cell survival promoted by the ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. doi: 10.1126/science.286.5443.1358. [DOI] [PubMed] [Google Scholar]
- Bregman B.S. Kunkel-Bagden E. Schnell L. Dai H.N. Gao D. Schwab M.E. Recovery from spinal cord injury mediated by antibodies to neurite growth inhibitors. Nature. 1995;378:498–501. doi: 10.1038/378498a0. [DOI] [PubMed] [Google Scholar]
- Bütefisch C. Neurobiological bases of rehabilitation. Neurol. Sci. Suppl. 2006;1:S18–S23. doi: 10.1007/s10072-006-0540-z. [DOI] [PubMed] [Google Scholar]
- Carmichael S.T. Plasticity of cortical projections after stroke. Neuroscientist. 2003a;9:64–75. doi: 10.1177/1073858402239592. [DOI] [PubMed] [Google Scholar]
- Carmichael S.T. Gene expression changes after focal stroke, traumatic brain and spinal cord injuries. Curr. Opin. Neurol. 2003b;16:699–704. doi: 10.1097/01.wco.0000102621.38669.77. [DOI] [PubMed] [Google Scholar]
- Clausen F. Lundqvist H. Ekmark S. Lewen A. Ebendal T. Hillered L. Oxygen free radical-dependent activation of extracellular signal-regulated kinase mediates apoptosis-like cell death after traumatic brain injury. J. Neurotrauma. 2004;21:1168–1182. doi: 10.1089/neu.2004.21.1168. [DOI] [PubMed] [Google Scholar]
- Conti A.C. Raghupathi R. Trojanowski J.Q. Mcintosh T.K. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J. Neurosci. 1998;18:5663–5672. doi: 10.1523/JNEUROSCI.18-15-05663.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtine G. Bunge M.B. Fawcett J.W. Grossman R.G. Kaas J.H. Lemon R. Maier I. Martin J. Nudo R.J. Ramon-Cueto A. Rouiller E.M. Schnell L. Wannier T. Schwab M.E. Edgerton V.R. Can experiments in nonhuman primates expedite the translation of treatments for spinal cord injury in humans? Nat. Med. 2007;13:561–566. doi: 10.1038/nm1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czigner A. Mihály A. Farkas O. Büki A. Krisztin-Péva B. Dobó E. Barzó P. Kinetics of the cellular immune response following closed head injury. Acta Neurochir. (Wien) 2007;149:281–289. doi: 10.1007/s00701-006-1095-8. [DOI] [PubMed] [Google Scholar]
- Dancause N. Barbay S. Frost S.B. Plautz E.J. Chen D. Zoubina E.V. Stowe A.M. Nudo R.J. Extensive cortical rewiring after brain injury. J. Neurosci. 2005;25:10167–10179. doi: 10.1523/JNEUROSCI.3256-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling W.G. Peterson C.R. Herrick J.L. McNeal D.W. Stilwell-Morecraft K.S. Morecraft R.J. Measurement of coordination of object manipulation in non-human primates. J. Neurosci. Methods. 2006;154:38–44. doi: 10.1016/j.jneumeth.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Darling W.G. Pizzimenti M.A. Rotella D.L. Peterson C.R. Ge J. Cline K. McNeal D.W. Stilwell-Morecraft K.S. Morecraft R.J. Volumetric effects of frontal motor cortex injury on recovery of dexterous movements. Exp. Neurol. 2009;220:90–108. doi: 10.1016/j.expneurol.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S. Fry E.J. López-Vales R. Novel roles for Nogo receptor in inflammation and disease. Trends Neurosci. 2008;31:221–226. doi: 10.1016/j.tins.2008.02.002. [DOI] [PubMed] [Google Scholar]
- Dietrich W. Alonso O. Halley M. Early microvascular and neuronal consequences of traumatic brain injury: A light and electron microscopic study in rats. J. Neurotrauma. 1994;11:289–301. doi: 10.1089/neu.1994.11.289. [DOI] [PubMed] [Google Scholar]
- Enomoto T. Osugi T. Satoh H. Mcintosh T.K. Nabeshima T. Pre-injury magnesium treatment prevents traumatic brain injury-induced hippocampal ERK activation, neuronal loss, and cognitive dysfunction in the radial-arm maze test. J. Neurotrauma. 2005;22:783–792. doi: 10.1089/neu.2005.22.783. [DOI] [PubMed] [Google Scholar]
- Faden A. Demediuk P. Panter S. Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- Felten D. Felten S. Bellinger D. Carlson S. Ackerman K. Madden K. Olschowki J. Livnat S. Noradrenergic sympathetic neural interactions with the immune system: Structure and function. Immunol. Rev. 1987;100:225–260. doi: 10.1111/j.1600-065x.1987.tb00534.x. [DOI] [PubMed] [Google Scholar]
- Fournier A.E. Grandpre T. Strittmatter S.M. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature. 2001;409:341–346. doi: 10.1038/35053072. [DOI] [PubMed] [Google Scholar]
- Fry E.J. Ho C. David S. A role for Nogo receptor in macrophage clearance from injured peripheral nerve. Neuron. 2007;53:649–662. doi: 10.1016/j.neuron.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Fukuda S. Del Zoppo G.J. Models of focal cerebral ischemia in the nonhuman primate. ILAR J. 2003;44:96–104. doi: 10.1093/ilar.44.2.96. [DOI] [PubMed] [Google Scholar]
- Gehrmann J. Matsumoto Y. Kreutzberg G.W. Microglia: Intrinsic immuneffector cell of the brain. Brain Res. Rev. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- GrandPré T. Li S. Strittmatter S.M. Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature. 2002;417:547–551. doi: 10.1038/417547a. [DOI] [PubMed] [Google Scholar]
- Hamo L. Stohlman S.A. Otto-Duessel M. Bergmann C.C. Distinct regulation of MHC molecule expression on astrocytes and microglia during viral encephalomyelitis. Glia. 2007;55:1169–1177. doi: 10.1002/glia.20538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge C.W. Mcgurk D. Thomas J.L. Cox A.L. Engle C.C. Castro C.A. Mild traumatic brain injury in U.S. Soldiers returning from Iraq. New Eng. J. Med. 2008;358:453–463. doi: 10.1056/NEJMoa072972. [DOI] [PubMed] [Google Scholar]
- Jara J.H. Singh B.B. Floden A.M. Combs C.K. Tumor necrosis factor alpha stimulates NMDA receptor activity in mouse cortical neurons resulting in ERK-dependent death. J. Neurochem. 2007;100:1407–1420. doi: 10.1111/j.1471-4159.2006.04330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karns L. Ng S. Freeman J. Fishman M. Cloning of complementary DNA for GAP-43, a neuronal growth-related protein. Science. 1987;236:597–600. doi: 10.1126/science.2437653. [DOI] [PubMed] [Google Scholar]
- King C.E. Canty A.J. Vickers J.C. Alterations in neurofilaments associated with reactive brain changes and axonal sprouting following acute physical injury to the rat neocortex. Neuropathol. Appl. Neurobiol. 2001;27:115–126. doi: 10.1046/j.1365-2990.2001.00317.x. [DOI] [PubMed] [Google Scholar]
- Koura S. Doppenberg E. Marmarou A. Choi S. Young H. Bullock R. Relationship between excitatory amino acid release and outcome after severe human head injury. Acta Neurochir. Suppl. 1998;71:244–246. doi: 10.1007/978-3-7091-6475-4_70. [DOI] [PubMed] [Google Scholar]
- Kuhtz-Buschbeck J.P. Hoppe B. Gölge M. Dreesmann M. Damm-Stünitz U. Ritz A. Sensorimotor recovery in children after traumatic brain injury: Analyses of gait, gross motor, and fine motor skills. Dev. Med. Child Neurol. 2003;45:821–828. doi: 10.1017/s001216220300152x. [DOI] [PubMed] [Google Scholar]
- Langlois J.A. Rutland-Brown W. Thomas K.E. Atlanta, GA: Centers for Disease Control and Prevention, National Center for Injury Prevention and Control; 2004. Traumatic brain injury in the United States: Emergency department visits, hospitalizations, and deaths. [Google Scholar]
- Leker R.R. Shohami E. Cerebral ischemia and trauma--different etiologies yet similar mechanisms: Neuroprotective opportunities. Brain Res. Brain Res. Rev. 2002;39:55–73. doi: 10.1016/s0165-0173(02)00157-1. [DOI] [PubMed] [Google Scholar]
- Lemon R.N. Griffiths J. Comparing the function of the corticospinal system in different species: Organizational differences in motor function? Muscle Nerve. 2005;32:261–279. doi: 10.1002/mus.20333. [DOI] [PubMed] [Google Scholar]
- Li S. Strittmatter S.M. Delayed systemic Nogo-66 receptor antagonist promotes recovery from spinal cord injury. J. Neurosci. 2003;23:4219–4227. doi: 10.1523/JNEUROSCI.23-10-04219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B.P. Fournier A. GrandPré T. Strittmatter S.M. Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science. 2002;297:1190–1193. doi: 10.1126/science.1073031. [DOI] [PubMed] [Google Scholar]
- López-Redondo F. Nakajima K. Honda S. Kohsaka S. Glutamate transporter GLT-1 is highly expressed in activated microglia following facial nerve axotomy. Mol. Brain Res. 2000;76:429–435. doi: 10.1016/s0169-328x(00)00022-x. [DOI] [PubMed] [Google Scholar]
- Lotze M. Grodd W. Rodden F.A. Gut E. Schonle P.W. Kardatzki B. Cohen L.G. Neuroimaging patterns associated with motor control in traumatic brain injury. Neurorehabil. Neural Repair. 2006;20:14–23. doi: 10.1177/1545968305282919. [DOI] [PubMed] [Google Scholar]
- Mark V.W. Taub E. Morris D.M. Neuroplasticity and constraint-induced therapy. Eura. Medicophy. 2006;42:269–284. [PubMed] [Google Scholar]
- Matute C. Alberdi E. Domercq M. Sánchez-Gómez M.V. Pérez-Samartín A. Rodríguez-Antigüedad A. Pérez-Cerdá F. Excitotoxic damage to white matter. J. Anat. 2007;210:693–702. doi: 10.1111/j.1469-7580.2007.00733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matute C. Sánchez-Gómez M.V. Martínez-Millán L. Miledi R. Glutamate receptor-mediated toxicity in optic nerve oligodendrocytes. Proc. Natl. Acad. Sci. USA. 1997;94:8830–8835. doi: 10.1073/pnas.94.16.8830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeal D.W. Darling W.G. Ge J. Stilwell-Morecraft K.S. Solon K. Hynes S.M. Pizzimenti M.A. Rotella D.L. T V. Morecraft R.J. Selective long-term reorganization of the corticospinal projection from the supplementary motor cortex following recovery from lateral motor cortex injury. J. Comp. Neurol. 2010;518:586–621. doi: 10.1002/cne.22218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morecraft R.J. Cipolloni P. Stilwell-Morecraft K. Gedney M. Pandya D. Cytoarchitecture and cortical connections of the posterior cingulate and adjacent somatosensory fields in the rhesus monkey. J. Comp. Neurol. 2004;469:37–69. doi: 10.1002/cne.10980. [DOI] [PubMed] [Google Scholar]
- Morecraft R.J. Geula C. Mesulam M. Cytoarchitecture and neural afferents of orbitofrontal cortex in the brain of the monkey. J. Comp. Neurol. 1992;323:341–358. doi: 10.1002/cne.903230304. [DOI] [PubMed] [Google Scholar]
- Morecraft R.J. Herrick J.L. Stilwell-Morecraft K.S. Louie J.L. Schroeder C.M. Ottenbacher J.G. Schoolfield M.W. Localization of arm representation in the corona radiata and internal capsule in the non-human primate. Brain. 2002;125:176–198. doi: 10.1093/brain/awf011. [DOI] [PubMed] [Google Scholar]
- Morecraft R.J. Louie J.L. Herrick J.L. Stilwell-Morecraft K.S. Cortical innervation of the facial nucleus in the non-human primate: A new interpretation of the effects of stroke and related subtotal brain trauma on the muscles of facial expression. Brain. 2001;124:176–208. doi: 10.1093/brain/124.1.176. [DOI] [PubMed] [Google Scholar]
- Morecraft R.J. McNeal D. Stilwell-Morecraft K. Dvanajscak Z. Ge J. Schneider P. Localization of arm representation in the cerebral peduncle of the non-human primate. J. Comp. Neurol. 2007;504:149–167. doi: 10.1002/cne.21438. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossman M.C. Lenzlinger P.M. Hans V. Stahel P. Csuka E. Ammann E. Stocker R. Trentz O. Kossmann T. Production of cytokines following brain injury: Beneficial and deleterious for the damaged tissue. Mol. Psychiatry 2, 1997:133. doi: 10.1038/sj.mp.4000227. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossman M.C. Satgunaseelan L. Bye N. Kossmann T. Modulation of immune response by head injury. Injury. 2007;38:1392–1400. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Nagamoto-Combs K. McNeal D.W. Morecraft R.J. Combs C.K. Prolonged microgliosis in the rhesus monkey central nervous system after traumatic brain injury J. Neurotrauma. 2007;24:1719–1742. doi: 10.1089/neu.2007.0377. [DOI] [PubMed] [Google Scholar]
- Newcomb J.K. Zhao X. Pike B.R. Hayes R.L. Temporal profile of apoptotic-like changes in neurons and astrocytes following controlled cortical impact injury in the rat. Exp. Neurol. 1999;158:76–88. doi: 10.1006/exnr.1999.7071. [DOI] [PubMed] [Google Scholar]
- Nudo R. Recovery after damage to motor cortical areas. Curr. Opin. Neurobiol. 1999;9:740–747. doi: 10.1016/s0959-4388(99)00027-6. [DOI] [PubMed] [Google Scholar]
- Okie S. Traumatic brain injury in the war zone. New Eng. J. Med. 2005;352:2043–2047. doi: 10.1056/NEJMp058102. [DOI] [PubMed] [Google Scholar]
- Otani N. Nawashiro H. Fukui S. Nomura N. Shima K. Temporal and spatial profile of phosphorylated mitogen-activated protein kinase pathways after lateral fluid percussion injury in the cortex of the rat brain. J. Neurotrauma. 2002a;19:1587–1596. doi: 10.1089/089771502762300247. [DOI] [PubMed] [Google Scholar]
- Otani N. Nawashiro H. Fukui S. Nomura N. Yano A. Miyazawa T. Shima K. Differential activation of mitogen-activated protein kinase pathways after traumatic brain injury in the rat hippocampus. J. Cereb. Blood Flow Metab. 2002b;22:327–334. doi: 10.1097/00004647-200203000-00010. [DOI] [PubMed] [Google Scholar]
- Otani N. Nawashiro H. Fukui S. Ooigawa H. Ohsumi A. Toyooka T. Shima K. Role of the activated extracellular signal-regulated kinase pathway on histological and behavioral outcome after traumatic brain injury in rats. J. Clin. Neurosci. 2007;14:42–48. doi: 10.1016/j.jocn.2005.11.044. [DOI] [PubMed] [Google Scholar]
- Ouardouz M. Coderre E. Zamponi G.W. Hameed S. Yin X. Trapp B.D. Stys P.K. Glutamate receptors on myelinated spinal cord axons: II. AMPA and GluR5 receptors. Ann. Neurol. 2009;65:160–166. doi: 10.1002/ana.21539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer A.M. Marion D.W. Botscheller M.L. Swedlow P.E. Styren S.D. Dekosky S.T. Traumatic brain injury-induced excitotoxicity assessed in a controlled cortical impact model. J. Neurochem. 1993;61:2015–2024. doi: 10.1111/j.1471-4159.1993.tb07437.x. [DOI] [PubMed] [Google Scholar]
- Persson M. Brantefjord M. Hansson E. Rönnbäck L. Lipopolysaccharide increases microglial GLT-1 expression and glutamate uptake capacity in vitro by a mechanism dependent on TNF-alpha. Glia. 2005;51:111–120. doi: 10.1002/glia.20191. [DOI] [PubMed] [Google Scholar]
- Pizzimenti M.A. Darling W.G. Rotella D.L. Mcneal D.W. Herrick J.L. Ge J. Stilwell-Morecraft K.S. Morecraft R.J. Measurement of reaching kinematics and prehensile dexterity in nonhuman primates. J. Neurophysiol. 2007;98:1015–1029. doi: 10.1152/jn.00354.2007. [DOI] [PubMed] [Google Scholar]
- Popovich P.G. Wei P. Stokes B.T. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J. Comp. Neurol. 1997;377:443–464. doi: 10.1002/(sici)1096-9861(19970120)377:3<443::aid-cne10>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Raghupathi R. Muir J.K. Fulp C.T. Pittman R.N. Mcintosh T.K. Acute activation of mitogen-activated protein kinases following traumatic brain injury in the rat: Implications for posttraumatic cell death. Exp. Neurol. 2003;183:438–448. doi: 10.1016/s0014-4886(03)00166-3. [DOI] [PubMed] [Google Scholar]
- Rao V.L. Baskaya M.K. Dogan A. Rothstein J.D. Dempsey R.J. Traumatic brain injury down-regulates glial glutamate transporter (GLT-1 and GLAST) proteins in rat brain. J. Neurochem. 1998;70:2020–2027. doi: 10.1046/j.1471-4159.1998.70052020.x. [DOI] [PubMed] [Google Scholar]
- Redwine J.M. Buchmeier M.J. Evans C.F. In vivo expression of major histocompatibility complex molecules on oligodendrocytes and neurons during viral infection. Am. J. Pathol. 2001;159:1219–1224. doi: 10.1016/S0002-9440(10)62507-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rink A. Fung K.M. Trojanowski J.Q. Lee V.M. Neugebauer E. Mcintosh T.K. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am. J. Pathol. 1995;147:1575–1583. [PMC free article] [PubMed] [Google Scholar]
- Rose M.E. Huerbin M.B. Melick J. Marion D.W. Palmer A.M. Schiding J.K. Kochanek P.M. Graham S.H. Regulation of interstitial excitatory amino acid concentrations after cortical contusion injury. Brain Res. 2002;935:40–46. doi: 10.1016/s0006-8993(02)02445-9. [DOI] [PubMed] [Google Scholar]
- Satoh J. Onoue H. Arima K. Yamamura T. Nogo-A and Nogo receptor expression in demyelinating lesions of multiple sclerosis. J. Neuropathol. Exp. Neurol. 2005;64:129–138. doi: 10.1093/jnen/64.2.129. [DOI] [PubMed] [Google Scholar]
- Sbordone R. Liter J. Pettler-Jennings P. Recovery of function following severe traumatic brain injury: A retrospective 10-year follow-up. Brain Inj. 1995;9:285–299. doi: 10.3109/02699059509008199. [DOI] [PubMed] [Google Scholar]
- Schnell L. Schwab M.E. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature. 1990;343:269–272. doi: 10.1038/343269a0. [DOI] [PubMed] [Google Scholar]
- Schwab M.E. Nogo and axon regeneration. Curr. Opin. Neurobiol. 2004;14:118–124. doi: 10.1016/j.conb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Stagi M. Dittrich P.S. Frank N. Iliev A.I. Schwille P. Neumann H. Breakdown of axonal synaptic vesicle precursor transport by microglial nitric oxide. J. Neurosci. 2005;25:352–362. doi: 10.1523/JNEUROSCI.3887-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll M. Capper D. Dietz K. Warth A. Schleich A. Schlaszus H. Meyermann R. Mittelbronn M. Differential microglial regulation in the human spinal cord under normal and pathological conditions. Neuropathol. Appl. Neurobiol. 2006;32:650–661. doi: 10.1111/j.1365-2990.2006.00774.x. [DOI] [PubMed] [Google Scholar]
- Straub R.H. Westermann J. Schölmerich J. Falk W. Dialogue between the CNS and the immune system in lymphoid organs. Immunol. Today. 1998;19:409–413. doi: 10.1016/s0167-5699(98)01297-3. [DOI] [PubMed] [Google Scholar]
- Streit W. Microglial response to brain injury: A brief synopsis. Toxicol. Pathol. 2000;28:28–30. doi: 10.1177/019262330002800104. [DOI] [PubMed] [Google Scholar]
- Streit W.J. Graeber M.B. Kreutzberg G.W. Expression of Ia antigen on perivascular and microglial cells after sublethal and lethal motor neuron injury. Exp. Neurol. 1989;105:115–126. doi: 10.1016/0014-4886(89)90111-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekkok S.B. Goldberg M.P. AMPA/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J. Neurosci. 2001;21:4237–4248. doi: 10.1523/JNEUROSCI.21-12-04237.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Landeghem F. Stover J. Bechmann I. Brück W. Unterberg A. Bührer C. Von Deimling A. Early expression of glutamate transporter proteins in ramified microglia after controlled cortical impact injury in the rat. Glia. 2001;35:167–179. doi: 10.1002/glia.1082. [DOI] [PubMed] [Google Scholar]
- Wade D. Wood V. Hewer R. Recovery after stroke – The first 3 months. J. Neurol. Neurosurg. Psychiatry. 1985;48:7–13. doi: 10.1136/jnnp.48.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xydakis M.S. Fravell M.D. Nasser K.E. Casler J.D. Analysis of battlefield head and neck injuries in Iraq and Afghanistan. Otolaryngol. Head Neck Surg. 2005;133:497–504. doi: 10.1016/j.otohns.2005.07.003. [DOI] [PubMed] [Google Scholar]
- Yaguchi M. Ohta S. Toyama Y. Kawakami Y. Toda M. Functional recovery after spinal cord injury in mice through activation of microglia and dendritic cells after IL-12 administration. J. Neurosci. Res. 2008;86:1972–1980. doi: 10.1002/jnr.21658. [DOI] [PubMed] [Google Scholar]
- Yamamoto T. Rossi S. Stiefel M. Doppenberg E. Zauner A. Bullock R. Marmarou A. CSF and ECF glutamate concentrations in head injured patients. Acta Neurochir. Suppl. 1999;75:17–19. doi: 10.1007/978-3-7091-6415-0_4. [DOI] [PubMed] [Google Scholar]