

Abstract
Overexpression of β-catenin, a protein that functions in both cell adhesion and signaling, causes expansion of the cerebral cortical precursor population and cortical surface area enlargement. Here, we find that focal elimination of β-catenin from cortical neural precursors in vivo causes premature neuronal differentiation. Precursors within the cerebral cortical ventricular zone exhibit robust β-catenin-mediated transcriptional activation, which is downregulated as cells exit the ventricular zone. Targeted inhibition of β-catenin signaling during embryonic development causes cortical precursor cells to prematurely exit the cell cycle, differentiate into neurons, and migrate to the cortical plate. These results show that β-catenin-mediated transcriptional activation functions in the decision of cortical ventricular zone precursors to proliferate or differentiate during development, and suggest that the cell-autonomous signaling activity of β-catenin can control the production of cortical neurons and thus regulate cerebral cortical size.
Keywords: neural precursor, β-catenin, development, cerebral cortex, neurogenesis, cell cycle
Introduction
The mammalian cerebral cortex is characterized by a disproportionate increase in surface area during evolution. The growth of the surface area of the mammalian neocortex is thought to result from increases in the number of columnar functional units generated from a corresponding increased number of progenitor cells (Rakic, 1995). It has been proposed that subtle changes in the relative production of progenitor cells and neurons can lead to striking variations in neuronal number and thus cortical size (Caviness et al., 1995; Rakic, 1995).
Progenitor cell production and differentiation is regulated in a variety of developing tissues by the Wnt signaling pathway (Reya and Clevers, 2005). A crucial component of the Wnt signaling pathway is β-catenin, a multifunctional protein that can function in gene transcription and cell adhesion (Nelson and Nusse, 2004). Although transcriptional activation by β-catenin is believed to help drive cell proliferation, recent studies suggest that cell growth control by β-catenin may be more complex (Peifer and Tepass, 2000). Although the degree to which the signaling and adhesive functions of β-catenin interconnect remains less well understood, recent studies suggest that both signaling and adhesion function in the organization and maintenance of some stem cells (Song et al., 2002).
Wnt signaling and β-catenin play vital roles in mammalian neural development. Inactivation of specific Wnts (McMahon and Bradley, 1990; Lee et al., 2000), T cell factor (TCF)/lymphoid-enhancer factor (LEF) members (Galceran et al., 2000), or β-catenin (Brault et al., 2001) results in specific developmental brain defects. In the developing cerebral cortex, Wnt 7a and Wnt 7b promote the proliferation of neurogenic progenitors (Viti et al., 2003), and overexpression of a stabilized β-catenin in cortical neural precursors leads to increased cell cycle reentry and subsequent overproduction of neurons (Chenn and Walsh, 2002). Conditional deletion of β-catenin from cortical neural precursors results in disruptions in the neuroepithelium, loss of adherens junctions, impaired radial migration of neurons, and decreased cell proliferation (Machon et al., 2003), and alterations in dorsal–ventral patterning (Backman et al., 2005).
Traditional knock-out approaches eliminated β-catenin on a tissue-wide scale, making it difficult to determine whether the observed phenotypes might have been secondary to tissue disruption. Conditional deletion of β-catenin from precursors early in cortical development caused loss of epithelial integrity, delamination of neuroepithelial cells, and apoptosis (Junghans et al., 2005). The apparent lack of β-catenin transcriptional activation and detectable nuclear β-catenin, led to the speculation that the role of β-catenin in cell adhesion rather than signaling was important during early telencephalic development (Junghans et al., 2005). In contrast, similar conditional deletion experiments were seen to cause shifts in dorsal–ventral cell patterning (Backman et al., 2005), suggesting alterations in signaling. Because interpretation of tissue-level loss- or gain-of-function experiments is complicated by the importance of tissue architecture in cell growth control and proliferation (Bilder et al., 2000; Peifer, 2000), we chose to examine the cell-autonomous function of β-catenin in neural development by focal loss-of-function studies in a wild-type tissue background using in vivo electroporation approaches.
Materials and Methods
Animals.
All timed-pregnant Swiss-Webster mice (Charles River Laboratories, Wilmington, MA) were treated according to protocols reviewed and approved by the institutional animal care and use committees of Northwestern University. β-Cateninflox/flox mice (B6.129-Ctnnb1tm2Kem/KnwJ) were obtained from The Jackson Laboratory (Bar Harbor, ME). Mice were genotyped according to The Jackson Laboratory protocols available at http://jaxmice.jax.org/pub-cgi/protocols/protocols.sh?objtype=protocol&protocol_id=471.
Plasmids.
DN-TCF4 cDNA (Tetsu and McCormick, 1999) (a kind gift from Osamu Tetsu and Frank McCormick, University of California, San Francisco, CA) was subcloned into the pEGFP-N1 plasmid (Clontech, Cambridge, UK) for expression of N-terminal enhanced green fluorescent protein (eGFP)-tagged proteins. GFP-DN-TCF4 cDNA was subcloned into the pCAG electroporation vector, which directs subcloned gene expression in both precursors and postmitotic progeny (Niwa et al., 1991). The pCAG vector was modified by the addition of an internal ribosome entry site (IRES) upstream of the eGFP coding sequence (Clontech) to generate the pCAG-IRES-GFP vector, which directs gene expression from the CAG promoter and eGFP expression through the IRES. Inhibitor of catenin and TCF-4 (ICAT) cDNA [from Cara Gottardi (Northwestern University, Chicago, IL)] was subcloned into the pCAG-IRES-GFP vector. pTOP-d-GFP was from Randall Moon (University of Washington, Seattle, WA). pCAG-Cre-IRES2-EGFP was created by subcloning Cre with an N-terminal nuclear localization signal into pCAG-IRES2-EGFP. Cre was a gift from Mark Lewandoski (National Cancer Institute, Frederick, MD). ICAT binds β-catenin and competes for its interaction with TCF-4 (Tago et al., 2000; Daniels and Weis, 2002; Gottardi and Gumbiner, 2004a), and a truncated TCF-4 lacking β-catenin binding sites (DN-TCF-4) functions as a dominant-negative protein (Korinek et al., 1997).
In utero electroporation. For in utero injection, timed-pregnant mice at embryonic day 13.5 (E13.5) were deeply anesthetized with a ketamine–xylazine mixture (10:1), after which abdominal fur was removed, and the uterine horns were exposed through a midline laparotomy incision. DNA solution (2.5 μl) in PBS containing 0.01% fast green was injected through the uterine wall into the lateral ventricle of the embryos using a glass micropipette made from a microcapillary tube. After injection, Tweezer-trodes (BTX, Holliston, MA) were applied across the outside of the uterus (oriented in a way so as to flank the embryonic brain), and five 50 ms square pulses of 45 mV with 950 ms intervals were delivered by an electroporator (BTX 830). After injection and electroporation, the uterus is returned inside the abdomen and the abdominal muscle wall and skin sealed with sutures. For in vivo TOP-d-GFP signaling studies, 0.8 μg/μl DNA was used for the pTOP-d-GFP, pcDNA-lacZ, pcDNA-FLAG-ICAT, and pcDNA-DN-TCF4 plasmids, and 0.5 μg/μl DNA was used for the pCAG-mRFP plasmid. For the cell-cycle-exit and proliferation fraction studies, 0.8 μg/μl DNA was used for the pCAG-GFP, pcDNA-DN-TCF4, and pcDNA-FLAG-ICAT plasmids. For the distribution, and differentiation studies, 0.5 μg/μl DNA was used for the pCAG-GFP-DN-TCF4, pCAG-ICAT-IRES-GFP, pCAG-mRFP, and pCAG-GFP plasmids. For the TOP-d-GFP signaling studies, embryos were killed 30 h after electroporation. For the distribution and differentiation studies, embryos were killed 72 h after electroporation. Brains were fixed for 4 h in 4% paraformaldehyde, and then embedded in a 1% agarose cube, and again fixed for 4 h. An OTS-5000 vibrating tissue slicer (Electron Microscopy Sciences, Fort Washington, PA) was used to make 40 μm coronal sections.
Cortical disassociation studies.
β-Cateninflox/flox mice were crossed to create timed-pregnant females with all embryos carrying homozygous floxed β-catenin alleles. A plasmid mixture of pCAG-Cre-IRES2-EGFP (0.5 μg/μl) and pCAG-mRFP1 (1.0 μg/μl) was introduced into the dorsolateral cortex of these embryos at E13.5 via in utero electroporation. The embryos were killed 24 h (E14.5) or 48 h (E15.5) after electroporation, and the dorsolateral cortices were isolated by dissection. Cortices were disassociated with 0.25% trypsin and then plated on poly-d-lysine-treated coverslips in six-well plates at a density of 5 × 105 cells per well in complete DMEM. Cells were incubated at 37°C for 2 h, and then fixed with 4% paraformaldehyde for 10 min. Coverslips were then stained for β-catenin (BD Transduction Laboratories, Lexington, KY; mouse monoclonal), green fluorescent protein (GFP) (Invitrogen, Eugene, OR; rabbit polyclonal), and DNA (Hoechst 33342). Primary antibodies were detected with secondary antibodies from Invitrogen (goat anti-mouse Alexa 633, goat anti-rabbit Alexa 488). Images were obtained using the 40× objective on a Zeiss (Oberkochen, Germany) confocal microscope. ImageJ software and the LSM Reader Plug-in (http://rsb.info.nih.gov/ij/) were used to determine the mean pixel intensity for β-catenin immunofluorescence in the disassociated cells. Cells with pyknotic-appearing nuclei were removed from analysis by a blinded researcher.
In utero electroporation internal controls and analysis.
To address potential experimental variability caused by in utero surgery and electroporation, we developed a method using the simultaneous electroporation of an internal control expression plasmid for the red fluorescent protein (mRFP-1) (Bevis and Glick, 2002) along with our GFP-expressing experimental construct. Using this approach, we could thus compare the distribution and fate of cells expressing the GFP-tagged construct (any GFP+ cell) with control cells expressing red fluorescent protein (RFP) only. In this way, all experimental conditions (target tissue, surgical variability, electroporation location and effectiveness, timing, etc.) would be identical between cells expressing the experimental construct versus only the control mRFP.
Immunohistochemistry and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling analysis.
Brain sections were incubated with blocking solution (5% goat serum and 0.3% Triton X-100 in PBS) overnight at 4°C, and then incubated with primary antibodies diluted in blocking solution for 24–48 h at 4°C. Primary antibodies used were anti-GFP rabbit polyclonal antibody (1:1000; Invitrogen), anti-Nestin mouse monoclonal antibody (1:350; BD Biosciences, Mountain View, CA), anti-TuJ1 rabbit polyclonal antibody (1:2000; Covance, Princeton, NJ), anti-activated caspase-3 rabbit polyclonal antibody (1:200; Cell Signaling, Beverly, MA), anti-5-bromo-2′-deoxyuridine (BrdU) rat monoclonal antibody (1:250; Serotec, Oxford, UK), and anti-Ki67 rabbit polyclonal antibody (1:500; Novocastra Laboratories, Newcastle, UK). Secondary antibodies used were Alexa 555-conjugated goat anti-rabbit IgG antibody (1:500; Invitrogen), Alexa 647-conjugated goat anti-rat IgG antibody (1:500; Invitrogen), Alexa 647-conjugated goat anti-mouse IgG antibody (1:500; Invitrogen), and Alexa 647-conjugated goat anti-rabbit IgG antibody (1:500; Invitrogen). Fragmented DNA of apoptotic cells was labeled using a terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay following the manufacturer's protocols (Promega, Madison, WI) with the following modifications: biotinylated nucleotide incorporated at the 3′ end of damaged DNA was detected by Alexa 647-conjugated streptavidin (Invitrogen). Vibratome sections were fixed with 4% paraformaldehyde, permeabilized with proteinase K (20 μg/ml; 10–30 min at room temperature). Finally, immunofluorescent and TUNEL images were captured using a Zeiss UV LSM510 confocal microscope.
Cell-cycle-exit analysis.
For cell-cycle-exit studies, E13.5 embryos were electroporated with pCAG-GFP and either pcDNA-DN-TCF4 or pcDNA-FLAG-ICAT plasmids. Mother mice were injected intraperitoneally with BrdU (50 mg/g body weight) at E14.5, and 24 h after BrdU injection, embryos brains were fixed and processed as described above. Brain sections (40 μm) were incubated with 3.4N HCl for 20 min at room temperature to unmask antigen, followed by three washes in 0.5× Tris-borate-EDTA and three washes with PBS to rescue GFP fluorescence. Brain sections were then incubated with blocking solution (as above) and subsequently incubated with anti-BrdU rat monoclonal antibody and anti-Ki67 rabbit polyclonal antibody in blocking solution for 48 h at 4°C. Next, sections were incubated with Alexa 555-conjugated goat anti-rabbit IgG and Alexa 647-conjugated goat anti-rat IgG antibodies in PBS (as described above). To determine the fraction of cells that had exited the cell cycle within comparable electroporated sections, GFP and BrdU double-labeled cells were counted first, and then Ki67 expression was determined as either positive or negative. Cell-cycle-exit index was calculated as the fraction of BrdU pulse-labeled, electroporated cells that exited the cell cycle (GFP+, BrdU+, and Ki67−), over the total number of electroporated cells that had been pulse-labeled with BrdU (GFP+ and BrdU+) (Chenn and Walsh, 2002).
Cell culture and luciferase assays.
Primary cortical cultures were generated from E14.5 embryos using minor modifications to the method described by Murphy et al. (1989). After dissection, cells were plated in DMEM (Invitrogen) with 2 mm l-glutamine, 1 mm N-acetyl-cysteine, and 1 mm sodium pyruvate, and B27 and N2 supplement (final 1×), with 25 ng/ml FGF2 on poly-d-lysine-treated tissue culture plates and transfected immediately. Cells were transfected using Lipofectamine 2000 following the manufacturer's protocol (Invitrogen). Luciferase activity was determined 24 h after transfection, using the Dual Luciferase Reporter System (Promega), which allows the combined detection of Luciferase and Renilla internal control for transfection efficiency. Amounts of DNA/transfection are 0.5 μg of Super8TOP/FOPFLASH, 0.17 μg of pCDNA-EGFP, and 0.033 μg of TK Renilla. For studies of inhibition by DN-TCF4 or ICAT, CMV-EGFP was used to normalize for total quantity of transfected DNA, and Pol III–Renilla was used to normalize for transfection efficiency.
Results
β-catenin signaling in developing cerebral cortex
To examine β-catenin-mediated transcriptional signaling in the ventricular zone (VZ), we used a reporter construct that expresses a destabilized GFP variant under the control of a β-catenin responsive promoter (TOPdGFP) (Dorsky et al., 2002). We used a method of in utero coelectroporation of multiple expression constructs to incorporate a red fluorescent protein (mRFP-1) internal control construct in each experimental brain (see Materials and Methods). Coelectroporation of TOPdGFP and mRFP-1 into E13.5 cortical precursors shows that mouse cortical ventricular zone precursors express GFP, representing activation of β-catenin signaling, within 24 h of electroporation limited primarily to precursor cells in the ventricular zone (Fig. 1).
Figure 1.

β-Catenin signaling in cortical neural precursors. β-Catenin signaling in cortical ventricular zone revealed by expression of destabilized GFP controlled by TOP reporter elements. E13.5 precursors were electroporated with pTOP-dGFP and pCAG-RFP with either CMV stuffer plasmid (A) or DN-TCF4 (B), or ICAT (C), and examined after 24 h. Right, Higher magnification confocal images from the same sections shown in the left panels. Green signal representing activation of signaling by β-catenin is limited to cells in ventricular zone and eliminated by coelectroporation of DN-TCF4 or ICAT. Scale bars: left, 200 μm; right, 20 μm. IZ, Intermediate zone; CP, cortical plate.
After electroporation of equal amounts of TOPdGFP and mRFP plasmids, we observe that the vast majority of electroporated cells in the ventricular zone were double-labeled with both RFP and GFP (Fig. 2). An identical fraction of cells were labeled exclusively with either mRFP or GFP indicating that TOPdGFP does not selectively label subpopulations of ventricular zone precursors. This contrasts with continuing expression of RFP in cells beginning to migrate from the ventricular zone in the same brains (Figs. 1, 2).
Figure 2.

β-Catenin signaling does not characterize a specific subpopulation of ventricular zone precursors. E13.5 ventricular zone precursors were electroporated by equal amounts of pCAGmRFP and pTOPdGFP plasmid. Twenty-four hours after electroporation, the vast majority of electroporated cells in the VZ were double-labeled with both RFP and GFP (representing activation of β-catenin signaling). An identical fraction of cells were labeled exclusively with either mRFP or GFP, indicating that TOPdGFP does not selectively label subpopulations of ventricular zone precursors. Cells exiting the ventricular zone [subventricular zone (SVZ)/intermediate zone (IZ)] retain RFP while downregulating GFP expression. The proportions of cells in the VZ exclusively labeled with RFP or GFP were not significantly different (n = 4 brains, 504 cells; ANOVA, p < 0.0001, with p > 0.05 for RFP-only cells compared with GFP-only cells by Tukey's post hoc test) for VZ cells. For non-VZ cells (n = 195), all proportions are significantly different from each other (ANOVA, p < 0.0001, with p < 0.001 for RFP-only cells compared with GFP-only cells). Error bars indicate SEM.
Examination of brains after longer intervals after electroporation revealed that β-catenin signaling changes dynamically during neuronal differentiation. At 24 and 48 h after electroporation at E13.5, GFP expression, representing β-catenin signaling, is present predominantly only in the ventricular zone. In contrast, at 24 h, newly migrating neurons entering the intermediate zone continue to express RFP, with an increasing number evident in the intermediate zone at 48 h (Fig. 3). At 72 h, young neurons continuing to express RFP begin to arrive at the cortical plate; meanwhile, GFP expression remains absent from the intermediate zone and cortical plate, and is only found rarely in the ventricular zone. At 96 h after electroporation, cortical plate neurons appear to reexpress the GFP reporter plasmid. Together, these observations suggest that ventricular zone precursors exhibit β-catenin signaling that is downregulated as cells exit the ventricular zone as they begin migrating to the cortical plate. Interestingly, 24 h after cells enter the cortical plate, they begin to reexpress the reporter plasmid, suggesting a second phase of β-catenin signaling as young neurons mature in the cortical plate.
Figure 3.
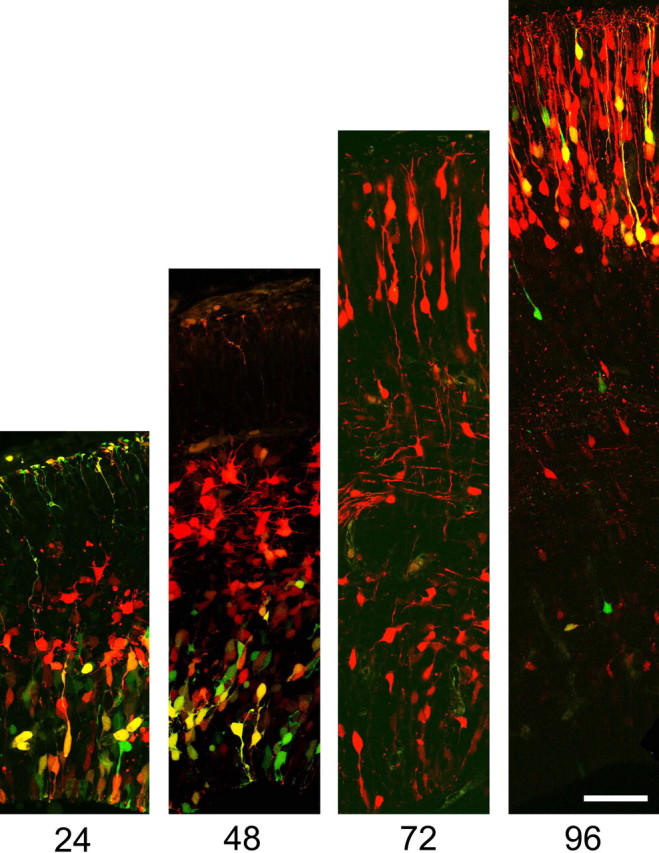
β-Catenin signaling undergoes dynamic changes during neuronal differentiation. E13.5 ventricular zone precursors coelectroporated with equal amounts of pCAG-mRFP or pTOPdGFP were analyzed after different intervals. At 24 h, GFP expression, representing β-catenin signaling, is restricted to the ventricular zone. At 48 h, some postmitotic neurons have begun to migrate from the ventricular zone and continue to express RFP, although they have downregulated GFP as they enter the intermediate zone. Ventricular zone cells continue to express GFP. At 72 h, young neurons continuing to express RFP begin to arrive at the cortical plate. GFP expression is absent from the intermediate zone and cortical plate, and is only found rarely in the ventricular zone. At 96 h after electroporation, cortical plate neurons begin to reexpress GFP. Scale bar, 50 μm.
Coelectroporation of inhibitors of β-catenin signaling [dominant-negative TCF-4 (DNTCF-4) (Korinek et al., 1997) or ICAT (Tago et al., 2000)] eliminated expression from the TOP-dGFP reporter plasmid, confirming the specificity of the TOPdGFP reporter for β-catenin signaling (Fig. 1 B,C).
Focal elimination of β-catenin causes early exit from the cortical ventricular zone
The importance of tissue architecture in cell growth control and proliferation (Bilder et al., 2000) complicates interpretations of traditional knock-out approaches of β-catenin on a tissue-wide scale (Backman et al., 2005; Junghans et al., 2005). To examine the cell-autonomous requirement for β-catenin in cortical development, we performed focal loss-of-function studies in a wild-type tissue background using in utero electroporation approaches. We observe that β-catenin protein is significantly reduced in electroporated cells 24 h (Fig. 4 A) and 48 h (Fig. 4 B) after electroporation of a Cre recombinase expression plasmid (pCAG-cre-IRES-GFP) into cortical precursors of floxed β-catenin (Ctnnb1tm2Kem) (Brault et al., 2001) E13.5 mouse embryos. Although reduction of total β-catenin immunofluorescence is modest (∼43% reduction) even after 48 h, relatively small changes in total β-catenin can have dramatic phenotypic consequences (Cox et al., 1996; Sanson et al., 1996), likely because β-catenin resides in two distinct pools in cells (Nelson and Nusse, 2004). Although the vast majority of β-catenin is bound to cadherin, active signaling β-catenin is likely only a small fraction of total β-catenin (Gottardi and Gumbiner, 2004b).
Figure 4.

β-Catenin protein is reduced after Cre-electroporation in floxed β-catenin mouse cortex. Floxed β-catenin cortical cells electroporated with Cre-Ires2-GFP have decreased immunoreactivity for β-catenin after 24 h (A) and 48 h (B). E13.5 timed-pregnant Flox βCat mice were electroporated with Cre-IRES2-EGFP (0.5 μg/μl) and mRFP1 (1.0 μg/μl). Twenty-four hours after surgery (E14.5), mice were killed and brains were removed from embryos, disassociated with trypsin, and plated on coverslips in six-well plates (5 × 106 cells/well). Cells were fixed 2 h after plating. Cells were stained for GFP and β-catenin (BCat) with antibodies. Immunofluorescent intensity for β-catenin was quantified, and cells expressing Cre-IRES2-EGFP were compared with cells exclusively expressing RFP. *p = 0.0312, ***p < 0.0001 by Student's t test. Error bars indicate SEM. Scale bar, 10 μm.
In vivo, we observed that an increased number of cre-electroporated cells leave the ventricular zone versus control cells. In electroporated brains, Cre-electroporated cells are visualized after staining with antibodies to GFP and are either green only or yellow (reflecting coelectroporation with RFP control plasmid). Reduction of β-catenin protein in ventricular zone precursors caused an increased fraction of cre-electroporated cells (61.1%) to migrate to the cortical plate compared with control cells expressing RFP only (22.4%) (Fig. 5). Correspondingly, whereas 33.6% of control cells were found in the ventricular zone, only 10.6% of cre-expressing cells remained in the ventricular zone, suggesting that, without β-catenin, cells prematurely exit the ventricular zone.
Figure 5.
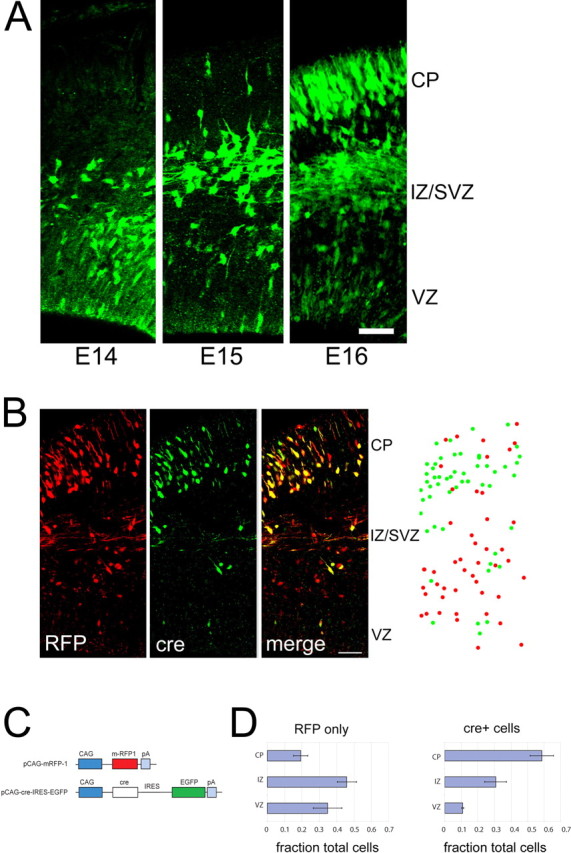
Focal elimination of β-catenin causes premature exit from the ventricular zone. A, Cortical precursors from homozygous floxed β-catenin mice (Ctnnb1tm2Kem) were electroporated in utero with pCAG-cre-IRES-EGFP at E13.5. Positions of electroporated cells were identified by the presence of EGFP after 1, 2, and 3 d. B, Left, Confocal images of EGFP and mRFP-1 expressing cells in sections through brains 3 d after coelectroporation of cre-IRES-EGFP and mRFP-1 at E13.5. Right, Example of analysis of positions of cre-expressing cells with or without RFP expression (green dots) versus control cells expressing RFP exclusively (no EGFP) (red dots), recorded from confocal stacks. C, Electroporation constructs. Constructs use the CAG promoter (Niwa et al., 1991). For detection of cre, construct uses EGFP driven by an IRES. D, Histograms, Cre-expressing cells show increased exit from the ventricular zone and migration to the cortical plate compared with control cells expressing RFP exclusively. There was a total of 342 cells from four brains. Error bars indicate 1 SEM. Scale bars, 50 μm. CP, Cortical plate; IZ, intermediate zone.
Focal inhibition of β-catenin signaling causes premature neuronal differentiation
Our observations that β-catenin signaling is downregulated as cells exit the ventricular zone and the behavior of cre-electroporated cells with reduced (but not absent) β-catenin protein in electoporated cells suggests that β-catenin signaling (vs its adhesive function) may regulate whether neural precursors remain in the ventricular zone as precursors. To examine whether inactivating β-catenin-dependent transcription alone could regulate maintenance of cortical precursors in the ventricular zone, we overexpressed DN-TCF-4 or ICAT in E13.5 cortical precursors by in utero electroporation. Analysis at E16.5 revealed that inhibition of β-catenin signaling caused cells to exit the ventricular zone when compared with RFP-transfected internal controls (Fig. 6 A,B). Similar to the findings when β-catenin was eliminated by cre electroporation, an increased fraction of transfected cells (49.9% DN-TCF-4; 57.9% ICAT) were found in the cortical plate, compared with controls (19.9% DNTCF-4 control and 17.9% ICAT control). Correspondingly, whereas only 8.1% (DNTCF-4) and 10.6% (ICAT) cells were found in the ventricular zone, 38.5% (DNTCF-4) and 42.6% (ICAT) of control cells remained in the ventricular zone. Thus, inhibition of β-catenin signaling in a cell-autonomous manner appears to increase the proportion of cells that exit the ventricular zone.
Figure 6.

Inhibition of β-catenin signaling causes premature exit from the ventricular zone. E13.5 brains coelectroporated with pCAG-GFP-DN-TCF4 and pCAG-mRFP (A) or pCAG-ICAT-IRES-EGFP and pCAG-mRFP (B) and analyzed at E16.5. A greater fraction of cells expressing DN-TCF4 (green and yellow cells) (A) or ICAT (green and yellow cells) (B) have migrated from the ventricular zone to the cortical plate compared with control cells expressing RFP exclusively. C, Histograms from five brains, 1883 cells (DN-TCF4) and from three brains, 2275 cells (ICAT), with error bars indicating 1 SEM. D, Electroporation constructs. Constructs use the CAG promoter (Niwa et al., 1991). GFP-DN-TCF4 is a deletion mutant lacking the N-terminal 30 amino acids (Korinek et al., 1997), to which we fused EGFP to the N terminus. For detection of ICAT, construct uses EGFP driven by an IRES. CP, Cortical plate; IZ, intermediate zone. Scale bars, 50 μm.
To determine whether precursors differentiated into neurons or remained as precursors after inhibition of β-catenin signaling, we examined the expression of a marker of newly postmitotic neurons (TuJ1) and a precursor marker Nestin. After electroporation of inhibitors of β-catenin signaling, an increased fraction of electroporated cells expressed TuJ1 compared with control GFP-electroporated cells [DNTCF4 (57.4 vs 31.9% control; p = 0.0067, t test) and ICAT (58.6 vs 31.0% control; p = 0.0329, t test)]. The fraction of electroporated cells expressing Nestin was reduced after inhibition of β-catenin signaling (DNTCF-4, 5.96 vs 39.5% control; p = 0.0017, t test; ICAT, 13.0 vs 42.6%; p < 0.0001, t test) (Fig. 7). Together, these studies suggest that inhibition of β-catenin signaling increases the proportion of cells that exit the ventricular zone and differentiate into neurons.
Figure 7.

Inhibition of β-catenin/TCF signaling causes neuronal differentiation. A, B, Sections through E16.5 cerebral cortex electroporated at E13.5 with DN-TCF4 (A) or ICAT (B) (green) and RFP expression plasmids (red) and stained with the TuJ1 antibody (blue) reveal that inhibition of β-catenin signaling causes neuronal differentiation (increases the fraction of electroporated cells that express TuJ1) (DNTCF-4, p = 0.0067, t test; n = 3 brains, 918 cells; ICAT, p = 0.0329, t test; n = 2 brains, 827 cells). C, D, Sections through E16.5 cerebral cortex electroporated at E13.5 with DN-TCF4 (C) or ICAT (D) (green) and RFP expression plasmids (red) and stained with the nestin antibody (blue) reveal inhibition of β-catenin signaling decreases the fraction of cells that express nestin (described in Results) (DNTCF-4, p = 0.0017, t test; n = 3 brains, 965 cells; ICAT, p < 0.0001, t test; n = 3 brains, 1065 cells). Scale bars, 100 μm (in all panels).
Loss of β-catenin signaling causes increased cell cycle exit
Neuronal differentiation in the developing cerebral cortex is coupled closely to terminal cell cycle exit (Takahashi et al., 1996), with the time of cell cycle exit (or “birthdate”) correlating with the final laminar position of cortical neurons (McConnell, 1988; Rakic, 1988; Caviness et al., 1995; Takahashi et al., 1999). To determine whether the relative increase in neurons generated results from a shift in the fraction of precursors that differentiate instead of remaining as precursors, we examined cell cycle exit and reentry by determining the fraction of cells that remain dividing after pulse labeling with BrdU 24 h earlier. We found an ∼60% increase in cell cycle exit in cells expressing DNTCF-4 or ICAT versus control cells expressing solely GFP (Fig. 8). This finding suggests that inhibiting β-catenin signaling leads to increased cell cycle exit. In light of these observations, the increased neuronal differentiation after inhibition of β-catenin signaling appears to be directly related to increased cell cycle exit, rather than an additional effect on neuronal differentiation. Whether cell cycle exit lies upstream of neuronal differentiation or vice versa remains unclear.
Figure 8.

Cell cycle exit increased by inhibition of β-catenin/TCF signaling. A, Animals were electroporated with expression plasmids for GFP alone or coelectroporated with DNTCF-4 or ICAT, and then exposed to a single pulse label of BrdU 24 h before killing. Sections were stained with antibodies to BrdU (blue) and Ki67 (red). The exit fraction is represented by the fraction of electroporated cells labeled only with BrdU (GFP+/Brdu+/Ki67−; no longer dividing) 24 h after pulse label, compared with GFP+/BrdU+/Ki67+ (triple-labeled; reentered cell cycle). Approximately 1.6-fold more cells leave the cell cycle when β-catenin/TCF signaling is inhibited by electroporation of DN-TCF4 or ICAT, compared with GFP-electroporated controls [ANOVA, p = 0.0055, Tukey's post hoc test shows both DN-TCF4 and ICAT differ significantly from GFP control (* signifies p < 0.01 for both pairwise comparisons).]. Error bars indicate SEM. CP, Cortical plate; IZ, intermediate zone.
Because programmed cell death (apoptosis) occurs during normal development of the CNS (Kuan et al., 2000), we examined whether inhibition of β-catenin signaling changed rates of apoptosis. Apoptotic cell death was examined using TUNEL staining and expression of activated caspase 3 (cleaved caspase-3). Positive cells were confirmed by verifying condensed nuclei labeled with the DNA binding dye Hoechst 33342. Counts of total numbers of labeled cells revealed that cell death was not substantially changed by inhibition of β-catenin signaling both within electroporated regions as well as surrounding regions (Fig. 9). Together, these findings suggest that cell-autonomous inhibition of β-catenin signaling causes cortical precursors to exit the cell cycle and differentiate into neurons.
Figure 9.

Inhibition of β-catenin signaling does not increase apoptotic cell death. A, E13.5 electroporation with pCAG-EGFP alone (left), pCAG-mEGFP and pcDNA-DN-TCF4 (middle), or pCAG-EGFP and pcDNA-ICAT (right). DN-TCF4 and ICAT do not affect apoptotic cell death, with similar numbers of activated caspase-3-labeled cells (red) in all constructs in electroporated as well as adjacent nonelectroporated fields 24 h after electroporation (p > 0.5, ANOVA). DNA is counterstained with Hoechst 33342. B, C, E13.5 electroporation with pCAG-mRFP alone (left), pCAG-mRFP and pCAG-GFP-DN-TCF (middle), or pCAG-mRFP and pCAG-ICAT-IRES-GFP (right). Shown is an analysis 72 h after electroporation. DN-TCF4 and ICAT do not affect apoptotic cell death, with similar numbers of TUNEL-positive cells (B; green) or activated caspase-3-labeled cells (C; green) for all constructs (p > 0.5; ANOVA). Scale bars, 200 μm. Error bars indicate SEM.
Discussion
By interfering with β-catenin signaling using focal electroporation techniques, we observe that that precursor proliferation in the cerebral cortical ventricular zone requires cell-autonomous β-catenin signaling. Focal elimination of β-catenin by cre-mediated gene excision or inhibition of β-catenin signaling with ICAT and dominant-negative TCF-4 leads to premature cell cycle withdrawal and neuronal differentiation.
It has been proposed that the number of neural precursor cells generated during development controls the size of the cerebral cortex (Rakic, 1995). Although our previous work showed that overexpression of β-catenin led to increased numbers of cortical precursors and a subsequent expansion of cortical surface area (Chenn and Walsh, 2002), the cell-autonomous requirement for β-catenin in the normal regulation of cortical precursor number remained unexplored. Conditional knock-out studies suggested a role for β-catenin in cell proliferation in the cortex, but interpretation of β-catenin function was complicated by the severe architectural abnormalities of the tissue (Machon et al., 2003). Although recent studies using overexpression of Wnts or dominant-negative Wnts have suggested a similar function for Wnt-β-catenin signaling in hippocampal progenitors (Lie et al., 2005), the methodology did not permit the examination of cell-autonomous function. The electroporation approach allows us to investigate cell-autonomous function of β-catenin in the absence of architectural disruption, and demonstrates that β-catenin signaling function is required to maintain progenitor proliferation. Together, the gain- and loss-of-function studies suggest a model in which high levels of β-catenin signaling expand the neural precursor population, whereas inhibition of signaling causes withdrawal from the cell cycle and depletion of the precursor pool.
During cortical development, cell cycle exit and neuronal differentiation are closely linked (Caviness et al., 1995), and cells that continue to cycle while initiating neuronal development exhibit increased cell death (Lee et al., 1992). Recent studies of the cyclin-dependent kinase inhibitor p27Kip1 suggest that neuronal differentiation and cell cycle exit may be independently regulated, because p27Kip1 can promote neuronal differentiation and radial migration of cortical projection neurons separable from its role in cell cycle regulation (Nguyen et al., 2006). Although β-catenin can regulate cell cycle through transcriptional regulation of cyclin D1 and c-myc (Peifer and Polakis, 2000) as well as promote neuronal differentiation by regulating Neurogenin1 (Israsena et al., 2004), our methods do not provide sufficient resolution to distinguish whether β-catenin independently regulates cell cycle and neuronal differentiation. Although inhibition of β-catenin signaling leads both to increased cell cycle exit and neuronal differentiation (at the expense of precursor number), we did not observe evidence that neuronal differentiation occurred at rates different from would be predicted by cell cycle exit. Regardless of whether cell cycle exit and differentiation are independently regulated by β-catenin, our findings suggest that inhibition of β-catenin signaling results in depletion of the precursor pool.
From the perspective of proliferation and differentiation, the size of the precursor pool can be regulated by the types of cell division that occur. During development, neural progenitors can undergo three types of cell divisions with regard to cell cycle reentry and exit (for review, see Caviness et al., 1995): (1) symmetric divisions that generate two additional progenitors serve to expand the progenitor pool. (2) Asymmetric divisions generate one daughter that exits the cell cycle and one daughter that reenters the cell cycle to divide again. These asymmetric divisions maintain the progenitor population while generating postmitotic neuronal progeny during cortical development. (3) Symmetric terminal divisions generate two postmitotic daughters, and these divisions will ultimately deplete the progenitor population.
The existence and relative contributions of these types of divisions have been suggested by population studies of proliferating cells (Takahashi et al., 1996; Miyama et al., 1997) and time-lapse microscopy (Chenn and McConnell, 1995; Adams, 1996; Noctor et al., 2001; Haydar et al., 2003). Recent studies modeling retroviral lineage tracing suggest that different proportions of asymmetric, symmetric terminal, and symmetric nonterminal cell divisions coexist during cortical development period (Cai et al., 2002), supporting the notion that changes in the relative proportions of these three types of divisions would influence the production of neurons from the progenitor population (Takahashi et al., 1996). Our experiments showing that inhibition of β-catenin signaling increases cell cycle exit cannot distinguish between the relative contributions of these different types of divisions, and multiple possibilities can generate similar outcomes. For example, increasing the fraction of symmetric terminal divisions at the expense of asymmetric divisions is difficult to distinguish on a population level from increasing the relative fraction of asymmetric divisions at the expense of symmetric proliferative divisions, and time-lapse imaging would provide definitive evidence of the relative contributions of cell divisions with varying symmetry.
Although we did not perform direct observation of changes in division symmetry, the localization of β-catenin at adherens junctions suggests a testable hypothesis: asymmetric localization of β-catenin may serve a role in regulation of signaling after asymmetric division. Although the factors that inhibit β-catenin signaling in the ventricular zone remain unknown, the observation that the bulk of β-catenin is asymmetrically localized at the adherens junctions of dividing precursors (Chenn et al., 1998) suggests that one way of regulating signaling is through unequal inheritance of β-catenin after a vertically oriented cell division. Recent studies suggest that asymmetric inheritance of the apical membrane appears sufficient to confer differences to two daughters after mitosis (Kosodo et al., 2004). Mitotic cleavage planes only slightly oblique from vertical were capable of distributing the apical membrane preferentially to one daughter (Kosodo et al., 2004). We noted that such divisions could lead to the asymmetric segregation of the bulk of the adherens complex to only one daughter as well. It has been proposed that the cadherin-bound pool of β-catenin might serve as a reservoir for signaling competent β-catenin (Cox et al., 1996; Barth et al., 1997), and recent studies provide support for cross talk between the cadherin bound and signaling pools of β-catenin (Klingelhofer et al., 2003). Therefore, after an asymmetric cleavage (as defined by asymmetric inheritance of apical membrane) (Kosodo et al., 2004), the cell adjacent to the ventricular lumen (the apical cell) will inherit the adherens complex and β-catenin, and thus may be influenced to remain in the cell cycle because of maintained β-catenin signaling. In contrast, the basal daughter after an asymmetric cleavage may be predisposed to exit the cell cycle as a consequence of receiving less β-catenin. Our model would suggest that inhibition of β-catenin signaling could potentially influence previously asymmetric divisions to become symmetric and terminal divisions, and symmetric proliferative divisions might also be influenced to become symmetric terminal divisions.
However, additional complexity is introduced by recent experiments that indicate that secondary proliferative populations identified in the subventricular zone (SVZ) (Takahashi et al., 1995) contribute significantly to neuronal production during cortical development (Noctor et al., 2004). Whereas progenitor divisions in the ventricular zone can be asymmetric, generating one daughter that exits the cell cycle and one daughter that reenters the cell cycle, asymmetric VZ divisions can also generate one daughter that moves to the SVZ (to divide again), and one daughter that remains in the ventricular zone. Although this mode of division is symmetric in the sense that it generates two daughters that reenter the cell cycle, these resultant daughters are distinct, in both physical location and behavior. Furthermore, symmetric divisions in the VZ could also generate two daughters that move to the SVZ, which then divide again. In the SVZ, the majority of cell divisions (∼90%) that subsequently occur in the SVZ are symmetrically terminal, generating two postmitotic neurons that migrate to the cortical plate (Noctor et al., 2004). Our findings that inhibition of β-catenin signaling increase cell cycle exit and neuronal differentiation cannot distinguish between effects on cell cycle exit in the ventricular zone versus the subventricular zone; although β-catenin signaling could conceivably regulate overall cell cycle exit by altering the frequency of asymmetric ventricular divisions that generate SVZ precursors, it could also influence the patterns of subsequent SVZ divisions as well. Our observation that β-catenin signaling is reduced in the SVZ suggests that such changes in SVZ behavior may be a later consequence of β-catenin inhibition in the VZ rather than a direct role for β-catenin in SVZ divisions.
Our studies target a period approximating the midpoint of cortical neurogenesis in the mouse (Takahashi et al., 1999). Although we have not yet determined whether β-catenin signaling occurs in precursors before the onset of neurogenesis, we suspect based on gain-of-function studies (Backman et al., 2005) that β-catenin functions similarly early during development. Nevertheless, our current studies do not provide information on whether other aspects of cell fate choice are affected by β-catenin (for example, choice of cortical lamina, or decision to generate glia). Although these choices normally appear to be regulated by when a cell exits the cell cycle (McConnell, 1989), we do not know whether forcing a precursor to exit the cell cycle early will cause it to adopt a relatively earlier phenotype. For example, although we observe β-catenin signaling later in cortical development (G. J. Woodhead and A. Chenn, in preparation), it remains unexplored whether loss of β-catenin signaling will lead to increased differentiation into non-neuronal cells appropriate for that period of development.
Recent studies provide support for the notion that mitotic spindle orientation plays a crucial role in neural precursor proliferation and brain size (for review, see Woods et al., 2005). The most common cause of primary autosomal recessive microcephaly is mutation in the abnormal spindle-like microcephaly associated (ASPM) gene. ASPM is highly expressed in neural precursors and the protein is localized to mitotic spindle poles (Kouprina et al., 2005), and its Drosophila counterpart, Asp, is an asymmetrically localized centrosomal protein required for mitotic spindle integrity (do Carmo Avides and Glover, 1999). Similarly, knock-down of Aspm in telencephalic neuroepithelial cells causes alterations in cleavage plane orientation and leads to an increased frequency of asymmetric division (Fish et al., 2006). Furthermore, recent studies in mouse have also shown the importance of Doublecortin-like kinase and G-proteins in the regulation of the mitotic spindle and the symmetric/asymmetric mode of division (Sanada and Tsai, 2005; Shu et al., 2006).
The model proposed above places β-catenin signaling downstream of mitotic spindle orientation, as an apical factor that can be distributed symmetrically or asymmetrically to daughter cells depending on the mitotic cleavage angle. However, our data cannot rule out a role for β-catenin signaling in the organization of mitotic spindle orientation, and recent studies suggest that multiple Wnt/β-catenin signaling pathways serve to orient the mitotic spindle in early Caenorhabditis elegans embryos (Walston et al., 2004).
β-Catenin is likely to function at multiple times during the lifetime of a developing neuron, and the function of β-catenin in promoting neuronal differentiation has been suggested by previous studies. Gain-of-function studies in neurosphere cultures suggest that developmental context and presence of other growth factors (FGF2) influence whether β-catenin signaling promotes proliferation or differentiation (Israsena et al., 2004), and β-catenin overexpression increases neuronal production from embryonic stem cells differentiated in vitro (Otero et al., 2004). Although our studies suggest that one key function of β-catenin is to maintain precursor proliferation in vivo, they do not exclude subsequent roles for β-catenin in promoting neuronal differentiation. Indeed, our observations that β-catenin signaling reappears in newly arriving neurons of the cortical plate support later functions for β-catenin signaling in neuronal differentiation. Although additional studies are necessary to determine what function this later signaling plays in neuronal development, early aspects of cortical neuronal differentiation (such as development of long-distance callosal projections) appear grossly normal in the absence of β-catenin (data not shown).
What is the role of β-catenin signaling versus cell adhesion in the developing cortex? Here, we have shown by electroporation of a reporter construct responsive to β-catenin signaling that β-catenin signaling exists in cortical precursors during cortical neurogenesis. Focal deletion of β-catenin leads to premature ventricular zone exit, and furthermore, two different specific inhibitors of β-catenin signaling (that do not affect adhesion) lead to premature cell cycle exit and neuronal differentiation. Although these approaches provide multiple lines of evidence that β-catenin signaling function is required for cortical precursor proliferation, the role of β-catenin cell adhesion in neural precursors is more difficult to examine directly.
Although β-catenin has distinct functions in signaling and cell adhesion, the protein domain of β-catenin required for signaling through TCF/LEF binding domains overlaps significantly with the cadherin binding domains (Huber et al., 1997). This substantial overlap may underlie alterations in cell proliferation that result when epithelial architecture is disrupted by mutation or deletion of other adherens junction proteins (Lien et al., 2006). Loss of adherens junction integrity may allow for the redistribution and accumulation of previously membrane-localized β-catenin leading to increased signaling and proliferation. Although the role of β-catenin-mediated cell adhesion in neuronal differentiation remains unclear, our studies suggest that β-catenin signaling is essential to maintain neural precursor proliferation during cortical development.
Our observations that robust β-catenin signaling is found primarily in the ventricular zone support a role for localized signaling in cortical precursor maintenance. It is likely that neuronal differentiation and exit from the ventricular zone are linked, because in vivo, the early cortical ventricular zone is notably devoid of differentiated neurons (Menezes and Luskin, 1994). Our findings that inhibiting β-catenin signaling in vivo causes exit from the ventricular zone suggest (1) inhibition of signaling is sufficient to lead to cell cycle withdrawal and VZ exit, and (2) exit from the ventricular zone may ensure continued inhibition of signaling. A role for cell adhesion to physically maintain cortical precursors in the ventricular zone remains an attractive, but unexplored possibility.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grant R01NS04719, Searle Scholars Program, and Sontag Foundation Distinguished Scientist Award (A.C.). G.J.W. was supported by Ruth L. Kirschstein National Research Service Award Predoctoral Fellowship F30NS053303 and Katten Muchin Rosenman Travel Scholarship. We thank R. Tsien (University of California San Diego, La Jolla, CA) for mRFP-1, R. Moon (University of Washington, Seattle, WA) for pSUPER-8-TOP/FOPFLASH, and R. Dorsky (University of Utah, Salt Lake City, UT) for pTOP-dGFP; C. Gottardi (Northwestern University, Chicago, IL) for pcDNA-FLAG-ICAT; W. Tourtellotte (Northwestern University) for pIRES2-eGFP plasmid; and G. Hammer (University of Michigan, Ann Arbor, MI) for DN-TCF4. We thank Jack Kessler for comments and data interpretation, and J. A. Brzezinski, C. J. Gottardi, K. Haldar, H. McNeill, D. A. Sipkins, G. A. Smith, and C. A. Wrobel for comments on this manuscript.
References
- Adams RJ. Metaphase spindles rotate in the neuroepithelium of rat cerebral cortex. J Neurosci. 1996;16:7610–7618. doi: 10.1523/JNEUROSCI.16-23-07610.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backman M, Machon O, Mygland L, van den Bout CJ, Zhong W, Taketo MM, Krauss S. Effects of canonical Wnt signaling on dorso-ventral specification of the mouse telencephalon. Dev Biol. 2005;279:155–168. doi: 10.1016/j.ydbio.2004.12.010. [DOI] [PubMed] [Google Scholar]
- Barth AI, Nathke IS, Nelson WJ. Cadherins, catenins and APC protein: interplay between cytoskeletal complexes and signaling pathways. Curr Opin Cell Biol. 1997;9:683–690. doi: 10.1016/s0955-0674(97)80122-6. [DOI] [PubMed] [Google Scholar]
- Bevis BJ, Glick BS. Rapidly maturing variants of the Discosoma red fluorescent protein (DsRed) Nat Biotechnol. 2002;20:83–87. doi: 10.1038/nbt0102-83. [DOI] [PubMed] [Google Scholar]
- Bilder D, Li M, Perrimon N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science. 2000;289:113–116. doi: 10.1126/science.289.5476.113. [DOI] [PubMed] [Google Scholar]
- Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128:1253–1264. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- Cai L, Hayes NL, Takahashi T, Caviness VS, Jr, Nowakowski RS. Size distribution of retrovirally marked lineages matches prediction from population measurements of cell cycle behavior. J Neurosci Res. 2002;69:731–744. doi: 10.1002/jnr.10398. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Jr, Takahashi T, Nowakowski RS. Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995;18:379–383. doi: 10.1016/0166-2236(95)93933-o. [DOI] [PubMed] [Google Scholar]
- Chenn A, McConnell SK. Cleavage orientation and the asymmetric inheritance of Notch1 immunoreactivity in mammalian neurogenesis. Cell. 1995;82:631–641. doi: 10.1016/0092-8674(95)90035-7. [DOI] [PubMed] [Google Scholar]
- Chenn A, Walsh CA. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science. 2002;297:365–369. doi: 10.1126/science.1074192. [DOI] [PubMed] [Google Scholar]
- Chenn A, Zhang YA, Chang BT, McConnell SK. Intrinsic polarity of mammalian neuroepithelial cells. Mol Cell Neurosci. 1998;11:183–193. doi: 10.1006/mcne.1998.0680. [DOI] [PubMed] [Google Scholar]
- Cox RT, Kirkpatrick C, Peifer M. Armadillo is required for adherens junction assembly, cell polarity, and morphogenesis during Drosophila embryogenesis. J Cell Biol. 1996;134:133–148. doi: 10.1083/jcb.134.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels DL, Weis WI. ICAT inhibits beta-catenin binding to Tcf/Lef-family transcription factors and the general coactivator p300 using independent structural modules. Mol Cell. 2002;10:573–584. doi: 10.1016/s1097-2765(02)00631-7. [DOI] [PubMed] [Google Scholar]
- do Carmo Avides M, Glover DM. Abnormal spindle protein, Asp, and the integrity of mitotic centrosomal microtubule organizing centers. Science. 1999;283:1733–1735. doi: 10.1126/science.283.5408.1733. [DOI] [PubMed] [Google Scholar]
- Dorsky RI, Sheldahl LC, Moon RT. A transgenic Lef1/beta-catenin-dependent reporter is expressed in spatially restricted domains throughout zebrafish development. Dev Biol. 2002;241:229–237. doi: 10.1006/dbio.2001.0515. [DOI] [PubMed] [Google Scholar]
- Fish JL, Kosodo Y, Enard W, Paabo S, Huttner WB. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc Natl Acad Sci USA. 2006;103:10438–10443. doi: 10.1073/pnas.0604066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galceran J, Miyashita-Lin EM, Devaney E, Rubenstein JL, Grosschedl R. Hippocampus development and generation of dentate gyrus granule cells is regulated by LEF1. Development. 2000;127:469–482. doi: 10.1242/dev.127.3.469. [DOI] [PubMed] [Google Scholar]
- Gottardi CJ, Gumbiner BM. Role for ICAT in beta-catenin-dependent nuclear signaling and cadherin functions. Am J Physiol. 2004a;286:C747–C756. doi: 10.1152/ajpcell.00433.2003. [DOI] [PubMed] [Google Scholar]
- Gottardi CJ, Gumbiner BM. Distinct molecular forms of beta-catenin are targeted to adhesive or transcriptional complexes. J Cell Biol. 2004b;167:339–349. doi: 10.1083/jcb.200402153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haydar TF, Ang E, Jr, Rakic P. Mitotic spindle rotation and mode of cell division in the developing telencephalon. Proc Natl Acad Sci USA. 2003;100:2890–2895. doi: 10.1073/pnas.0437969100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AH, Nelson WJ, Weis WI. Three-dimensional structure of the armadillo repeat region of beta-catenin. Cell. 1997;90:871–882. doi: 10.1016/s0092-8674(00)80352-9. [DOI] [PubMed] [Google Scholar]
- Israsena N, Hu M, Fu W, Kan L, Kessler JA. The presence of FGF2 signaling determines whether beta-catenin exerts effects on proliferation or neuronal differentiation of neural stem cells. Dev Biol. 2004;268:220–231. doi: 10.1016/j.ydbio.2003.12.024. [DOI] [PubMed] [Google Scholar]
- Junghans D, Hack I, Frotscher M, Taylor V, Kemler R. Beta-catenin-mediated cell-adhesion is vital for embryonic forebrain development. Dev Dyn. 2005;233:528–539. doi: 10.1002/dvdy.20365. [DOI] [PubMed] [Google Scholar]
- Klingelhofer J, Troyanovsky RB, Laur OY, Troyanovsky S. Exchange of catenins in cadherin-catenin complex. Oncogene. 2003;22:1181–1188. doi: 10.1038/sj.onc.1206245. [DOI] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science. 1997;275:1784–1787. doi: 10.1126/science.275.5307.1784. [DOI] [PubMed] [Google Scholar]
- Kosodo Y, Roper K, Haubensak W, Marzesco AM, Corbeil D, Huttner WB. Asymmetric distribution of the apical plasma membrane during neurogenic divisions of mammalian neuroepithelial cells. EMBO J. 2004;23:2314–2324. doi: 10.1038/sj.emboj.7600223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouprina N, Pavlicek A, Collins NK, Nakano M, Noskov VN, Ohzeki J, Mochida GH, Risinger JI, Goldsmith P, Gunsior M, Solomon G, Gersch W, Kim JH, Barrett JC, Walsh CA, Jurka J, Masumoto H, Larionov V. The microcephaly ASPM gene is expressed in proliferating tissues and encodes for a mitotic spindle protein. Hum Mol Genet. 2005;14:2155–2165. doi: 10.1093/hmg/ddi220. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Roth KA, Flavell RA, Rakic P. Mechanisms of programmed cell death in the developing brain. Trends Neurosci. 2000;23:291–297. doi: 10.1016/s0166-2236(00)01581-2. [DOI] [PubMed] [Google Scholar]
- Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature. 1992;359:288–294. doi: 10.1038/359288a0. [DOI] [PubMed] [Google Scholar]
- Lee SM, Tole S, Grove E, McMahon AP. A local Wnt-3a signal is required for development of the mammalian hippocampus. Development. 2000;127:457–467. doi: 10.1242/dev.127.3.457. [DOI] [PubMed] [Google Scholar]
- Lie DC, Colamarino SA, Song HJ, Desire L, Mira H, Consiglio A, Lein ES, Jessberger S, Lansford H, Dearie AR, Gage FH. Wnt signalling regulates adult hippocampal neurogenesis. Nature. 2005;437:1370–1375. doi: 10.1038/nature04108. [DOI] [PubMed] [Google Scholar]
- Lien WH, Klezovitch O, Fernandez TE, Delrow J, Vasioukhin V. alphaE-catenin controls cerebral cortical size by regulating the hedgehog signaling pathway. Science. 2006;311:1609–1612. doi: 10.1126/science.1121449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machon O, van den Bout CJ, Backman M, Kemler R, Krauss S. Role of beta-catenin in the developing cortical and hippocampal neuroepithelium. Neuroscience. 2003;122:129–143. doi: 10.1016/s0306-4522(03)00519-0. [DOI] [PubMed] [Google Scholar]
- McConnell SK. Development and decision-making in the mammalian cerebral cortex. Brain Res Rev. 1988;13:1–23. doi: 10.1016/0165-0173(88)90002-1. [DOI] [PubMed] [Google Scholar]
- McConnell SK. The determination of neuronal fate in the cerebral cortex. Trends Neurosci. 1989;12:342–349. doi: 10.1016/0166-2236(89)90041-6. [DOI] [PubMed] [Google Scholar]
- McMahon AP, Bradley A. The Wnt-1 (int-1) proto-oncogene is required for development of a large region of the mouse brain. Cell. 1990;62:1073–1085. doi: 10.1016/0092-8674(90)90385-r. [DOI] [PubMed] [Google Scholar]
- Menezes JR, Luskin MB. Expression of neuron-specific tubulin defines a novel population in the proliferative layers of the developing telencephalon. J Neurosci. 1994;14:5399–5416. doi: 10.1523/JNEUROSCI.14-09-05399.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyama S, Takahashi T, Nowakowski RS, Caviness VS., Jr A gradient in the duration of the G1 phase in the murine neocortical proliferative epithelium. Cereb Cortex. 1997;7:678–689. doi: 10.1093/cercor/7.7.678. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- Nelson WJ, Nusse R. Convergence of Wnt, beta-catenin, and cadherin pathways. Science. 2004;303:1483–1487. doi: 10.1126/science.1094291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L, Besson A, Heng JI, Schuurmans C, Teboul L, Parras C, Philpott A, Roberts JM, Guillemot F. p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev. 2006;20:1511–1524. doi: 10.1101/gad.377106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Dammerman RS, Kriegstein AR. Neurons derived from radial glial cells establish radial units in neocortex. Nature. 2001;409:714–720. doi: 10.1038/35055553. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Martinez-Cerdeno V, Ivic L, Kriegstein AR. Cortical neurons arise in symmetric and asymmetric division zones and migrate through specific phases. Nat Neurosci. 2004;7:136–144. doi: 10.1038/nn1172. [DOI] [PubMed] [Google Scholar]
- Otero JJ, Fu W, Kan L, Cuadra AE, Kessler JA. Beta-catenin signaling is required for neural differentiation of embryonic stem cells. Development. 2004;131:3545–3557. doi: 10.1242/dev.01218. [DOI] [PubMed] [Google Scholar]
- Peifer M. Cell biology. Travel bulletin—traffic jams cause tumors. Science. 2000;289:67–69. doi: 10.1126/science.289.5476.67. [DOI] [PubMed] [Google Scholar]
- Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis—a look outside the nucleus. Science. 2000;287:1606–1609. doi: 10.1126/science.287.5458.1606. [DOI] [PubMed] [Google Scholar]
- Peifer M, Tepass U. Cell biology. Which way is up? Nature. 2000;403:611–612. doi: 10.1038/35001187. [DOI] [PubMed] [Google Scholar]
- Rakic P. Specification of cerebral cortical areas. Science. 1988;241:170–176. doi: 10.1126/science.3291116. [DOI] [PubMed] [Google Scholar]
- Rakic P. A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci. 1995;18:383–388. doi: 10.1016/0166-2236(95)93934-p. [DOI] [PubMed] [Google Scholar]
- Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–850. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- Sanada K, Tsai LH. G protein betagamma subunits and AGS3 control spindle orientation and asymmetric cell fate of cerebral cortical progenitors. Cell. 2005;122:119–131. doi: 10.1016/j.cell.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Sanson B, White P, Vincent JP. Uncoupling cadherin-based adhesion from wingless signalling in Drosophila . Nature. 1996;383:627–630. doi: 10.1038/383627a0. [DOI] [PubMed] [Google Scholar]
- Shu T, Tseng HC, Sapir T, Stern P, Zhou Y, Sanada K, Fischer A, Coquelle FM, Reiner O, Tsai LH. Doublecortin-like kinase controls neurogenesis by regulating mitotic spindles and M phase progression. Neuron. 2006;49:25–39. doi: 10.1016/j.neuron.2005.10.039. [DOI] [PubMed] [Google Scholar]
- Song X, Zhu CH, Doan C, Xie T. Germline stem cells anchored by adherens junctions in the Drosophila ovary niches. Science. 2002;296:1855–1857. doi: 10.1126/science.1069871. [DOI] [PubMed] [Google Scholar]
- Tago K, Nakamura T, Nishita M, Hyodo J, Nagai S, Murata Y, Adachi S, Ohwada S, Morishita Y, Shibuya H, Akiyama T. Inhibition of Wnt signaling by ICAT, a novel beta-catenin-interacting protein. Genes Dev. 2000;14:1741–1749. [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS., Jr Early ontogeny of the secondary proliferative population of the embryonic murine cerebral wall. J Neurosci. 1995;15:6058–6068. doi: 10.1523/JNEUROSCI.15-09-06058.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS., Jr The leaving or Q fraction of the murine cerebral proliferative epithelium: a general model of neocortical neuronogenesis. J Neurosci. 1996;16:6183–6196. doi: 10.1523/JNEUROSCI.16-19-06183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Goto T, Miyama S, Nowakowski RS, Caviness VS., Jr Sequence of neuron origin and neocortical laminar fate: relation to cell cycle of origin in the developing murine cerebral wall. J Neurosci. 1999;19:10357–10371. doi: 10.1523/JNEUROSCI.19-23-10357.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- Viti J, Gulacsi A, Lillien L. Wnt regulation of progenitor maturation in the cortex depends on Shh or fibroblast growth factor 2. J Neurosci. 2003;23:5919–5927. doi: 10.1523/JNEUROSCI.23-13-05919.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walston T, Tuskey C, Edgar L, Hawkins N, Ellis G, Bowerman B, Wood W, Hardin J. Multiple Wnt signaling pathways converge to orient the mitotic spindle in early C. elegans embryos. Dev Cell. 2004;7:831–841. doi: 10.1016/j.devcel.2004.10.008. [DOI] [PubMed] [Google Scholar]
- Woods CG, Bond J, Enard W. Autosomal recessive primary microcephaly (MCPH): a review of clinical, molecular, and evolutionary findings. Am J Hum Genet. 2005;76:717–728. doi: 10.1086/429930. [DOI] [PMC free article] [PubMed] [Google Scholar]