

Abstract
Background and Objectives
The complement system is a biochemical cascade composed of several plasma proteins that can interact with endothelial cells and blood cells, including platelets. In order to investigate the effect of the complement system on platelets, we studied platelet function in C3-deficient mice that lack complement activity.
Method and Results
Tail-cut bleeding time was prolonged and platelet aggregation in response to protease activated receptor-4 (PAR4) peptide was decreased in C3-deficient mice compared to wild-type littermates. Platelet aggregation in response to other agonists (ADP and collagen) was similar between C3-deficient mice and their normal littermates. Isolated platelets from wild-type mice aggregate less in C3-deficient plasma than in normal plasma and conversely, addition of plasma from wild-type mice or plasma-purified C3 improved aggregation of C3-deficient platelets. We also monitored formation of murine arteriole or venule thrombi in an intravital microscopy thrombosis model. We found that C3-deficient mice had a significantly delayed thrombotic response in arterioles compared to their wild-type littermates. Furthermore, thrombi in C3-deficient mice were less stable and embolized more frequently than those in wild-type mice.
Conclusions
Platelets of C3-deficient mice have subnormal function resulting in a prolonged tail-cut bleeding time and delayed thrombosis after vessel wall injury.
Keywords: platelet, complement system, thrombosis
Introduction
The interaction between platelets and the complement system has been studied for a long time (1;2). Platelet activation results in activation of the complement system (3–5) and complement activation results in activation of platelets (6). This interaction might be responsible for thrombotic complications in disorders characterized by complement dysregulation, such as paroxysmal nocturnal hemoglobinuria and atypical hemolytic uremic syndrome. In these disorders, a hyperreactive complement system results in circulating activated platelets (7–9). Whether the lack of a functional complement system would have the reverse effect, and decrease platelet responsiveness, is unknown. In order to investigate the role of the platelet-complement interaction in hemostasis, we studied platelet aggregation in C3-deficient mice (a kind gift from Dr. Rick Wetsel, The University of Texas Health Science Center at Houston), using both in vitro and in vivo assays.
Materials and Methods
All animal procedures were performed according to protocols approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine and the handling was performed following the regulation of the ethical committee.
Mice
C3-deficient mice were on a C57BL/6 background and we used interbreeding heterozygous C3-deficient mice to generate littermates with C3−/−, C3+/−, and C3+/+ genotypes.
Bleeding Time
Mice were anaesthetized by intraperitoneal injection of a standard mixture of ketamine, xylazine, and acepromazine. The terminal 3mm of the tails of mice were severed and immediately immersed in 1.5 mL tubes containing PBS at 37°C. The tail was immersed in successive tubes every 30 seconds until bleeding stopped. The time interval between the cut and cessation of bleeding by gross visual inspection of the tubes was defined as the bleeding time.
Platelet Studies
Mice were anesthetized with isofluorane. Blood was collected from the inferior vena cava using a 1 ml syringe containing 100 μl of 3.8% sodium citrate and then diluted with an equal volume of calcium- and magnesium-free Tyrode’s buffer (138 mM sodium chloride, 5.5 mM glucose, 12 mM sodium bicarbonate, 2.9 mM potassium chloride, and 0.36 mM sodium phosphate dibasic, pH 7.4). Platelet-rich plasma (PRP) was obtained from diluted blood by differential centrifugation at 68 × g for 10 min. Platelet count from PRP was measured using Vet abc hematology analyzer (SCIL Animal Care Company, Gurnee, IL, USA) and then corrected to 2.5 × 105/μL with Tyrode’s-diluted plasma. Platelet aggregation in response to different concentrations of agonists (ADP, collagen, and PAR4 activating peptide) was measured in 225 μL of PRP at 37°C in a four-channel Bio/Data PAP-4C aggregometer (Bio/Data corporation, Pennsylvania, USA). In some experiments, plasma-purified C3 (Complement Technologies, Tyler, Texas, USA) was added to PRP at a final concentration of 0.4 mg/ml prior to addition of platelet agonist. In order to remove protein preservatives that might affect platelet aggregation, purified C3 was dialyzed in PBS for 2 hours using Slide-A-Lyzer dialysis cassettes (Thermo Scientific, Rockford, IL, U.S.A). To isolate platelets, first, PRP was obtained from blood collected in 100 μL of acid-citrate-dextrose, and then PRP was centrifuged at 754 × g for 15 minutes. The platelet pellet was resuspended in plasma from either C3-deficient or wild-type mice.
In vivo thrombosis model
Microvascular thrombosis was assessed in cremaster venules and arterioles, using a photochemical injury model under intravital microscopy, as described previously (10). Briefly, pentobarbital-anesthetized mice underwent exteriorization of the cremaster microvascular bed; following a 30 minute equilibration period, FITC-labeled dextran (150kd, 5 ml/kg of a 5% solution) was injected intravenously and subsequently a 100 μm length of vessel was exposed to epi-illumination (0.5 W/cm2), according the procedures described in detail previously (10;11). Time of onset of platelet thrombus formation, time to flow cessation, and the thrombus size were monitored by an independent observer, blinded to the genotype of the animals. Data were analyzed by ANOVA, a p < 0.05 was considered statistically significant.
Results
Baseline complete blood counts
The baseline hematologic parameters are shown in Table 1. There was no statistically significant difference in hematocrits, white blood cell counts, or platelet counts among mice with different C3 genotypes.
Table 1.
Complete Blood Counts in mice with different C3 genotypes
| C3−/− | C3+/− | C3+/+ | |
|---|---|---|---|
| Hematocrit (%) | 29 ± 2 | 29 ± 1 | 30 ± 1 |
| WBC (× 103/μl) | 3.2 ± 0.5 | 2.5 ± 0.6 | 2.9 ± 0.5 |
| Platelet (× 103/μl) | 706 ± 92 | 653 ± 197 | 746 ± 189 |
Bleeding Time
C3−/− mice had a longer bleeding time from a tail cut compared to that of C3+/+ or C3+/− mice (Figure 1). Bleeding stopped after 86 ± 48 sec in C3+/+ mice and 113 ± 51 sec in C3+/− mice; however, C3−/− mice continued to bleed for more than 10 minutes. An interesting observation was that C3−/− mice continued to bleed intermittently from the tail cut following the initial cessation of bleeding. The same phenomenon was not observed in either C3+/+ or C3+/− mice.
Figure 1. Bleeding time.
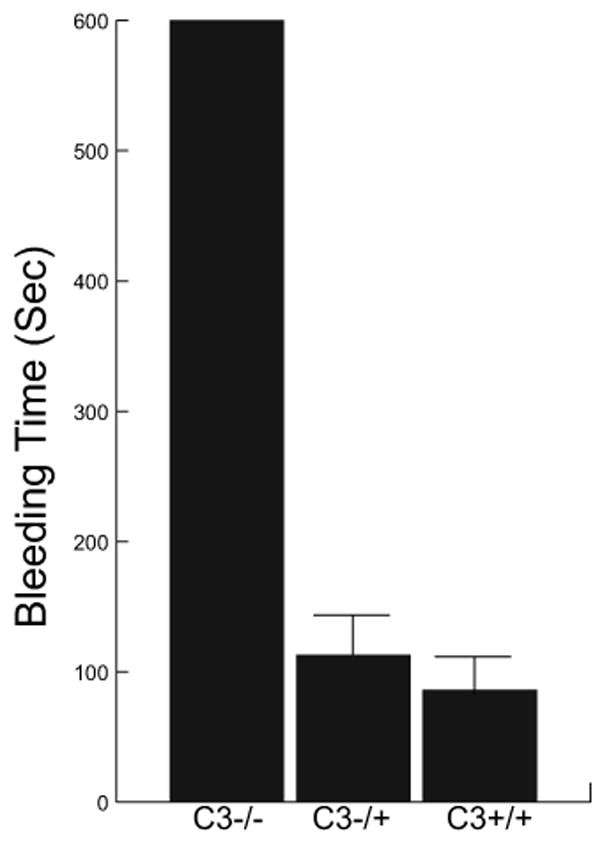
Bleeding time from a tail-cut according to the C3 genotype of mice (4 mice in each group). In addition to prolonged bleeding time, C3−/− mice rebled after temporary cessation of blood loss from the tail cut.
Platelet aggregation studies
We measured platelet aggregation in response to ADP, collagen and PAR4 peptide (Figure 2A) and found an agonist-specific abnormal platelet aggregation in C3-deficient mice. ADP at concentrations of 2.5 μM and 5 μM, and collagen at concentrations of 0.5 and 2.0 μg/ml induced similar extent of platelet aggregation in PRP samples obtained from C3-deficient, heterozygous, and wild-type mice (Figure 2B). However, in response to PAR4 peptide, platelets from C3−/− mice persistently aggregated less than those of C3+/− and C3+/+ mice (Figure 2A and 2B). PAR4 peptide at a concentration of 400 μM induced 27% ± 1.8%, 56% ± 1.6%, and 49% ± 1.5% platelet aggregation in C3−/−, C3+/−, and C3+/+ mice, respectively (15 mice in each group). At a higher concentration of 600 μM, PAR4 peptide induced 50% ± 1.7%, 70% ± 2.7%, and 67% ± 2% platelet aggregation in C3−/−, C3+/−, and C3+/+ mice, respectively (4 mice in each group). To determine if this observation was due to an intrinsic platelet abnormality or to the effect of plasma C3, we prepared samples using platelets from either C3−/− or wild type mice resuspended in plasma from either genotype (Figure 3). C3−/− platelets aggregate more in C3+/+ plasma than in C3−/− plasma (68% ± 4% vs 47% ± 2%; p=0.04, n=3). Conversely, C3+/+ platelets aggregate less in C3−/− plasma compared to that in C3+/+plasma (54% ± 2% vs 71% ± 3%; p=0.006). We also studied the effect of C3 on platelet aggregation by adding purified C3 to PRP obtained from C3−/− mice, and comparing the PAR4 peptide-induced platelet aggregation in C3-supplemented samples with that of C3-deficient and wild-type mice (Figure 4). Addition of purified C3 improved aggregation of platelets in C3−/− PRP samples (42% ± 3% without C3 vs 61% ± 4% with C3; p=0.03, n=4). There was no statistical significance between platelet aggregation in C3−/− PRP samples with added purified C3 and those in C3+/+ PRP samples (61% ± 4 and 69% ± 3, respectively; p=0.09).
Figure 2. Platelet aggregation.
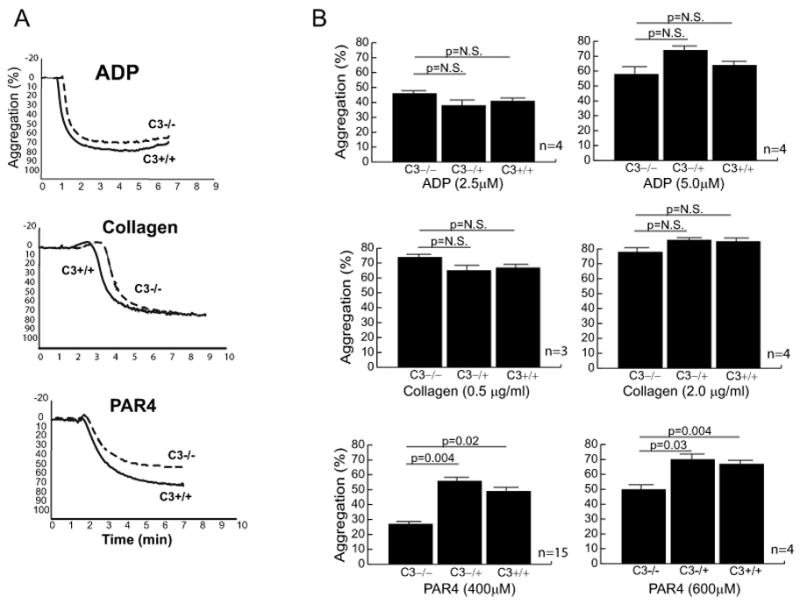
Using a light transmittance aggregometry and different agonists (ADP, collagen, and PAR4 peptide), platelet aggregation was compared between C3-deficient mouse and its wild-type littermates. For each experiment, 225 μl of platelet-rich plasma (normalized to 2.5 × 105/μL platelets) was used. (A) Representative curves of the aggregation studies using ADP (5 μM), collagen (0.5 μg/ml), and PAR4 peptide (600 μM). (B) The results of several platelet aggregation studies are summarized as bargraphs showing the average ± standard error values. P values were calculated using paired t-test analysis (two-tailed).
Figure 3. Mixing studies with plasma and platelet aggregation.
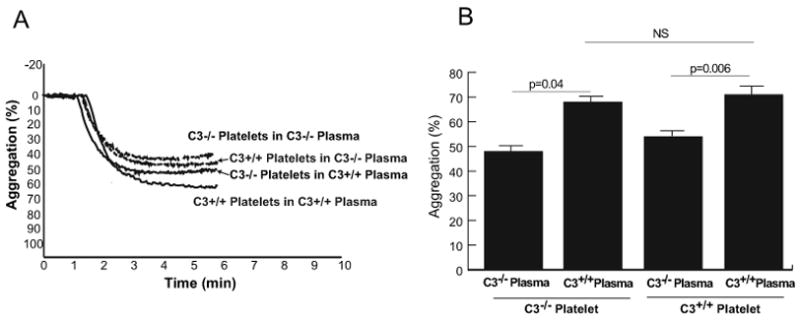
Platelets from C3-deficient mice or wild-type mice were suspended in plasma from either C3-deficient or wild-type mice to a concentration of 2.5 × 105/μL, and activated with PAR4 peptide (400 μM). (A) The aggregation tracing of one experiment. (B) The aggregation data from 3 experiments (3 mice in each group) are summarized as means ± standard errors.
Figure 4. Mixing studies with plasma- purified C3 and platelet aggregation.
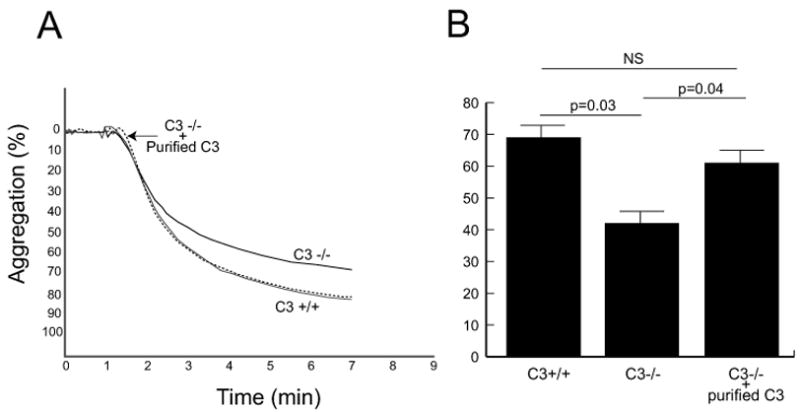
Platelet aggregation in PRP samples from C3-deficient mice in the presence or absence of 0.4 mg/ml of purified C3 was induced by adding PAR4 peptide (600 μM) and compared to that of wild type mice. (A) Aggregation tracing from one experiment. (B) Summary of aggregation data from 4 experiments (4 mice in each group) are summarized as bar graphs (means ± standard errors).
Intravital microscopy and in vivo thrombosis model
In order to investigate the in vivo effect of complement deficiency on platelet function, we used a mouse thrombosis model (10). We monitored the initiation and progression of thrombosis in the cremasteric microvessels of mice after photochemical injury to vessel wall. The onset of thrombosis and its progression to a complete occlusion of photochemically-injured microvessels (arterioles and venules) were recorded and compared between different genotypes (Figure 5). The time necessary for complete cessation of the blood flow in arterioles (mean diameter 37.5 ± 0.5 μm) was significantly prolonged in C3−/− compared to C3+/+ mice (28.9 min ± 2.5 and 22.5 min ± 1.7, respectively, p<0.05) (Figure 5A). The onset of thrombosis and the time interval required for the complete occlusion of venules (mean diameter 44.7 ± 0.6 μm) were similar in C3−/− and C3+/+ mice (Figure 5B).
Figure 5. Intravital microscopy in a murine thrombosis model.
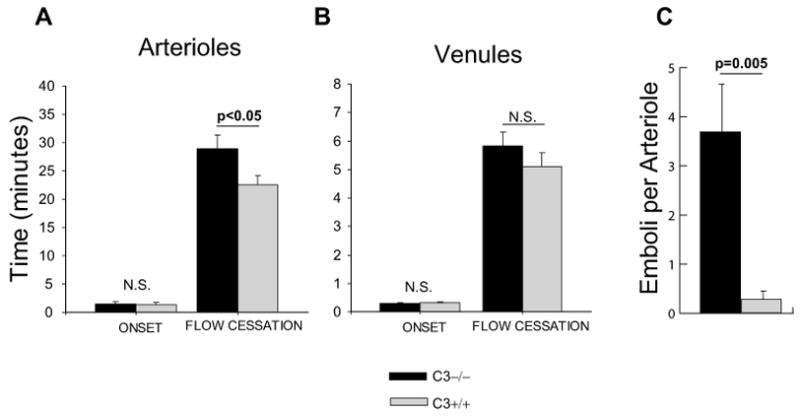
The time to onset and completion of thrombosis in the cremastric (A) arterioles and (B) venules of C3-deficient mice are compared to that of its wild-type littermates. (C) The average number of emboli detached from the thrombus in each arteriole was compared between C3-deficient and wild-type mice. Data are shown as average ± standard deviation and each bar represents the results obtained from 10 mice in each group.
The delayed time to flow cessation in arterioles of C3−/− mice was associated with thrombus instability, as has been reported in photochemical thrombosis of other mouse strains (12). The majority of arterioles in C3−/− mice revealed formation of large thrombi (with diameter more than half the vascular diameter), which embolized prior to flow cessation. In contrast, in littermate controls most large thrombi progressed to vessel occlusion and embolization occurred infrequently. On average, C3−/− mice had 3.7 ± 1.0 emboli per arteriole, as compared to 0.3 ± 0.1 in wild-type littermates (p < 0.005) (Figure 5C).
Discussion
The complement system shares several structural and functional similarities with the coagulation cascade (13). Blood coagulation can activate the complement system through a platelet-dependent mechanism (14;15). Similarly, activation of the complement system increases procoagulant activity of platelets (16) and accelerates blood coagulation (17). Whether the complement system has a role in the baseline hemostatic function of platelets is unknown. It has been reported that C6-deficient rabbits have a bleeding diathesis (18) and abnormal serotonin release in response to platelet activation (19). However, humans deficient in C6 or C7 did not show any evidence of excessive bleeding or abnormal platelet function (20;21). We studied platelet function in C3-deficient mice and found that platelets of C3−/− mice were hypoactive in comparison to that of C3+/+ platelets. The decrease in platelet aggregation in C3-deficient mice was agonist-specific. ADP and collagen-induced platelet aggregation were similar between C3-deficient and wild-type mice. However, PAR4 peptide caused less aggregation in C3-deficient mice compared to their littermates. This might be a clue that abnormal aggregation of C3−/− platelets in response to PAR4 peptide is not due to an intrinsic platelet abnormality, because ADP and collagen could induce normal aggregation. To further investigate this point, we isolated C3−/− platelets and resuspended them in either C3-deficient plasma or normal plasma (Figure 3). C3−/− platelets aggregated less in C3−/− plasma compared to C3−/− platelets in normal plasma. C3+/+ platelets also aggregated less in C3−/− plasma than C3+/+ plasma. Furthermore, addition of purified C3 improved in vitro platelet aggregation in C3−/− mice (Figure 4). These findings are consistent with the possibility that lack of C3 in plasma (or absence of the complement activity) is the reason for reduced platelet activity in C3−/− mice.
In order to investigate the in vivo effect of complement deficiency on platelet function, we used a mouse thrombosis model (10). We monitored the initiation and progression of thrombosis in the cremasteric microvessels of mice after photochemical injury to vessel wall. The onset of thrombosis and progression of it to a complete occlusion of photochemically-injured microvessels (arterioles and venules) were recorded and compared between different genotypes (Figure 5). The time necessary for complete cessation of the blood flow in arterioles was significantly prolonged in C3−/− compared to C3+/+ mice (Figure 5A). The delayed arteriolar closure time in C3−/− mice might be due to the lack of thrombus stability and more frequent detachment of emboli from thrombi in these mice compared to the wild-type mice (Figure 5C).
Decreased platelet aggregation and delayed arteriolar thrombosis are consistent with the presence of abnormal platelet function in C3-deficient mice. In order to explain this finding, we put forward the following hypothesis. Activation of platelets would result in deposition of C3b on platelets and C3b can bind to P-selectin (4). C3b on activated platelets can act as a ligand for P-selectin on the other activated platelets and mediate platelet aggregation. Lack of C3 (and as a result lack of C3b) would destabilize platelet aggregates and could explain a prolonged bleeding time, recurrent bleeding from a tail cut, and thrombus instability in C3−/− mice. Another possible explanation for our finding is that under physiologic conditions, platelet activation and complement activation would reinforce each other, and lack of a functional complement system would break this co-activation loop and results in fewer activated platelets.
Acknowledgments
This work is supported by NHLBI grant HL-079368 (R.E.R.), AHA Scientist Development Grant 0630180N (F.G.), and Department of Veteran Affairs Merit Review Grant (V.A-K and R.E.R).
Footnotes
Authorship: F.C. Gushiken: Performed research, designed experiments, analyzed data
H. Han: performed research, analyzed data, wrote the paper
J. Li: Performed research
R.E. Rumbaut: Performed research, designed experiments analyzed data, wrote paper
V. Afshar-Kharghan: performed research, designed experiments, analyzed data, wrote the paper
Reference List
- 1.Zimmerman TS, Muller-Eberhard HJ. Complement-induced platelet protein alterations. Science. 1973;180:1183–1185. doi: 10.1126/science.180.4091.1183. [DOI] [PubMed] [Google Scholar]
- 2.Polley MJ, Nachman RL. Ultrastructural lesions on the surface of platelets associated with either blood coagulation or with antibody-mediated immune injury. J Exp Med. 1975;141:1261–1268. doi: 10.1084/jem.141.6.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Polley MJ, Nachman R. The human complement system in thrombin-mediated platelet function. J Exp Med. 1978;147:1713–1726. doi: 10.1084/jem.147.6.1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Del Conde I, Cruz MA, Zhang H, Lopez JA, Afshar-Kharghan V. Platelet activation leads to activation and propagation of the complement system. J Exp Med. 2005;201:871–879. doi: 10.1084/jem.20041497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peerschke EI, Yin W, Grigg SE, Ghebrehiwet B. Blood platelets activate the classical pathway of human complement. J Thromb Haemost. 2006;4:2035–2042. doi: 10.1111/j.1538-7836.2006.02065.x. [DOI] [PubMed] [Google Scholar]
- 6.Sims PJ, Wiedmer T. The response of human platelets to activated components of the complement system. Immunol Today. 1991;12:338–342. doi: 10.1016/0167-5699(91)90012-I. [DOI] [PubMed] [Google Scholar]
- 7.Gralnick HR, Vail M, McKeown LP, Merryman P, Wilson O, Chu I, Kimball J. Activated platelets in paroxysmal nocturnal haemoglobinuria. Br J Haematol. 1995;91:697–702. doi: 10.1111/j.1365-2141.1995.tb05371.x. [DOI] [PubMed] [Google Scholar]
- 8.Stahl AL, Vaziri-Sani F, Heinen S, Kristoffersson AC, Gydell KH, Raafat R, Gutierrez A, Beringer O, Zipfel PF, Karpman D. Factor H dysfunction in patients with atypical hemolytic uremic syndrome contributes to complement deposition on platelets and their activation. Blood. 2008;111:5307–5315. doi: 10.1182/blood-2007-08-106153. [DOI] [PubMed] [Google Scholar]
- 9.Afshar-Kharghan V. Unleashed platelets in aHUS. Blood. 2008;111:5266. doi: 10.1182/blood-2008-03-140905. [DOI] [PubMed] [Google Scholar]
- 10.Rumbaut RE, Randhawa JK, Smith CW, Burns AR. Mouse Cremaster Venules are Predisposed to Light/Dye-Induced Thrombosis Independent of Wall Shear Rate, CD18, ICAM-1, or P-Selectin. Microcirculation. 2004;11:239–247. doi: 10.1080/10739680490425949. [DOI] [PubMed] [Google Scholar]
- 11.Rumbaut RE, Bellera RV, Randhawa JK, Shrimpton CN, Dasgupta SK, Dong JF, Burns AR. Endotoxin enhances microvascular thrombosis in mouse cremaster venules via a TLR4-dependent, neutrophil-independent mechanism. Am J Physiol Heart Circ Physiol. 2006;290:H1671–H1679. doi: 10.1152/ajpheart.00305.2005. [DOI] [PubMed] [Google Scholar]
- 12.Bonnefoy A, Daenens K, Feys HB, De VR, Vandervoort P, Vermylen J, Lawler J, Hoylaerts MF. Thrombospondin-1 controls vascular platelet recruitment and thrombus adherence in mice by protecting (sub)endothelial VWF from cleavage by ADAMTS13. Blood. 2006;107:955–964. doi: 10.1182/blood-2004-12-4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Markiewski MM, Nilsson B, Ekdahl KN, Mollnes TE, Lambris JD. Complement and coagulation: strangers or partners in crime? Trends Immunol. 2007;28:184–192. doi: 10.1016/j.it.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Kalowski S, Howes EL, Jr, Margaretten W, McKay DG. Effects of intravascular clotting on the activation of the complement system: The role of the platelet. Am J Pathol. 1975;78:525–536. [PMC free article] [PubMed] [Google Scholar]
- 15.Siraganian RP. Platelet requirement in the interaction of the complement and clotting systems. Nat New Biol. 1972;239:208–210. doi: 10.1038/newbio239208a0. [DOI] [PubMed] [Google Scholar]
- 16.Sims PJ, Faioni EM, Wiedmer T, Shattil SJ. Complement proteins C5b-9 cause release of membrane vesicles from the platelet surface that are enriched in the membrane receptor for coagulation factor Va and express prothrombinase activity. J Biol Chem. 1988;263:18205–18212. [PubMed] [Google Scholar]
- 17.Zimmerman TS, Muller-Eberhard HJ. Blood coagulation initiation by a complement-mediated pathway. J Exp Med. 1971;134:1601–1607. doi: 10.1084/jem.134.6.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zimmerman TS, Arroyove CM, Muller-Eberhard HJ. A blood coagulation abnormality in rabbits deficient in the sixth component of complement (C6) and its correction by purified C6. J Exp Med. 1971;134:1591–1600. doi: 10.1084/jem.134.6.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henson PM, Cochrane CG. Immunological induction of increased vascular permeability. II. Two mechanisms of histamine release from rabbit platelets involving complement. J Exp Med. 1969;129:167–184. doi: 10.1084/jem.129.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heusinkveld RS, Leddy JP, Klemperer MR, Breckenridge RT. Hereditary deficiency of the sixth component of complement in man. II. Studies of hemostasis. J Clin Invest. 1974;53:554–558. doi: 10.1172/JCI107589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boyer JT, Gall EP, Norman ME, Nilsson UR, Zimmerman TS. Hereditary deficiency of the seventh component of complement. J Clin Invest. 1975;56:905–913. doi: 10.1172/JCI108170. [DOI] [PMC free article] [PubMed] [Google Scholar]