

Abstract
Hybridization in isolated populations can lead either to hybrid breakdown and extinction or in some cases to speciation. The basis of hybrid breakdown lies in genetic incompatibilities between diverged genomes. In social Hymenoptera, the consequences of hybridization can differ from those in other animals because of haplodiploidy and sociality. Selection pressures differ between sexes because males are haploid and females are diploid. Furthermore, sociality and group living may allow survival of hybrid genotypes. We show that hybridization in Formica ants has resulted in a stable situation in which the males form two highly divergent gene pools whereas all the females are hybrids. This causes an exceptional situation with large-scale differences between male and female genomes. The genotype differences indicate strong transmission ratio distortion depending on offspring sex, whereby the mother transmits some alleles exclusively to her daughters and other alleles exclusively to her sons. The genetic differences between the sexes and the apparent lack of multilocus hybrid genotypes in males can be explained by recessive incompatibilities which cause the elimination of hybrid males because of their haploid genome. Alternatively, differentiation between sexes could be created by prezygotic segregation into male-forming and female-forming gametes in diploid females. Differentiation between sexes is stable and maintained throughout generations. The present study shows a unique outcome of hybridization and demonstrates that hybridization has the potential of generating evolutionary novelties in animals.
Keywords: Haldane’s rule, hybrid speciation, hybridization, Hymenoptera, sex-specific allele transmission
Hybridization between two species often has negative consequences that result in reduced fertility or viability (1–3). A common consequence is transmission ratio distortion (TRD) whereby genetic incompatibilities, either nuclear or cytonuclear, cause allelic segregation to depart from the Mendelian 1:1 ratio (4). Occasionally, hybridization can lead to hybrid vigor in which a hybrid offspring performs well. Yet, recombination and allelic segregation can lead to hybrid breakdown in the next generation. When hybridization is successful, it can contribute to adaptation and speciation (5). Introgression may transfer adaptations from one species into another (6) or hybridization may lead to new evolutionary lineages (7). The formation of new species by hybridization has been widely documented in plants (8), but evidence of homoploid hybrid speciation (i.e., formation of a hybrid lineage without a change in chromosome number) in animals is lacking. Recently, hybridization has been recognized as a potentially creative evolutionary process by which new species and evolutionary novelties can also arise in animals (9–12).
Consequences of hybridization in ants can differ from those in other animals because of haplodiploidy and sociality. Social insects live in colonies and display division of labor in reproduction. Only a small percentage of the individuals within a nest reproduce whereas others, the workers, take care of a range of tasks and do not usually produce any offspring. Because of this, sterility of hybrid workers would have no or a minimal fitness cost on the colony level (13). In fact, hybridization may even be an advantage in social insects provided that the workers show hybrid vigor. In haplodiploid Hymenoptera, males arise from unfertilized eggs and are haploid whereas females, both queens and workers, are diploid and produced biparentally. Allospecific mating is thus not necessarily an evolutionary dead end for a queen, because she can still produce nonhybrid males by laying unfertilized eggs (13). Even though F1 hybrid workers commonly occur in ants (14), there are few or no records on fertile hybrid queens or males (15).
We studied the genetic consequences of hybridization in a wood ant population originating from an ancient hybridization event. We report a unique outcome of hybridization in animals and show, by using 17 microsatellite markers and 81 amplified fragment length polymorphism (AFLP) loci, that hybridization in Formica ants has resulted in a stable situation with large-scale genetic differences between male and female genomes within a species. We hypothesize that the genetic differentiation between sexes has arisen because of genetic incompatibilities resulting from hybridization and that differentiation is maintained by sex-specific allele transmission.
Results
Population Is a Hybrid Between Two Wood Ant Species.
The discriminant analysis shows that all of the workers and gynes (newly produced sexual females) in the study population are morphologically intermediate between the species Formica aquilonia and Formica polyctena (Fig. S1). Cross-validation in the data set including the reference material rejects only 3.8% and 1.3% of the classifications of workers and gynes, respectively, but there was no rejection in the material from the study population. The morphological data thus indicate a hybrid identity of our study population. Both F. aquilonia and F. polyctena are known to have highly polygynous nests (i.e., nests with many queens), and accordingly several tens of reproductive queens within single nests were observed in the study population. Only one mitochondrial haplotype was obtained from queens (n = 3), males (n = 9), and workers (n = 8) of the study population, and it was identical to that of F. polyctena.
Population Consists of Two Genetic Groups.
New reproductive individuals, gynes and males, are produced once per year in the spring. We found that the pair-wise AFLP differences among the haploid males (n = 97) had a bimodal distribution with peaks at d values of 0.26 and 0.37, corresponding to 21 and 30 differences, respectively (Fig. S2). Bayesian mixture analysis performed by BAPS software found that the best partitioning of the data were two clusters. Almost half of the AFLP markers (38 of 81) showed a diagnostic difference between the two genetic groups. Twenty-four alleles were diagnostic for one group and 14 for the other. An allele was classified as diagnostic if it was missing in one group and the frequency exceeded 10% in the other. The two genetic groups are hereon referred to as groups W and R. The distribution of pair-wise AFLP differences (Fig. S2) showed that group W had significantly less variation than group R. The mean pair-wise distances within group W (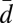 = 0.26, SD = 0.05), within group R (
= 0.26, SD = 0.05), within group R (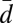 = 0.33, SD = 0.06), and between the groups (
= 0.33, SD = 0.06), and between the groups (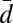 = 0.40, SD = 0.06) all differed significantly from each other (t test, using the number of individuals as sample size, in all comparisons t > 6, P < 0.01). Next, microsatellites (17 loci) from the same individuals were genotyped. The genetic difference between groups W and R was further demonstrated by the microsatellites as almost half of the alleles (13 alleles in both W and R groups of the total of 55) were specific and thus diagnostic to either group. Accordingly, the best partitioning of male microsatellite genotypes by mixture analysis was two genetic groups, which were identical to the W and R groups formed as described earlier.
= 0.40, SD = 0.06) all differed significantly from each other (t test, using the number of individuals as sample size, in all comparisons t > 6, P < 0.01). Next, microsatellites (17 loci) from the same individuals were genotyped. The genetic difference between groups W and R was further demonstrated by the microsatellites as almost half of the alleles (13 alleles in both W and R groups of the total of 55) were specific and thus diagnostic to either group. Accordingly, the best partitioning of male microsatellite genotypes by mixture analysis was two genetic groups, which were identical to the W and R groups formed as described earlier.
We genotyped diploid gynes (n = 95) to compare them to the males. As there were two genetic groups among males, we also divided the gyne microsatellite genotypes in two clusters with BAPS. The groups thus obtained were identical with those formed by another Bayesian clustering program, Structure. Computing the ΔK statistic for Structure results indicates that the most likely number of genetic groups is two in the gyne data. When comparing the two female groups to the groups found in males, gynes of one cluster are clearly assigned to the male group W [likelihood ratio (LR) ranging from 18.7 to 46.5] and those of the other cluster to the male group R (LR ranging from 0.3 to −22.1; Fig. S3). Thus, two genetic groups (R and W) were also formed from the gynes. Whereas ΔK statistic indicated two genetic clusters in the gyne data, BAPS suggested that the optimal number is four by dividing the W gynes further into three subgroups. It is not evident whether these subgroups are biologically relevant or caused by departures from the expected Hardy-Weinberg genotype frequencies. Diagnostic alleles were also found in the W and R females. However, alleles diagnostic to the male groups were not all diagnostic in the females. The female groups differed from each other also at the AFLP loci, even though the differences were less clear because of the dominant nature of AFLP markers.
Combining the two sexes, approximately one third of all of the markers were diagnostic and specific to one of the genetic groups, namely 25 AFLP loci and 19 microsatellite alleles (Table 1 and Table S1). Furthermore, several other alleles showed significant frequency differences between the groups. Group W consisted of 62 males and 70 females and group R 35 males and 25 females. The result was consistent in both microsatellite and AFLP markers genotyped from the same individuals. Group R was less frequent in the population and always coexisted in the same nests with group W. The worker genotypes in the population were similar to the gynes, as they could be classified into the two groups and had the same diagnostic alleles.
Table 1.
The number of alleles in different categories and their occurrence in the two groups (W and R) and sexes (gynes and males)
| Allele category | Total no. of alleles | Mean number per individual | |||
| W males | W females | R females | R males | ||
| Diagnostic to W | 8 | 3.3 | 4.1 | — | — |
| Diagnostic to R | 11 | — | — | 4.1 | 4.7 |
| Admixed from W to R | 5 | 2.0 | 5.0 | 3.0 | — |
| Admixed from R to W | 3 | — | 0.8 | 2.6 | 0.9 |
| Female-specific | 3 | 0.0 | 0.9 | 1.2 | — |
| Shared | 24 | 11.5 | 23.0 | 22.8 | 11.3 |
The numbers of alleles per individual were calculated over the microsatellite loci. A dash indicates that no allele was observed and 0.0 indicates that the number of observed alleles was less than 0.05 per individual. “Shared” refers to alleles observed in gynes and males of both groups even though there were frequency differences. One rare allele was omitted as it could not be classified.
We next genotyped old reproductive queens and the contents of their spermathecae (i.e., the sperm of their mates) with the nine most informative microsatellite loci to establish if mating had taken place between or within groups. The results indicated that the two groups and their genetic characteristics are stable, as the old reproductive queens showed the same characteristics as the newly produced daughters (i.e., new gynes and workers). The old reproductive queens were divided into the two groups with the same diagnostic alleles as the newly produced gynes. The 56 old queens had mated with at least 77 males in total. This is a minimum estimate, because it is based on the spermathecal content and the maximal detectable mating number equals the largest number of alleles present at any locus. The male alleles inferred from the spermathecae showed that the haplotypes of the mates matched the R and W genotypes, and that a clear majority of matings had taken place within the groups. Based on the alleles that were diagnostic to the two male groups, only five of the inferred matings (6.5%) had occurred between the two groups.
Both Groups Have Genetic Differences Between Sexes.
The most striking finding was that both genetic groups W and R had large genetic differences between the sexes. The males represented two highly diverged gene pools (DA= 0.39; from microsatellites), whereas the females, even though clearly separating into two groups, were to some extent intermediate between the male gene pools. The distance between the two female groups was DA= 0.16. In addition, the females had many alleles which the males in their group lacked. Bayesian admixture analysis of the microsatellite data showed that, on average, 8% of the W group female genome had come from the gene pool of the R group (as defined by the R males) and, on average, 36% of the R group female genome had come from the W group (as defined by the W males; Fig. 1). Within a group, genetic differences between the sexes arise because the females are admixed and carry alleles originating from the other group.
Fig. 1.

Admixture results showing introgression in females. The males were used to define the two gene pools (W and R) and the admixture proportions were estimated for each gyne (n = 95) and old queen (Q; n = 23) by using the Bayesian method implemented in BAPS. Each individual is represented by a vertical bar. The black part indicates the fraction of the genome originating from the W gene pool and the white part the fraction from the R gene pool.
Segregation of Alleles Is Not Random.
The microsatellite alleles could be divided into four categories based on their occurrence in the two genetic groups and sexes (Table 1 and Tables S1 and S2). Individual genotypes were examined to infer the transmission and origin of different allele types. The first allele category included the diagnostic alleles (35% of all the alleles) mentioned earlier (Table 1). Eleven alleles (20%) were specific to group R. All these alleles were only observed as heterozygotes in the R females, on average 4.1 alleles per single female (SD = 1.6). The expected number of such diagnostic alleles in a haploid male should equal that in a random gamete produced by a female. Yet, the number per single R male (mean = 4.7; SD = 1.2) was significantly larger than expected in the randomly produced gametes (P < 10−6 based on 1 million randomly sampled haploid genomes from the female genotype pool) and even larger than in an average diploid female. If mating within a group and segregation of alleles were random, many homozygous females would also be expected but were not observed. The situation in group W was somewhat similar but not identical. There were eight alleles (15%) diagnostic to group W. An average male carried 3.3 such alleles (SD = 1.3) and a female 4.2 (SD = 1.5). Again, the number of these alleles observed per single male was significantly larger than per haploid genome within the females (P < 10−6). For seven of these W-diagnostic alleles, some homozygous females were found, but the frequencies of these alleles were always smaller in the females (mean = 0.28) than in the males (mean = 0.44; sign test, P ≈ 0.016).
The second category included admixed alleles (15% of all), i.e., alleles that were shared by the two female groups but were missing from the males of one group (Table 1). The alleles that were not found in a male group always existed only as heterozygotes in the females of that group, whereas homozygous females existed in the other group. Yet, the frequency of such heterozygous females could be high. For example, the five microsatellite alleles that were missing from R males had an average frequency of 0.30 in R females (significant frequency differences between the sexes for each allele, P ≤ 0.01). Particularly, all of the R females were heterozygous for an allele, Fe13198, that was not found in R males (P = 10−7). Likewise, three alleles that were missing from W males had frequencies ranging from 0.07 to 0.18 in W females (P ≤ 0.02 for each).
The third category was formed by the female-specific alleles (5%; Table 1). Three microsatellite alleles and three additional AFLP alleles were shared by the two female groups with frequencies ranging from 0.08 to 0.38, but were missing from the males. Four of these alleles were completely absent from all of the males and two were carried only by an occasional male (frequency of 0.01–0.02 in the males; difference between the sexes, P ≤ 1.5 × 10−4; Table S3). For example, more than half the R females were heterozygous for the allele Fe756, which was never observed in any male. The remaining 44% of the alleles made up the fourth category with alleles shared by the genetic groups and the sexes, even though there could be frequency differences.
Discussion
Our results demonstrate that the females in the study population did not represent the same gene pools as the males of their respective group, but all of the females had a hybrid background both morphologically and genetically. They were not simple F1 hybrids, as the admixture analysis indicated that the two male gene pools were unequally represented in the female genomes. Furthermore, individual female genotypes did not agree with the expected frequencies of F1 hybrids, as the females were at the same time homozygous for some alleles of their own group and heterozygous for alleles originating from the other group. The two groups and both sexes shared the same mtDNA haplotype, which means that the males also had the same maternal origin and must have an old hybrid background, even though this was not seen in the present-day data. Our conclusions on possible hybrid males are restricted because the males were initially used to define the two gene pools. However, the large genetic difference between the male gene pools shows that recent introgression between them has been limited. There are only a few known examples showing that ant hybrids can develop into queens (15), and our results present a unique case in which hybrid females regularly become queens. Normally, hybrid female ants develop into workers whether hybridization is local and sporadic (13, 14) or widespread and systematic as in the Pogonomyrmex harvester ants (16).
The comparison of male and female genotypes within groups W and R suggests TRD, which strongly depends on the sex of the offspring. TRD is a common consequence of hybridization (4), but sex-specific TRD is unusual. There are three arguments supporting the conclusion that a Formica queen transmits some alleles exclusively to her daughters and other alleles exclusively to her sons (Fig. 2). (i) Females were largely or exclusively heterozygous for alleles that were specific to their own genetic group, even though these alleles were common in the males. The frequencies of such alleles among the females were approximately half of those among the males of the same group. It thus seems that the females had inherited these alleles from their fathers and transmitted them to their sons. (ii) Females were frequently heterozygous for alleles that had apparently been introgressed from the other genetic group (our second category of alleles). Yet, these females seemed to transfer these alleles only to their daughters as the alleles were not found among the males of that group. (iii) There were several alleles that existed in females of both groups but were never detected in any males. These alleles obviously always existed in a heterozygous condition in the females and seem to be transmitted only from mothers to daughters. All three points contribute to an overall excess of heterozygotes in females compared with the Hardy-Weinberg expectations. Such an excess was detected at 15 of 16 variable loci in group R and at 13 of 16 loci in W (exact test, P < 0.011).
Fig. 2.

Hypothesized model of allele transmission. The mother transmits her maternally inherited alleles (white circle) exclusively to her daughters and the paternally inherited alleles (black circle) to her sons. This sex-specific transmission could arise either from strong postzygotic selection eliminating incompatible genotypes or prezygotic segregation of alleles into female-forming and male-forming gametes.
Our finding raises immediately questions concerning the origin and maintenance of the two genetic groups and the genetic differences between the sexes within a group. Genetic distance between the two male groups was DA = 0.39. Assuming that the females have received the paternal haploid genome from a male of their own group, as was evident in most cases of the spermathecal analysis, the allele frequencies among the newly produced males also represent the frequencies among the fathers in the previous generation. The gyne genotypes can thus be used to estimate the allele frequencies among the maternal gametes. Based on these estimates (Table S4), the maternal contributions in R and W females were genetically closer to each other (DA = 0.08) than either was to the male gene pool of their own group (DA = 0.34 within R and DA = 0.16 within W). It is thus logical to conclude that the unusual segregation patterns within the groups descend from an ancestral hybrid population, and the common ancestry is still seen as the genetic similarity in that part of the genome that is transmitted along the female lineages (i.e., from mothers to daughters). This scheme suggests differences between maternally and paternally inherited haploid genomes. The maternally inherited haploid genome is admixed and contains some parts originating from the W group and some from the R group (Fig. 3). Consequently, an allele that is found only in the maternally transmitted haploid genome and not in the paternal counterpart (and vice versa) can exist only in a heterozygous condition in females of that genetic group. The following questions arise: How do the paternally and maternally received haploid genomes segregate and do they recombine?
Fig. 3.

Schematic presentation of male (haploid) and female (diploid) genotypes in groups R and W. Loci within each haploid genome are represented by rectangles and the color codes indicate the hypothesized origin of alleles. Different allele categories are indicated by letters: a, shared by various haploid genomes (gray); b, diagnostic to group W (red); c, admixed from W to R females (red); d, admixed from R to W females (blue); e, diagnostic to group R (blue); f, female specific (yellow). A diploid female genome consists of a paternal component (similar to males in that group) and maternal component (similar in the two female groups).
There was some indication of recombination between the postulated maternal and paternal haploid genomes. Six loci (three microsatellites and three AFLPs) had female-specific alleles, which thus must be associated with the maternally transmitted haploid genome. At two of these loci altogether, three allelic copies were found in males, two of them in the same male. That male also had an intermediate position in the AFLP analysis with an about equal distance to the W and R males. It is likely that the male was a recombinant between the maternal and paternal genomes in group W as all its diagnostic alleles were of type W.
Despite possible recombination, the system is stable as similar genotypic patterns characterized successive generations (old queens and the sperm in their spermathecae as well as newly produced sexuals and workers). The two genetic groups stay separate from each other as current-day mating happens mainly within the groups. The spermathecal analysis showed a small fraction of mating between the groups, but it is not known if these result in fertile offspring. Furthermore, genetic differences between the sexes were observed in the same population already two decades earlier in a study of six allozyme loci (17). In that study, three allozyme loci (Me, Pgk, Gpi) had a large fraction of heterozygous queens (36–54%), yet the less frequent alleles were completely missing from newly produced males (131 males) and had very low frequencies (0.3–3%) among the fathers inferred from mother-offspring analyses. Two other enzyme loci (Pep, Est) also showed large allele frequency differences between the sexes, the allele frequency in the queens being clearly smaller than in the males (0.11 vs. 0.23) or larger (0.48 vs. 0.19). The sixth enzyme locus (Pgm) showed significant TRD. These observations fit the patterns detected in our present data and show that segregation distortion and genetic differences between the sexes have stayed in the population for decades.
What mechanisms then explain the unusual pattern of allele transmission and the lack of hybrid features in males and their persistence in females? The observed pattern could result from strong postzygotic selection eliminating incompatible genotype combinations (1–3), or alternatively prezygotic segregation could separate paternally and maternally inherited chromosomes into different eggs, the former producing haploid sons and the latter new females after having been fertilized. Strong selection against hybrid males could create large-scale differentiation between sexes, because recessive incompatibilities can be masked in diploid heterozygous females but not in haploid males. Consequently the hybrid males die, but females can survive. The fact that alleles missing from males were always heterozygous in females of the same group is in agreement with the incompatibility hypothesis. In addition to selection against hybrid males, strong heterosis is required for maintenance of hybrid features in females. If the allelic combinations within and between loci were produced at random, selection would be required to eliminate a large proportion of offspring (e.g., 89% of R males and 37% of W males) to produce the observed genotype patterns (Table S5). It should be noted that selection against unfit genotypes cannot alone explain why many of the putatively ancestral group-specific alleles did not exist as homozygotes in females.
Another alternative is that hybridization has led to the formation of two independently segregating allelic sets: the maternal set consisting of alleles transmitted from mothers exclusively to daughters and the paternal set consisting of alleles transmitted from mothers exclusively to sons. If maternal and paternal allelic sets segregate at the prezygotic level and are transmitted to the offspring as blocks with low recombination, the number of segregating genotype combinations and the required mortality are smaller. When the eggs with the paternal set remain unfertilized and develop into sons and those with the maternal set are fertilized and produce daughters, the females remain hybrids and the male groups separate. Two independently segregating allelic sets could be created because of chromosomal rearrangements. Chromosomal rearrangements can contribute to speciation by reducing recombination (18) and have been documented between and within species. In Oenothera primroses, several reciprocal translocations have led to formation of two chromosome sets that are inherited as blocks, one block through microspores and the other one through megaspores (19). The system has resulted in a stable genome-wide heterozygosity, a condition that characterizes the diploid females in our data as well. It has also been shown that genomic imprinting can lead to TRD (20). Furthermore, these phenomena are associated with kin conflicts, and imprinting is predicted to be particularly strong in male haploid Hymenoptera (21). Imprinting could provide a potential mechanism for distinction of maternal and paternal chromosome sets as has been suggested for the primrose case (22).
Stable differences between male and female genomes in a sexually reproducing animal are unique. The closest resemblance to such sex differences can be found in little fire ants (Wasmannia auropunctata), which demonstrate a similar outcome but a different underlying mechanism. In little fire ants, both females and males reproduce clonally, causing systematic differentiation between male and female genomes (23, 24). Similar situation is also found in the ant Vollenhovia emeryi (25). These examples, together with the current study of Formica ants, demonstrate the flexibility of the reproductive system in male haploid social insects (26).
In addition to the hypothesized transmission patterns, it is of considerable interest that the population has two genetically different groups coexisting in the same nests. Two coexisting lineages of hybrid origin are also known in Pogonomyrmex harvester ants. There the queens and males represent normal Mendelian populations of pure lineages whereas workers are hybrids between the lineages (27, 28). The situation in Formica ants is fundamentally different with evident introgression in females but not in males. The coexistence of the two genetic groups in the same Formica colonies also complicates the genetic conflicts predicted by the kin selection theory (29) and raises questions concerning recognition of the lineages and potential for nepotism (30, 31).
Our findings have important implications for the genetic background of reproductive isolation and thus for speciation. If hybrid sterility and inviability are caused by accumulation of complementary genes (32), severity of incompatibilities and the number of genes involved are expected to increase faster than linearly with time (33). Still, evidence for this snowball effect is lacking (34). The mtDNA sequences of F. aquilonia and F. polyctena differ by only 1.3% (35), but one half of the nuclear markers (AFLP and microsatellites) show diagnostic differences between the two male groups despite the hybrid nature of the females. If this differentiation reflects genetic incompatibilities, a large fraction of the genome must be involved. Thus, our data suggest that gene flow has multiple barriers that are scattered throughout the genome.
According to the Haldane’s rule, the heterogametic sex (i.e., XY or ZW) is likely to suffer more from genetic incompatibilities (36). Recently, Koevoets and Beukeboom (37) extended the Haldane’s rule to haplodiploid organisms, suggesting that the incompatibilities affect the hemizygous (i.e., haploid) sex. Thus, the negative effect of hybridization in ants would be strongest in the haploid males and would be seen on the scale of the entire genome. This is exactly what was observed in our study population, in which genetic differentiation was by far largest between the two male gene pools demonstrating the extended Haldane’s rule in a natural population. If the Formica females with apparent and complex hybrid characteristics represent viable, independently evolving units, the results also call for reconsideration of the importance of hybridization in speciation.
Materials and Methods
Sampling.
Samples were collected from a population situated in Tvärminne, Southern Finland, at a time period between 2004 and 2008 (Table S6). The population is located in a 10-ha area in a small cape in a coastal area. New reproductives, gynes (n = 95) and males (n = 97), were collected for genetic analyses as pupae from four nests in spring 2004. They were brought to the laboratory to eclose. Workers (n = 134) were collected as adults from all 24 nests of the population in spring 2005. Old reproductive queens from two nests were collected in 2007 (n = 23) and from seven nests in 2008 (n = 50). Workers (n = 54) and gynes (n = 18) were collected for morphometric analyses in spring 2007. The reference material for morphological identification included samples from the entire Palaearctic range, in total 410 workers from 74 F. aquilonia nests and from 50 F. polyctena nests, and 29 queens of each species. Voucher specimens are deposited in the entomological collection of the Senckenberg Museum of Natural History in Görlitz, Germany.
Genotyping.
DNA was extracted with the DNAeasy Tissue Kit (Qiagen) using the manufacturer's protocol designed for insects. Seventeen microsatellite markers, 81 variable AFLPs, and a 690-bp fragment of the mitochondrial cytochrome b sequence were used. Microsatellite loci used were FE7, FE38, FE13, FE19, FE37, FE17, FE42, FE51 (38); FL12, FL20, FL21, FL29 (39); and FY10, FY12, FY13, FY15, FY3 (40). Loci were amplified by PCR as described in the original reports with only small modifications. Annealing temperature and magnesium concentration were optimized for each locus individually (available upon request). AFLP reactions were conducted with AFLP Plant Mapping Kit supplied by Perkin-Elmer (Applied Biosystems) using 16 of all 64 possible primer combinations and the protocol designed for small genomes as described by the manufacturer. Cytochrome b PCR amplification was performed with primers Cyt-Fe-F (41) and CB11449 (42) and, in addition to these, sequenced with primers CB11059 (42) and CB11178 (42) as described previously (42). Genotypes were resolved by gel or capillary electrophoresis (ABI Prism 377 DNA sequencer or 3730 DNA Analyzer, Applied Biosystems) and scored with Genotyper or Genemapper v. 4.0 software (Applied Biosystems). Phenotypes at one allozyme locus (Gpi) were resolved by horizontal starch gel electrophoresis as described previously (17) and recorded with a digital camera.
Morphotyping.
Morphological identification of gynes and workers was carried out by discriminant analysis of 25 and 16 quantitative phenotypic characters, respectively, recorded by direct high-resolution stereomicroscopy as an extension of the methods used by Seifert (43). Confidence of clustering was confirmed by leave-one-out cross-validation (44).
Genetic Data Analysis.
The basic analyses (e.g., allele frequencies) of the microsatellite data were done by the program GENEPOP (45). Bayesian clustering of genotypes was carried out by the mixture analysis using BAPS software (46) and in some cases Structure (47) and the ΔK method (48). The Bayesian clustering algorithms were run with k ranging from 1 to 10 and in Structure admixture model with correlated allele frequencies (10 runs for each k, burn-in 20,000 and run length 80,000) was used. The assignment of gynes into groups W and R is based on microsatellites and was done by calculating for each female the likelihood (LW and LR) that its genotype arises from the respective gene pool as defined by the males. The likelihood was calculated as a probability that the female's multilocus genotype derives from the allele frequencies of either of the gene pools. Frequency of 0.02 was used for missing alleles. The LR was calculated as ln(LW/LR). The admixture analysis was carried out in BAPS by allowing the male allele frequencies define the parental populations. The genetic distances were estimated from the microsatellite data as the Nei DA (49) and the SEs were obtained by jackknifing over the loci. Other statistical tests involved t test, exact tests, and comparisons against a simulated distribution.
Supplementary Material
Acknowledgments
We thank R. Jokela for technical assistance and L. Sundström and the reviewers for valuable comments on an earlier version of the manuscript. Financial support was provided by the Finnish Graduate School of Population Genetics (J.K.), Biocenter Oulu (J.K.), and Academy of Finland Grant 122210 (to P.P.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0912409107/DCSupplemental.
References
- 1.Dobzhansky T. Studies on hybrid sterility. II. Localization of sterility factors in Drosophila pseudoobscura hybrids. Genetics. 1936;21:113–135. doi: 10.1093/genetics/21.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Muller HJ. Isolating mechanisms, evolution and temperature. Biol Symp. 1942;6:71–125. [Google Scholar]
- 3.Brideau NJ, et al. Two Dobzhansky-Muller genes interact to cause hybrid lethality in Drosophila. Science. 2006;314:1292–1295. doi: 10.1126/science.1133953. [DOI] [PubMed] [Google Scholar]
- 4.Nakazato T, Jung M-K, Housworth EA, Rieseberg LH, Gastony GJ. A genomewide study of reproductive barriers between allopatric populations of a homosporous fern, Ceratopteris richardii. Genetics. 2007;177:1141–1150. doi: 10.1534/genetics.107.076851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burke JM, Arnold ML. Genetics and the fitness of hybrids. Annu Rev Genet. 2001;35:31–52. doi: 10.1146/annurev.genet.35.102401.085719. [DOI] [PubMed] [Google Scholar]
- 6.Lewontin RC, Birch LC. Hybridization as a source of variation for adaptation to new environments. Evolution. 1966;20:315–336. doi: 10.1111/j.1558-5646.1966.tb03369.x. [DOI] [PubMed] [Google Scholar]
- 7.Rieseberg LH, Van Fossen C, Desrochers AM. Hybrid speciation accompanied by genomic reorganization in wild sunflowers. Nature. 1995;375:313–316. [Google Scholar]
- 8.Rieseberg LH. Hybrid origins of plant species. Annu Rev Ecol Syst. 1997;28:359–389. [Google Scholar]
- 9.Mallet J. Hybrid speciation. Nature. 2007;446:279–283. doi: 10.1038/nature05706. [DOI] [PubMed] [Google Scholar]
- 10.Mavárez J, Linares M. Homoploid hybrid speciation in animals. Mol Ecol. 2008;17:4181–4185. doi: 10.1111/j.1365-294x.2008.03898.x. [DOI] [PubMed] [Google Scholar]
- 11.Gompert Z, Fordyce JA, Forister ML, Shapiro AM, Nice CC. Homoploid hybrid speciation in an extreme habitat. Science. 2006;314:1923–1925. doi: 10.1126/science.1135875. [DOI] [PubMed] [Google Scholar]
- 12.Helms Cahan S, Keller L. Complex hybrid origin of genetic caste determination in harvester ants. Nature. 2003;424:306–309. doi: 10.1038/nature01744. [DOI] [PubMed] [Google Scholar]
- 13.Umphrey GJ. Sperm parasitism in ants: Selection for interspecific mating and hybridization. Ecology. 2006;87:2148–2159. doi: 10.1890/0012-9658(2006)87[2148:spiasf]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 14.Seifert B. Interspecific hybridisations in natural populations of ants by example of a regional fauna. (Hymenoptera, Formicidae) Insect Soc. 1999;46:45–52. [Google Scholar]
- 15.Feldhaar H, Foitzik S, Heinze J. Review. Lifelong commitment to the wrong partner: Hybridization in ants. Phil Trans R Soc B. 2008;363:2891–2899. doi: 10.1098/rstb.2008.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwander T, Helms Cahan S, Keller L. Characterization and distribution of Pogonomyrmex harvester ant lineages with genetic caste determination. Mol Ecol. 2006;16:367–387. doi: 10.1111/j.1365-294X.2006.03124.x. [DOI] [PubMed] [Google Scholar]
- 17.Pamilo P. Polyandry and allele frequency differences between the sexes in the ant Formica aquilonia. Heredity. 1993;70:472–480. [Google Scholar]
- 18.Hoffmann AA, Rieseberg LH. Revisiting the impact of inversions in evolution: From population genetic markers to drivers of adaptive shifts and speciation. Annu Rev Ecol Evol Syst. 2008;39:21–42. doi: 10.1146/annurev.ecolsys.39.110707.173532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holsinger KE, Ellstrand NC. The evolution and ecology of permanent translocation heterozygotes. Am Nat. 1984;124:48–71. [Google Scholar]
- 20.Naumova AK, Greenwood CMT, Morgan K. Imprinting and deviation from Mendelian transmission ratios. Genome. 2001;44:311–320. doi: 10.1139/g01-013. [DOI] [PubMed] [Google Scholar]
- 21.Wild G, West SA. Genomic imprinting and sex allocation. Am Nat. 2009;173:E1–E14. doi: 10.1086/593305. [DOI] [PubMed] [Google Scholar]
- 22.Úbeda F, Haig D. On the evolutionary stability of Mendelian segregation. Genetics. 2005;170:1345–1357. doi: 10.1534/genetics.104.036889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fournier D, et al. Clonal reproduction by males and females in the little fire ant. Nature. 2005;435:1230–1234. doi: 10.1038/nature03705. [DOI] [PubMed] [Google Scholar]
- 24.Foucaud J, Estoup A, Loiseau A, Rey O, Orivel J. Thelytokous parthenogenesis, male clonality and genetic caste determination in the little fire ant: New evidence and insights from the lab. Heredity. 2010 doi: 10.1038/hdy.2009.169. 10.1038/hdy.2009.169. [DOI] [PubMed] [Google Scholar]
- 25.Ohkawara K, Nakayama M, Satoh A, Trindl A, Heinze J. Clonal reproduction and genetic caste differences in a queen-polymorphic ant, Vollenhovia emeryi. Biol Lett. 2006;2:359–363. doi: 10.1098/rsbl.2006.0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keller L. Uncovering the biodiversity of genetic and reproductive systems: Time for a more open approach. American Society of Naturalists E. O. Wilson Award winner address. Am Nat. 2007;169:1–8. doi: 10.1086/509938. [DOI] [PubMed] [Google Scholar]
- 27.Helms Cahan S, et al. Extreme genetic differences between queens and workers in hybridizing Pogonomyrmex harvester ants. Proc R Soc B Biol Sci. 2002;269:1871–1877. doi: 10.1098/rspb.2002.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anderson KE, et al. Distribution and evolution of genetic caste determination in Pogonomyrmex seed-harvester ants. Ecology. 2006;87:2171–2184. doi: 10.1890/0012-9658(2006)87[2171:daeogc]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 29.Ratnieks FLW, Foster KR, Wenseleers T. Conflict resolution in insect societies. Annu Rev Entomol. 2006;51:581–608. doi: 10.1146/annurev.ento.51.110104.151003. [DOI] [PubMed] [Google Scholar]
- 30.Hannonen M, Sundström L. Sociobiology: Worker nepotism among polygynous ants. Nature. 2003;421:910. doi: 10.1038/421910a. [DOI] [PubMed] [Google Scholar]
- 31.Wenseleers T. Nepotism absent in insect societies - or is it? Mol Ecol. 2007;16:3063–3065. doi: 10.1111/j.1365-294X.2007.03313.x. [DOI] [PubMed] [Google Scholar]
- 32.Coyne JA, Orr HA. The evolutionary genetics of speciation. Philos Trans R Soc Lond B Biol Sci. 1998;353:287–305. doi: 10.1098/rstb.1998.0210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orr HA. The population genetics of speciation: The evolution of hybrid incompatibilities. Genetics. 1995;139:1805–1813. doi: 10.1093/genetics/139.4.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gourbière S, Mallet J. Are species real? The shape of the species boundary with exponential failure, reinforcement, and the “missing snowball”. Evolution. 2010;64:1–24. doi: 10.1111/j.1558-5646.2009.00844.x. [DOI] [PubMed] [Google Scholar]
- 35.Goropashnaya AV, Fedorov VB, Pamilo P. Recent speciation in the Formica rufa group ants (Hymenoptera, Formicidae): Inference from mitochondrial DNA phylogeny. Mol Phylogenet Evol. 2004;32:198–206. doi: 10.1016/j.ympev.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 36.Haldane JBS. Sex ratio and unisexual sterility in hybrid animals. J Genet. 1922;12:101–109. [Google Scholar]
- 37.Koevoets T, Beukeboom LW. Genetics of postzygotic isolation and Haldane's rule in haplodiploids. Heredity. 2009;102:16–23. doi: 10.1038/hdy.2008.44. [DOI] [PubMed] [Google Scholar]
- 38.Gyllenstrand N, Gertsch PJ, Pamilo P. Polymorphic microsatellite DNA markers in the ant Formica exsecta. Mol Ecol Notes. 2002;2:67–69. [Google Scholar]
- 39.Chapuisat M. Characterization of microsatellite loci in Formica lugubris B and their variability in other ant species. Mol Ecol. 1996;5:599–601. doi: 10.1111/j.1365-294x.1996.tb00354.x. [DOI] [PubMed] [Google Scholar]
- 40.Hasegawa E, Imai S. Characterization of microsatellite loci in red wood ants Formica (s.str.) spp. and the related genus Polyergus. Mol Ecol Notes. 2004;4:200–203. [Google Scholar]
- 41.Liautard C, Keller L. Restricted effective queen dispersal at a microgeographic scale in polygynous populations of the ant Formica exsecta. Evolution. 2001;55:2484–2492. doi: 10.1111/j.0014-3820.2001.tb00763.x. [DOI] [PubMed] [Google Scholar]
- 42.Goropashnaya AV, Fedorov VB, Seifert B, Pamilo P. Limited phylogeographical structure across Eurasia in two red wood ant species Formica pratensis and F. lugubris (Hymenoptera, Formicidae) Mol Ecol. 2004;13:1849–1858. doi: 10.1111/j.1365-294X.2004.02189.x. [DOI] [PubMed] [Google Scholar]
- 43.Seifert B. The “Hippie Ant” – a case of extreme intranidal polymorphism in Fennoscandian Formica lugubris Zetterstedt 1838 (Hymenoptera:Formicidae) Sociobiology. 2003;42:285–297. [Google Scholar]
- 44.Lesaffre E, Willems JL, Albert A. Estimation of error rate in multiple group logistic discrimination – the approximate leaving-one-out method. Comm Statist Theory Methods. 1989;18:2989–3007. [Google Scholar]
- 45.Raymond M, Rousset F. GENEPOP (version 1.2): Population genetics software for exact tests and ecumenicism. J Heredity. 1995;86:248–249. [Google Scholar]
- 46.Corander J, Marttinen P. Bayesian identification of admixture events using multilocus molecular markers. Mol Ecol. 2006;15:2833–2843. doi: 10.1111/j.1365-294X.2006.02994.x. [DOI] [PubMed] [Google Scholar]
- 47.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Evanno G, Regnaut S, Goudet J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol Ecol. 2005;14:2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x. [DOI] [PubMed] [Google Scholar]
- 49.Nei M, Tajima F, Tateno Y. Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J Mol Evol. 1983;19:153–170. doi: 10.1007/BF02300753. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.