

Abstract
The Hippo pathway controls organ size and suppresses tumorigenesis in metazoans by blocking cell proliferation and promoting apoptosis. The TEAD1-4 proteins (which contain a DNA-binding domain but lack an activation domain) interact with YAP (which lacks a DNA-binding domain but contains an activation domain) to form functional heterodimeric transcription factors that activate proliferative and prosurvival gene expression programs. The Hippo pathway inhibits the YAP-TEAD hybrid transcription factors by phosphorylating and promoting cytoplasmic retention of YAP. Here we report the crystal structure of the YAP-binding domain (YBD) of human TEAD2. TEAD2 YBD adopts an immunoglobulin-like β-sandwich fold with two extra helix-turn-helix inserts. NMR studies reveal that the TEAD-binding domain of YAP is natively unfolded and that TEAD binding causes localized conformational changes in YAP. In vitro binding and in vivo functional assays define an extensive conserved surface of TEAD2 YBD as the YAP-binding site. Therefore, our studies suggest that a short segment of YAP adopts an extended conformation and forms extensive contacts with a rigid surface of TEAD. Targeting a surface-exposed pocket of TEAD might be an effective strategy to disrupt the YAP-TEAD interaction and to reduce the oncogenic potential of YAP.
Keywords: Hippo pathway, oncogene, crystallography
The Hippo signaling pathway forms a kinase cascade and controls organ size by coordinately regulating both cell proliferation and apoptosis (1–7). This pathway is best characterized in the fruit fly, Drosophila. Through genetic screens in Drosophila for mutants with defects in imaginal disk growth, several tumor suppressors were identified as the core components of this pathway, including two kinase complexes, the Ste20 family kinase Hippo (Hpo) in complex with the adaptor protein Salvador (Sav) and the nuclear Dbf2-related (NDR) family kinase Warts (Wts) bound to its activator Mats (8, 9). Upon activation by extracellular stimuli or cell-cell contact, the Hpo-Sav complex phosphorylates and activates the Wts-Mats complex. The activated Wts-Mats complex in turn phosphorylates the transcriptional coactivator Yorki (Yki) on S168 and creates a binding site for 14-3-3 proteins. Binding of 14-3-3 causes the cytoplasmic sequestration and inactivation of Yki (10–14). When the Hippo pathway is inactivated, Yki is dephosphorylated by an unknown phosphatase and translocates into the nucleus. Because Yki does not contain a DNA-binding domain, it interacts with the transcription factor Scalloped (Sd) (which contains a sequence-specific DNA-binding domain) to form a functional heterodimeric transcription factor (15–18). The Yki-Sd hybrid transcription factor activates the transcription of important target genes, such as the cell cycle gene cyclin E, the antiapoptotic gene Diap1, and the microRNA Bantam, thereby promoting cell growth and proliferation and inhibiting apoptosis (19, 20). By restraining the activities of Yki-Sd, the Hippo pathway controls organ size.
The Hippo pathway is conserved in mammals (1–7). All core components in fly have human homologs, including MST1/2 for Hpo, WW45 for Sav, LATS1/2 for Wts, and MOB1 for Mats. Expression of mammalian MST2, LATS1, and MOB1 genes in fly functionally rescues the phenotypes of the corresponding Hpo, Wts, and Mats mutants (8–12, 16, 21). Similar to the fly pathway, the effectors of the mammalian Hippo pathway are transcription factors, including YAP (Yes-associated protein), and TEAD, the mammalian homologs of Yki and Sd, respectively (16, 18, 22, 23). There are four closely related human TEAD proteins, TEAD1-4. All TEAD proteins contain an N-terminal DNA-binding TEA (TEF-1, TEC1, and AbaA) domain and a C-terminal YAP-binding domain (YBD) (Fig. 1A). YAP has an N-terminal TEAD-binding domain and a C-terminal transcriptional activation domain. Through direct physical interaction, TEAD YBD recruits YAP to promoters of target genes, where it turns on gene expression. When the Hippo pathway is activated, LATS1/2 phosphorylates YAP on S127 and promotes its association with 14-3-3 and cytoplasmic retention, thus inhibiting the activities of the hybrid transcription factors formed between YAP and TEAD1-4.
Fig. 1.

Structure of human TEAD2217–447. (A) Schematic drawing of the domain organization for human YAP and TEAD2 proteins. The residue numbers for different domain boundaries are labeled. TBD: TEAD-binding domain; AD: transcriptional activation domain; TEA: DNA-binding TEA domain; YBD: YAP-binding domain. (B) Ribbon diagram of TEAD2217–447 in two views. Strands are colored blue, helices are colored magenta, and loops are colored gray. The N/C termini and secondary structure elements are labeled. (C Left) Ribbon diagram of the PDEδ-Arl2 complex. The PDEδ molecule is colored in wheat and the Arl2 molecule is colored in green. (C Right ) Superposition of PDEδ and TEAD2217–447. All structural figures were generated with PyMOL.
The Hippo pathway has been implicated in human cancers. In particular, several lines of evidence indicate YAP as an oncogene. The YAP gene is amplified in several human cancers. The YAP protein is frequently overexpressed in human cancers (12, 24–26). Overexpression of YAP in nontransformed human MCF10A cells induces the epithelial-mesenchymal transition, a hallmark of tumorigenic transformation (24). Overexpression of YAP in mouse liver causes dramatic liver overgrowth and eventually tumor formation (11, 27). The TEAD proteins are major partners of YAP and collaborate with it to regulate the expression of genes that promote cell proliferation and inhibit apoptosis. Thus, understanding how TEAD interacts with YAP will provide insights into how the Hippo pathway regulates the YAP-TEAD transcription factors and may ultimately lead to strategies that can disrupt the tumor-promoting YAP-TEAD interactions.
Toward this goal, we have determined the crystal structure of the YAP-binding domain of human TEAD2. We have further analyzed the interactions between TEAD and YAP using in vitro binding assays, in vivo luciferase assays, and NMR spectroscopy. Our studies pinpoint a surface-exposed pocket of TEAD YBD that is critical for YAP binding. Targeting this pocket chemically might be an effective strategy to disrupt the YAP-TEAD interactions and to attenuate the function of YAP.
Results and Discussion
Structure Determination of the YAP-Binding Domain of TEAD.
The structure of the TEA DNA-binding domain of TEAD proteins has been determined previously (28). The structure of the YBD of TEAD is unknown, however. We have determined the structure of human TEAD2 YBD containing residues 217–447 by the single anomalous dispersion (SAD) method from crystals of the seleno-methionine-labeled TEAD2217–447 protein (Table 1). TEAD2 YBD adopts an immunoglobulin (IgG)-like fold with two β-sheets packing against each other to form a β-sandwich (Fig. 1B). One β-sheet contains five antiparallel strands, including β1, β2, β5, β8, and β9, while the other contains seven parallel and antiparallel strands, including β3, β4, β6, β7, and β10–12. In addition to the two main β-sheets, TEAD2 YBD contains two helix-turn-helix motifs that are absent in the IgG-like fold. One helix-turn-helix motif consists of αA and αB and connects β3 and β4. This motif along with the β2-β3 loop encircles the C-terminal β12 strand, forming an unusual pseudoknot structure. The second helix-turn-helix motif consists of αC and αD and connects β9 and β10. This motif caps the opening at one end of the β-sandwich.
Table 1.
Data collection, structure determination, and refinement
| Data collection | |
| Crystal | SeMet (peak)* |
| Space group | C2 |
| Energy (eV) | 12,655.6 |
| Resolution range (Å) | 50.0–2.00 (2.03–2.00) |
| Unique reflections | 34,385 (1,250) |
| Multiplicity | 4.9 (3.8) |
| Data completeness (%) | 96.1 (70.9) |
| Rmerge (%)† | 10.1 (71.1) |
| I/σ(I) | 10.5 (1.8) |
| Wilson B value, Å2 | 31.9 |
| Phase determination | |
| Anomalous scatterer | Selenium (8 of 8 possible sites) |
| Figure of merit, 50–2.00 Å | 0.36 |
| Refinement statistics | |
| Resolution range, Å | 33.74–2.00 (2.05–2.00) |
| No. of reflections Rwork/Rfree | 32,458/1,878 (1,571/88) |
| Data completeness, % | 90.6 (60.0) |
| Atoms (non-H protein/solvent/other) | 3,277/160/18 |
| Rwork (%) | 18.8 (29.2) |
| Rfree (%) | 24.1 (34.3) |
| rmsd bond length (Å) | 0.01 |
| rmsd bond angle (°) | 1.05 |
| Mean B value (Å2) (protein/solvent/other) | 47.9/54.8/69.8 |
| Ramachandran plot (%) (favored/additional/disallowed)‡ | 97.9/2.1/0.0 |
| Maximum likelihood coordinate error | 1.75 |
| Missing residues | Chain A: 217–221, 240–247, 257–265, 309–324, 447; Chain B: 217–220, 257–262, 309–324, 447. |
Data for the outermost shell are given in parentheses.
*Bijvoet pairs were kept separate for data processing.
†Rmerge = 100 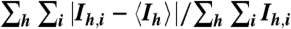 , where the outer sum (h) is over the unique reflections and the inner sum (i) is over the set of independent observations of each unique reflection.
, where the outer sum (h) is over the unique reflections and the inner sum (i) is over the set of independent observations of each unique reflection.
‡As defined by the validation suite MolProbity.
Comparison between TEAD2 YBD and known structures in the Protein Data Bank (PDB) using the Dali server reveals that TEAD2 YBD is structurally most closely related to phosphodiesterase δ (PDEδ) (PDB ID code 1KSHB) with a Z score of 10.1 and a rmsd of 2.5 Å. Superposition of the TEAD2 YBD and PDEδ structures reveals that the main β-sheets of the two molecules overlay well (Fig. 1C). Their major difference is that PDEδ has an N-terminal α-helix, whereas TEAD2 YBD has two helix-turn-helix inserts. Interestingly, PDEδ binds to the small GTPase Arf-like 2 (Arl2) through an edge-on β-β interaction, with one of its edge strands paring with the edge strand in Arl2 (Fig. 1C) (29). Based on the structural similarity, TEAD2 YBD might also use its edge strands to interact with YAP or other yet unidentified binding partners.
The TEAD-Binding Domain (TBD) of YAP Is Natively Unfolded.
We next studied the interaction between TEAD2 YBD and YAP using NMR. Previous studies have mapped the TBD of YAP to a small N-terminal region containing residues 48–102 (Fig. 1A) (30, 31). We expressed a large fragment of YAP (residues 2–268) containing the TBD and the two WW domains, labeled it with 15N, and acquired a 2D 1H/15N heteronuclear single quantum correlation (HSQC) spectrum (Fig. 2A). The HSQC spectrum of YAP2–268 contains two sets of peaks. One set of peaks is well dispersed and has intensities comparable to the peaks of several tryptophan side chains. These peaks presumably belong to the two WW domains of YAP. The other set of peaks is strong in intensity and clusters around 7.8–8.5 ppm in the 1H dimension. These peaks belong to residues with flexible random coil conformations. Therefore, a segment of YAP2–268 is natively unfolded. We next prepared 15N-labeled YAP2–268 in complex with TEAD2 YBD and acquired an HSQC spectrum on it. An overlay of the HSQC spectra of free YAP2–268 and the YAP2–268-TEAD2 YBD complex reveals that the signals belonging to the WW domains of YAP remain largely unaffected by TEAD2 YBD binding (Fig. 2A). This indicates that the WW domains of YAP are not involved in TEAD binding. By contrast, about 30 sharp and strong signals of YAP2–268 either disappear or become much weaker upon TEAD binding. These residues likely adopt ordered conformation when bound to TEAD2 YBD. Therefore, TEAD binding induces localized conformational changes of YAP. A small segment of YAP is directly involved in TEAD binding.
Fig. 2.

Interactions between YAP and TEAD. (A) Superposition of the 15N/1H-HSQC spectra of YAP and the YAP-TEAD complex, whose cross-peaks are in black and red, respectively. The free YAP cross-peaks that disappear upon TEAD binding are indicated by arrows. (B) ITC analysis of the binding between the YAP61–100 peptide and TEAD2217–447. (C) ITC analysis of the binding between YAP2–268 and TEAD2217–447. The dissociation constant (Kd) and other related parameters of binding are indicated.
We thus synthesized two peptides within the TBD of YAP: One peptide contains residues 61–100 of YAP while the other contains residues 67–97 and measured their binding affinity to TEAD2 YBD using isothermal titration calorimetry (ITC) (Fig. 2B). Consistent with our NMR data, the YAP61–100 peptide bound to TEAD2 YBD with a dissociation constant (Kd) of 96 nM and a stoichiometry of 0.94. Therefore, a short 40mer YAP peptide is sufficient to bind TEAD2 YBD with relatively high affinity to form a 1∶1 complex. On the other hand, we could not detect appreciable binding between TEAD2 YBD and the smaller YAP67–97 peptide with ITC. Furthermore, the ITC experiment also revealed that YAP2–268 used in the NMR studies binds to TEAD2 YBD with a dissociation constant (Kd = 33 nM) and a stoichiometry (N = 0.97) similar to those of the YAP61–100 peptide (Fig. 2C). Thus, residues 61–100 represent the essential TEAD-binding domain of YAP.
Mapping the YAP-Binding Site of TEAD2 YBD.
Because we failed to obtain diffracting crystals of TEAD2 YBD bound to various YAP fragments containing the minimal TBD as defined above, we used structure-based mutagenesis to map the YAP-binding site on TEAD2 YBD. We first aligned the sequences of TEAD YBD from various species ranging from Caenorhabditis elegans to human and displayed the side chains of surface-exposed residues that are identical in TEAD YBD from all species on the ribbon diagram (Fig. 3). These conserved residues form a contiguous surface on one face of TEAD2 YBD that contains β7, αC, αD, and the back of stands β4, β11, and β12. The other face of TEAD2 YBD contains few conserved residues. A conserved tyrosine (Y442 in TEAD2 YBD) in human TEAD1 is mutated to histidine in patients with a rare eye disorder called Sveinsson’s chorioretinal atrophy (32). This mutation has been shown previously to disrupt the YAP-TEAD interaction (33). Intriguingly, Y442 is surface exposed and is located at the back of strand β12 (Fig. 3 B and C). Therefore, this conserved surface of TEAD2 YBD might be involved in YAP binding.
Fig. 3.

A conserved surface on TEAD. (A) Multiple sequence alignment of TEAD YBD from different species. The secondary structural elements of TEAD2 YBD are drawn above the sequences and colored the same as in the ribbon diagram. Residues identical in all sequences are shaded yellow. Surface-exposed residues in human TEAD2 subjected to mutagenesis in this study (see below) are labeled with asterisks. (B Left) Ribbon diagram of TEAD2 YBD with conserved identical residues shown as yellow sticks. (B Right) Molecular surface of TEAD2 YBD in the same orientation. (C) Same as in B but showing the TEAD molecule in a view that is related to the view in B by a 180° rotation along the y axis. Y442 in TEAD2, which corresponds to the conserved tyrosine in TEAD1 mutated in Sveinsson’s choriorentinal atrophy, is labeled in red.
We next systematically mutated these surface-exposed conserved residues of TEAD2 YBD in the context of full-length TEAD2 and tested the binding between these mutants and YAP2–268. To do so, we expressed and purified GST-YAP2–268, immobilized this fusion protein on glutathione-agarose beads, and incubated these beads with in vitro translated 35S-labeled TEAD2 mutants. After washing, the proteins bound to beads were resolved on SDS-PAGE and visualized using a phosphoimager (Fig. 4 A and B). As expected, wild-type (WT) TEAD2 bound specifically to GST-YAP2–268, but not to the GST control. Consistent with previous reports, the TEAD2 Y442H mutation abolished YAP binding. Mutations of the majority of these conserved residues weakened the binding of YAP to TEAD2, confirming the involvement of this conserved surface in YAP binding.
Fig. 4.

The YAP-binding surface of TEAD. (A) 35S-labeled TEAD2 WT or its mutants were incubated with beads bound to GST or GST-YAP2–268. After washing, proteins bound to beads were separated on SDS-PAGE and analyzed using a phosphoimager. Twelve percent of each protein used in the assay was included as input for comparison. (B) Binding of different TEAD2 proteins toward YAP tested in A was quantified and normalized against the wild-type TEAD2. (C) Ribbon diagram of TEAD2 YBD with residues that when mutated abolished YAP binding shown as yellow sticks. (D) The electrostatic surface of TEAD2 YBD in the same orientation as in C. The critical YAP-binding pocket is indicated by a dashed circle.
A total of 10 mutant constructs (including Y442H) reduced YAP binding to below 10% of that of the wild-type TEAD2. Nine out of these 10 mutant constructs affected residues that formed a surface-exposed pocket, including E267, I274, K277, L299/K301, W303, E404/N405, V427/E429, Y442, and L444 (Fig. 4C). Strands β4, β11, and β12 form the base of this pocket, whereas β3, αA, and αD form the walls. The residues in the center of the pocket are hydrophobic, whereas many residues lining the periphery of the pocket are polar or charged, including E267, K277, K301, E404, N405, and E429 (Fig. 4D). Therefore, despite having an extensive YAP-binding surface, this pocket on TEAD2 YBD represents a major anchoring point for YAP and contributes a large fraction of their binding energy. Strand β7 and the surface formed by αC and αD contribute the rest of the binding surface and further strengthen YAP binding.
As discussed above, YAP is an oncogene and is frequently overexpressed in human cancers. Inhibiting YAP might be an effective way to block the proliferation and to induce apoptosis in cancer cells. TEAD proteins are the major partners of YAP in transcriptional activation. Disrupting the YAP-TEAD interaction is expected to significantly weaken the transforming function of YAP. YAP binding to TEAD requires a surface pocket on TEAD that is formed at the back of the seven-stranded β-sheet and lined with αA and αD. This pocket contains both hydrophobic and charged residues, making it feasible to design or to screen for chemical compounds that selectively bind to this site. This pocket is highly conserved in TEAD1-4. These compounds might be able to disrupt the YAP-TEAD1-4 interactions and have therapeutic potential.
The YAP-TEAD Interaction Is Required for Transcriptional Activation.
To further confirm the validity of the YAP-binding site on TEAD2 in cells, we constructed plasmids encoding the wild-type or mutant TEAD2 proteins fused to the Gal4 DNA-binding domain at their N termini and transfected 293 cells with these plasmids together with a Gal4-driven luciferase reporter plasmid in the presence or absence of YAP (Fig. 5). Gal4 DNA-binding domain or Gal4-TEAD2 WT alone did not activate the expression of the luciferase reporter. Cotransfection of YAP and Gal4-TEAD2 WT greatly increased the luciferase activity. By contrast, cotransfection of YAP and the Gal4-TEAD2 L299A/K301E, V427A/E429K, or Y442H mutants that lost their ability to bind YAP failed to activate the expression of the luciferase reporter. As a control, the Gal4-TEAD2 S349A/F350A mutant that retained YAP binding activated luciferase expression as did Gal4-TEAD2 WT. Therefore, these results indicate that the YAP-TEAD interaction is required for its ability to activate transcription. The YAP-binding site on TEAD2 YBD mapped by in vitro binding assays is also critical for YAP function in living cells.
Fig. 5.

The YAP-TEAD interaction is required for gene expression. (A) Schematic drawing of the experimental design. The Gal4-TEAD fusion protein recruits YAP to the UAS elements at the promoter of the luciferase reporter and turns on its expression. (B) Two hundred ninety-three cells were transfected with the indicated plasmids along with the Gal4-responsive luciferase reporter. Luciferase activity of Gal4-TEAD2 WT or Gal4-TEAD2 mutants with or without HA-YAP relative to that of Gal4 DBD alone was plotted. Experiments were performed in triplicates. Error bars represent standard deviations. (C) Cell lysates from B were blotted with anti-Gal4 and anti-HA antibodies to show the expression levels of the Gal4-TEAD fusion proteins and HA-YAP, respectively.
Conclusions
The Hippo pathway controls tissue homeostasis in multicellular organisms by restricting cell proliferation and promoting cell death. Malfunction of the Hippo pathway results in hyperplasia and cancer. One important output of this pathway is the inhibition of the YAP transcription coactivator, which involves the phosphorylation and cytoplasmic retention of YAP and the disruption of the binding of YAP to its major cellular partners, the TEAD proteins. In this paper, we have determined the structure of the YAP-binding domain of human TEAD2 and studied the YAP-TEAD interactions with a combination of biophysical, biochemical, and cellular assays. Our results suggest that a short natively unfolded segment of YAP binds to an extensive surface on TEAD and adopts an ordered conformation. Despite having an extensive interface, the YAP-TEAD interaction requires a surface pocket that has both hydrophobic and hydrophilic characters. Chemical compounds that selectively bind to this pocket may disrupt the YAP-TEAD interactions and may have therapeutic potential.
Materials and Methods
Plasmids, Protein Expression, and Purification.
The coding region of human TEAD2217–447 was amplified by PCR and cloned into a modified pET28 vector (EMD Biosciences) that also included a tobacco etch virus (TEV) protease cleavage site. The TEAD2 mutants were generated with the QuikChange mutagenesis kit (Stratagene). All constructs were verified by DNA sequencing. The pET28-TEAD2217–447 plasmid was transformed into the Escherichia coli strain BL21(DE3) to produce N-terminal His6-tagged TEAD2217–447 protein. TEAD2217–447 was purified with Ni2+-NTA agarose resin (Qiagen) and cleaved with TEV protease to remove the His6 tag. The protein was further purified by anion exchange chromatography followed by size exclusion chromatography (GE Healthcare). Purified TEAD2217–447 was concentrated to 12 mg/mL in a buffer containing 20 mM Tris (pH 8.0), 100 mM NaCl, 2 mM MgCl2, 1 mM Tris(2-carboxyethyl)phosphine (TCEP), and 5% glycerol. The seleno-methionine labeled TEAD2217–447 was produced using the methionine biosynthesis inhibition method (34).
To generate the pcDNAGal4DBD construct, the cDNA encoding the Gal4 DNA-binding domain (DBD) (1–147) was cloned into the pcDNA 3.1 vector. The wild-type or mutant TEAD2 sequences were inserted into the pcDNAGal4DBD vector in frame with Gal4DBD to generate Gal4-TEAD2 WT or Gal4-TEAD2 mutant constructs.
Crystallization, Data Collection and Structure Determination.
TEAD2217–447 was crystallized at 20 °C using the hanging-drop vapor-diffusion method with a reservoir solution containing 0.1 M Hepes (pH 7.4) and 2.8 M sodium formate. The crystals were cryoprotected with reservoir solution supplemented with 25% glycerol and then flash-cooled in liquid nitrogen. Crystals diffracted to a minimum Bragg spacing (dmin) of about 2.0 Å and exhibited the symmetry of space group C2 with cell dimensions of a = 121.1 Å, b = 61.6 Å, c = 80.5 Å, and β = 117.3° and contained two molecules per asymmetric unit.
Diffraction data were collected at beamline 19-BM (SBC-CAT) at the Advanced Photon Source (Argonne National Laboratory) and processed with HKL3000 (35). Phases were obtained from a selenium-SAD experiment using X-rays at an energy near the selenium K absorption edge. Phenix AutoSol was used to identify the selenium sites and calculate density modified experimental maps (36). A total of eight refined sites were found, and the experimental density map showed clear features of the protein backbone and well defined side chains. Automated building with Phenix AutoBuild resulted in a model containing 366 sequence assigned residues when refined against the experimental phases. The remaining 31 residues were manually built in COOT and refined in Phenix (37). The final model (Rwork = 18.8%, Rfree = 24.1%) contains 397 residues, 160 water molecules, and 3 glycerol molecules. MolProbity was used for structure validation and indicates all residues are in the Ramachandran favored/allowed regions (38). Data collection and structure refinement statistics are summarized in Table 1.
Isothermal Titration Calorimetry (ITC).
ITC was performed with a VP-ITC titration calorimeter (MicroCal Inc.) at 20 °C. Calorimetric measurements were carried out with purified TEAD217–447 and YAP2–268 plus two synthetic peptides corresponding to residues 61–100 (SETDLEALFNAVMNPKTANVPQTVPMRLRKLPDSFFKPPE) and 67–97 (ALFNAVMNPKTANVPQTVPMRLRKLPDSFFK) of human YAP. For each titration experiment, 2 mL of 6 μM TEAD protein in a buffer containing 20 mM Tris (pH 8.0), 100 mM KCl, 2 mM MgCl2, and 0.5 mM TCEP were added to the calorimeter cell. The YAP peptides (0.05–0.1 mM) in the same buffer were injected with 35 portions of 8 μL with an injection syringe. Binding parameters were evaluated using the Origin software package provided with the instrument.
NMR Spectroscopy.
All NMR spectra were acquired at 30 °C on a Varian Unity Inova 800-MHz spectrometer using H2O/D2O 95∶5 (vol/vol) as the solvent. Samples typically contained 0.1 mM 15N-labeled protein in the NMR buffer consisting of 20 mM sodium phosphate (pH 6.8), 100 mM KCl, 2 mM MgCl2, and 1 mM DTT.
In Vitro Protein Binding Assays.
To assay the binding between human TEAD2 and YAP proteins, the full-length WT or mutant TEAD2 was translated in reticulocyte lysate in the presence of 35S-methionine. Purified GST-YAP2–268 was bound to glutathione-agarose beads (GE Healthcare), incubated with 35S-labeled TEAD proteins, and washed three times with Tris buffered saline containing 0.05% Tween. The proteins retained on the beads were analyzed by SDS-PAGE followed by autoradiography. Glutathione-agarose beads bound to GST were used as controls.
Luciferase Assay.
For the luciferase assay, the Gal4-TEAD2 WT or Gal4-TEAD2 mutant plasmids were transfected in triplicates in 293 cells along with a luciferase reporter plasmid with or without YAP. The luciferase assay was performed using the Dual Luciferase Assay System (Promega) and a FLUOstar Luminometer (BMG Lab Technologies).
Acknowledgments.
We thank Xuewu Zhang for help in data collection and Hongtao Yu for discussion and critical reading of the manuscript. Results shown in this paper are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by University of Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under Contract DE-AC02-06CH11357. This work was supported in part by National Institutes of Health Grant GM085004 (to X.L.) and Grant EY015708 (to D.P.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3L15).
References
- 1.Edgar BA. From cell structure to transcription: Hippo forges a new path. Cell. 2006;124(2):267–273. doi: 10.1016/j.cell.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 2.Pan D. Hippo signaling in organ size control. Genes Dev. 2007;21(8):886–897. doi: 10.1101/gad.1536007. [DOI] [PubMed] [Google Scholar]
- 3.Harvey K, Tapon N. The Salvador-Warts-Hippo pathway—an emerging tumour-suppressor network. Nat Rev Cancer. 2007;7(3):182–191. doi: 10.1038/nrc2070. [DOI] [PubMed] [Google Scholar]
- 4.Zeng Q, Hong W. The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer Cell. 2008;13(3):188–192. doi: 10.1016/j.ccr.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Zhang L, Yue T, Jiang J. Hippo signaling pathway and organ size control. Fly (Austin) 2009;3(1):68–73. doi: 10.4161/fly.3.1.7788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao B, Lei QY, Guan KL. The Hippo-YAP pathway: New connections between regulation of organ size and cancer. Curr Opin Cell Biol. 2008;20(6):638–646. doi: 10.1016/j.ceb.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reddy BV, Irvine KD. The Fat and Warts signaling pathways: New insights into their regulation, mechanism and conservation. Development. 2008;135(17):2827–2838. doi: 10.1242/dev.020974. [DOI] [PubMed] [Google Scholar]
- 8.Wu S, Huang J, Dong J, Pan D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell. 2003;114(4):445–456. doi: 10.1016/s0092-8674(03)00549-x. [DOI] [PubMed] [Google Scholar]
- 9.Lai ZC, et al. Control of cell proliferation and apoptosis by mob as tumor suppressor, mats. Cell. 2005;120(5):675–685. doi: 10.1016/j.cell.2004.12.036. [DOI] [PubMed] [Google Scholar]
- 10.Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122(3):421–434. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 11.Dong J, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130(6):1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao B, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21(21):2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oh H, Irvine KD. In vivo regulation of Yorkie phosphorylation and localization. Development. 2008;135(6):1081–1088. doi: 10.1242/dev.015255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ren F, Zhang L, Jiang J. Hippo signaling regulates Yorkie nuclear localization and activity through 14-3-3 dependent and independent mechanisms. Dev Biol. 2010;337:303–312. doi: 10.1016/j.ydbio.2009.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goulev Y, et al. SCALLOPED interacts with YORKIE, the nuclear effector of the hippo tumor-suppressor pathway in Drosophila. Curr Biol. 2008;18(6):435–441. doi: 10.1016/j.cub.2008.02.034. [DOI] [PubMed] [Google Scholar]
- 16.Wu S, Liu Y, Zheng Y, Dong J, Pan D. The TEAD/TEF family protein Scalloped mediates transcriptional output of the Hippo growth-regulatory pathway. Dev Cell. 2008;14(3):388–398. doi: 10.1016/j.devcel.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 17.Zhang L, et al. The TEAD/TEF family of transcription factor Scalloped mediates Hippo signaling in organ size control. Dev Cell. 2008;14(3):377–387. doi: 10.1016/j.devcel.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao B, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22(14):1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson BJ, Cohen SM. The Hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell. 2006;126(4):767–774. doi: 10.1016/j.cell.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 20.Nolo R, Morrison CM, Tao C, Zhang X, Halder G. The bantam microRNA is a target of the hippo tumor-suppressor pathway. Curr Biol. 2006;16(19):1895–1904. doi: 10.1016/j.cub.2006.08.057. [DOI] [PubMed] [Google Scholar]
- 21.Tao W, et al. Human homologue of the Drosophila melanogaster lats tumour suppressor modulates CDC2 activity. Nat Genet. 1999;21(2):177–181. doi: 10.1038/5960. [DOI] [PubMed] [Google Scholar]
- 22.Ota M, Sasaki H. Mammalian Tead proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development. 2008;135(24):4059–4069. doi: 10.1242/dev.027151. [DOI] [PubMed] [Google Scholar]
- 23.Cao X, Pfaff SL, Gage FH. YAP regulates neural progenitor cell number via the TEA domain transcription factor. Genes Dev. 2008;22(23):3320–3334. doi: 10.1101/gad.1726608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Overholtzer M, et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci USA. 2006;103(33):12405–12410. doi: 10.1073/pnas.0605579103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zender L, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell. 2006;125(7):1253–1267. doi: 10.1016/j.cell.2006.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steinhardt AA, et al. Expression of Yes-associated protein in common solid tumors. Hum Pathol. 2008;39(11):1582–1589. doi: 10.1016/j.humpath.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Camargo FD, et al. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17(23):2054–2060. doi: 10.1016/j.cub.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 28.Anbanandam A, et al. Insights into transcription enhancer factor 1 (TEF-1) activity from the solution structure of the TEA domain. Proc Natl Acad Sci USA. 2006;103(46):17225–17230. doi: 10.1073/pnas.0607171103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hanzal-Bayer M, Renault L, Roversi P, Wittinghofer A, Hillig RC. The complex of Arl2-GTP and PDE delta: From structure to function. EMBO J. 2002;21(9):2095–2106. doi: 10.1093/emboj/21.9.2095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chan SW, et al. TEADs mediate nuclear retention of TAZ to promote oncogenic transformation. J Biol Chem. 2009;284(21):14347–14358. doi: 10.1074/jbc.M901568200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 2001;15(10):1229–1241. doi: 10.1101/gad.888601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fossdal R, et al. A novel TEAD1 mutation is the causative allele in Sveinsson’s chorioretinal atrophy (helicoid peripapillary chorioretinal degeneration) Hum Mol Genet. 2004;13(9):975–981. doi: 10.1093/hmg/ddh106. [DOI] [PubMed] [Google Scholar]
- 33.Kitagawa M. A Sveinsson’s chorioretinal atrophy-associated missense mutation in mouse Tead1 affects its interaction with the co-factors YAP and TAZ. Biochem Biophys Res Commun. 2007;361(4):1022–1026. doi: 10.1016/j.bbrc.2007.07.129. [DOI] [PubMed] [Google Scholar]
- 34.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229(1):105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- 35.Otwinowski Z, Minor W. Processing x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 36.Adams PD, et al. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr D. 2002;58(Pt 11):1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 37.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D. 2004;60(Pt 12):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 38.Davis IW, et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]