

Abstract
The relative importance of protein function change and gene expression change in phenotypic evolution is a contentious, yet central topic in evolutionary biology. Analyzing 5,199 mouse genes with recorded mutant phenotypes, we find that genes exclusively affecting morphological traits when mutated (dubbed “morphogenes”) are grossly enriched with transcriptional regulators, whereas those exclusively affecting physiological traits (dubbed “physiogenes”) are enriched with channels, transporters, receptors, and enzymes. Compared to physiogenes, morphogenes are more likely to be essential and pleiotropic and less likely to be tissue specific. Morphogenes evolve faster in expression profile, but slower in protein sequence and gene gain/loss than physiogenes. Thus, morphological and physiological changes have a differential molecular basis; separating them helps discern the genetic mechanisms of phenotypic evolution.
Keywords: evolutionary rate, gene expression, molecular evolution, phenotypic evolution
Nearly 35 years ago, King and Wilson remarked that, despite the large phenotypic difference, human and chimpanzee have virtually identical protein sequences, which prompted their proposal that gene expression change plays a more important role than protein function change in phenotypic evolution, including human origins (1). We now know that, between these two species, there are on average ∼2 amino acid differences per protein and >70% of their proteins are nonidentical (2, 3). Thus, contrary to King and Wilson’s assertion, protein sequence differences between human and chimpanzee are numerous, which potentially can account for many, if not all, of the phenotypic differences between the two species. Nonetheless, the role of gene expression change in phenotypic evolution has been documented in numerous case studies (4–6). But it remains unclear whether gene expression change is generally more important than protein function change (6–8). To address this question, two groups recently compiled cases of phenotypic evolution with known genetic mechanisms, but reached different conclusions (7, 8). Although these metaanalyses offer summaries of case studies, they may provide distorted pictures due to ascertainment bias caused by preferences for certain methods, phenotypes, genes, and types of mutations in research.
We here take a genomic approach to this question by identification of genes that affect classes of phenotypes when mutated, followed by an analysis of the properties and evolutionary patterns of these genes. We are particularly interested in examining whether a distinction exists in the genetic basis of morphological and physiological evolution, which was previously proposed on the basis of case studies and theoretical considerations (9). More specifically, by comparing the evolutionary rates of protein sequences, expression profiles, and cis-regulatory sequences between genes controlling morphological traits and those controlling physiological traits, we test the hypothesis that morphological evolution tends to involve gene expression changes caused by cis-regulatory mutations, whereas physiological evolution tends to occur by protein sequence changes or gene duplication/loss (9).
Results
Morphogenes and Physiogenes Have Distinct Molecular Functions.
We use the mouse Mus musculus as our focal organism because of the availability of its genome and transcriptome data as well as those of related species, presence of numerous well-characterized morphological and physiological traits (Table S1), and, most importantly, extensive documentation of its mutant phenotypes. At the time of our study, there were 5,199 mouse genes with recorded mutant phenotypes in the Mouse Genome Informatics (MGI) database, of which 821 affected only morphological traits and 912 affected only physiological traits (Materials and Methods). These genes are referred to as “morphogenes” and “physiogenes,” respectively (Table S2).
By definition, morphogenes and physiogenes differ in certain biological processes they participate in, such as “anatomical structure development” and “immune response” (Table S3). However, it is interesting to note that morphogenes are much more frequently associated with the biological process of “transcription” than physiogenes (P < E-29 after correction for multiple testing), although this is not expected a priori. In addition, the molecular function of “transcriptional regulator activity” is grossly overrepresented among morphogenes, whereas those of “ion transporter activity,” “channel or pore class transporter activity,” “receptor activity,” and “catalytic activity” are enriched among physiogenes (Fig. 1). Not unexpectedly, “structural molecule activity” is also much more prevalent among morphogenes than among physiogenes (Fig. 1). Table S3 lists all Gene Ontology (GO) categories identified by FatiGO (10) to be significantly differentially distributed among morphogenes and physiogenes.
Fig. 1.

Major differences in molecular function between mouse morphogenes and physiogenes. Shown here are the fractions of morphogenes and physiogenes belonging to selected large functional categories at Gene Ontology (GO) level 2 and level 3. P values have been corrected for multiple testing in FatiGO (http://www.fatigo.org/). The complete list of significant GO differences between morphogenes and physiogenes is in Table S3.
A gene is regarded as essential if its loss leads to infertility or death before puberty (11). We found that morphogenes are more likely to be essential than physiogenes (P = 6.00E-28, χ2 test, Fig. 2A). This difference cannot be entirely caused by potential underrecognition of physiological defects in prenatal/perinatal deaths, because even after removing genes with the phenotype of prenatal/perinatal lethality, the fraction of essential genes is still significantly higher among morphogenes (14.8%) than among physiogenes (7.5%) (P = 5.9E-6, χ2 test). We measure the pleiotropic level of a gene by the number of mutant phenotypes associated with the gene. Morphogenes are more pleiotropic than physiogenes (P = 4.70E-12, Mann–Whitney U test, Fig. 2B). This result is conservative, because the total number of morphological traits (129) considered in our analysis is smaller than that of physiological traits (183). Consistent with the pleiotropy difference, morphogenes are less tissue specific than physiogenes (P = 9.97E-5, U test, Fig. 2C), where tissue specificity is measured by the heterogeneity in gene expression level among tissues (Materials and Methods). Morphogenes are significantly more pleiotropic (P = 1.64E-4, U test, Fig. 2B) and less tissue specific (P = 8.32E-4, U test, Fig. 2C) than physiogenes even when only nonessential genes are considered. Because many proteins function by interacting with other proteins, we used human protein interaction data to infer the number (k) of protein interaction partners of each mouse gene (Materials and Methods). No significant difference in k is observed between morphogenes and physiogenes, regardless of whether all genes (P = 0.095, U test, Fig. 2D) or only nonessential genes are compared (P = 0.35, U test, Fig. 2D). We also examined the number of exons and the number of alternative splice forms of morphogenes and physiogenes, but did not find significant differences (P = 0.07 and 0.51, respectively).
Fig. 2.

Mouse morphogenes and physiogenes are significantly different in (A) the proportion of essential genes, (B) pleiotropy, and (C) tissue specificity in expression, but not in (D) the number of protein–protein interaction partners. The numbers in A indicate the number of genes. For B–D, the values of upper quartile, median, and lower quartile are indicated in each box, whereas the bars outside the box indicate semiquartile ranges. P values are from a χ2 test for A (P < E-27) and from a Mann–Whitney U test for B–D. Morpho, morphogenes; noness, nonessential; physio, physiogenes.
Physiogenes Evolve Faster Than Morphogenes in Protein Sequence.
After examining the basic properties of mouse morphogenes and physiogenes, we compared their rates of evolution. The rate of protein sequence evolution can be measured by the number of nonsynonymous substitutions per nonsynonymous site (dN) between mouse and human orthologs. Because mutation rate varies across the genome, a better measure of protein evolutionary rate is dN/dS, where dS is the number of synonymous substitutions per synonymous site between the orthologs and reflects the local mutation rate. We found the mean dN to be >50% greater for physiogenes than for morphogenes (P = 1.01E-14, U test, Fig. 3A). This difference cannot be explained by a higher mutation rate of physiogenes, because the mean dS differs only by ∼11% (P = 8.33E-10, U test, Fig. 3B). The significantly greater dN/dS of physiogenes (P = 1.60E-10, U test, Fig. 3C) indicates that their protein sequences experience lower purifying selection and/or stronger positive selection than those of morphogenes.
Fig. 3.

Evolutionary divergences between mouse and human orthologous genes measured by (A) nonsynonymous distance (dN), (B) synonymous distance (dS), (C) dN/dS, (D) dN controlled for tissue specificity, (E) expression-profile distance from GeneAtlas data, (F) expression-profile distance from ExonArrays data, and (G) expression-profile distance from GeneAtlas data controlled for tissue specificity. Error bars show one standard error of the mean. P values are from a Mann–Whitney U test. Morpho, morphogenes; noness, nonessential; physio, physiogenes.
The rate of protein sequence evolution is known to be correlated with several gene properties that show differences between morphogenes and physiogenes. First, mammalian nonessential genes have greater dN and dN/dS than essential genes (12). However, dN and dN/dS are still significantly greater for physiogenes than for morphogenes when only nonessential genes are considered (Fig. 3 A–C). Second, in yeast, dN is negatively correlated with the level of gene pleiotropy (13). In the present case, although there is a weak negative correlation between dN and pleiotropy (Pearson's r = −0.0834, P = 1.09E-3; Spearman's ρ = −0.0918, P = 3.25E-4), dN is still higher in physiogenes than in morphogenes after the control of pleiotropy (P = 1.65E-14 for all genes, P = 1.94E-10 for nonessential genes, partial rank correlation test). Third, mammalian tissue-specific genes are known to have greater dN than non-tissue-specific genes (14, 15). However, physiogenes have significantly greater dN than morphogenes even when tissue specificity is controlled for (Fig. 3D). Finally, even for genes of the same GO categories, physiogenes generally show significantly higher dN and dN/dS than morphogenes (Table S4). These results suggest that the difference in protein evolutionary rate between physiogenes and morphogenes is not entirely attributable to their differences in gene essentiality, pleiotropy, tissue specificity, and distribution among GO categories.
Morphogenes Evolve Faster Than Physiogenes in Expression.
We next investigate gene expression evolution using two independent datasets of human and mouse genomewide expression profiles in multiple tissues: GeneAtlas (16) and ExonArray (17). Expression-profile divergence between orthologs is measured by one minus Pearson’s correlation coefficient between the expression profiles in the two species (Materials and Methods). Both datasets show higher rates of expression divergence for morphogenes than for physiogenes (GeneAtlas, P = 1.57E-3, U test, Fig. 3E; ExonArray, P = 6.87E-4, U test, Fig. 3F). Comparing morphogenes and physiogenes of the same GO categories gives similar results (Table S4). Because essential genes are evolutionarily more constrained in gene expression (18), the difference in gene essentiality is unlikely to explain the faster expression evolution of morphogenes than physiogenes. Tissue-specific genes are more conserved than non-tissue-specific genes in expression profile (14). We found that even after the control for tissue specificity, morphogenes still have higher expression divergence than physiogenes, although the statistical support is weakened (Fig. 3G), suggesting that the faster expression evolution of morphogenes is in part related to their lower tissue specificity. Morphological evolution is believed to be caused by gene expression changes more than by protein function changes because protein function changes of morphogenes are thought to have pleiotropic effects more often than expression changes of the same genes and thus are more likely to be deleterious (4). This hypothesis implies that the difference in pleiotropy underlies the difference in the rate of expression evolution between morphogenes and physiogenes. Interestingly, controlling pleiotropy does not substantially reduce the difference in the rate of expression-profile evolution (all genes, P = 1.67E-3 and 6.43E-3 for before and after the control, respectively; nonessential genes, P = 2.40E-2 and 2.52E-2 for before and after the control, respectively), suggesting that pleiotropy contributes to this difference minimally.
Morphogenes Do Not Evolve Faster Than Physiogenes in Cis-Regulatory Sequences.
What molecular mechanisms may account for the faster expression evolution of morphogenes than physiogenes? The cis-regulatory hypothesis asserts that most morphological evolution is due to changes in cis-regulatory sequences (4, 5, 19), predicting faster cis-element turnover in morphogenes than in physiogenes. Because experimentally confirmed mammalian cis-elements are few, are likely to have been confirmed in only one species, and are potentially biased toward certain classes of genes, we tested the above hypothesis by using cis-elements that were predicted exclusively by motif sequence conservation among a set of vertebrate genome sequences and recorded in the cisRED database (20). In cisRED, 8,440 predicted mouse cis-elements and 7,688 predicted human cis-elements were found to be in the proximity of 586 mouse morphogenes and their human orthologs, respectively. Similarly, 7,082 mouse cis-elements and 7,215 human cis-elements were predicted for 621 physiogenes. Thus, morphogenes have significantly more cis-elements per gene than physiogenes (P = 1.51E-4, U test). Because cisRED predicted human and mouse cis-elements independently, we treated the human and mouse predictions as two separate datasets. When mouse is used as the reference species, we found a higher loss rate of cis-elements for physiogenes (1,752/7,082 = 24.7% have no orthologous motifs in human) than for morphogenes (1,660/8,440 = 19.7%) (P = 3.49E-14, χ2 test; Fig. 4A). The fractions of physiogenes (60.9%) and morphogenes (64.8%) that have lost at least one cis-element are not significantly different (P = 0.15, χ2 test). When human is used as the reference species, we again found a higher loss rate of cis-elements for physiogenes (2,346/7,215 = 32.5%) than for morphogenes (1,850/7,688 = 24.1%) (P = 2.44E-30, χ2 test; Fig. 4A). Furthermore, a significantly greater fraction of physiogenes (92.4%) than morphogenes (87.7%) have lost at least one cis-element (P = 0.008, χ2 test). We also examined known human single-nucleotide polymorphisms (SNPs) and found a higher fraction of SNPs in the predicted cis-elements of physiogenes (328/7,215 = 4.8%) than in those of morphogenes (293/7,688 = 4.0%) (P = 2.8E-2, χ2 test; Fig. 4A). Because the mutation rate indicated by dS appears slightly higher for physiogenes than for morphogenes (Fig. 3B), we repeated the above analysis while controlling dS. The results remain largely unchanged except that the SNP difference is no longer statistically significant (Fig. 4B).
Fig. 4.

Evolutionary divergences between mouse and human orthologous genes in cis-regulatory elements and noncoding regions upstream of the transcription start site. (A) Rates of loss of cis-elements within and between species. The interspecies comparison shows the fractions of mouse (or human) cis-elements that are lost in human (or mouse). The intraspecies comparison shows the fraction of human cis-elements that harbor SNPs among human individuals. (B) Rates of loss of cis-elements within and between species, controlled for local mutation rates by dS. (C) Sequence divergence (1 − identity) in upstream noncoding regions and (D) sequence divergence in upstream noncoding regions, controlled for local mutation rates by dS. The values of upper quartile, median, and lower quartile are indicated in each box, whereas the bars outside the box indicate semiquartile ranges. P values are from a Mann–Whitney U test.
Because cis-element prediction in mammals is far from complete, we also examined the divergence between human and mouse orthologous sequences in regions of 200, 500, and 1,000 nucleotides upstream of transcription start sites that presumably harbor cis-regulatory elements. Again, physiogenes generally have higher divergence in upstream noncoding sequences than morphogenes (Fig. 4C), although the difference is statistically significant only for the 200-nucleotide region after control for dS (Fig. 4D). Comparing noncoding regions upstream of start codons gave similar results (Fig. S1). We also compared transcription factors with nontranscription factors among morphogenes, but did not find them to be significantly different from each other or from nontranscription factor physiogenes in terms of upstream noncoding sequence divergence after the dS control (Fig. S2). Because the expression of a gene is influenced by its genomic environment (21, 22), we examined whether the frequency of relocation resulting in the change of neighboring genes is higher for morphogenes than for physiogenes. Contrary to expectation, we found the rate to be slightly higher for physiogenes (11.9%) than for morphogenes (9.7%) when the human and mouse gene maps were compared, although the difference is not significant (P = 0.16, χ2 test). Together, these results and the above analyses of predicted cis-elements fail to detect enhanced rates of cis-regulatory sequence evolution of morphogenes, despite their higher rates of expression evolution.
Faster Gene-Family-Size Changes for Physiogenes Than for Morphogenes.
Gene duplication is widely regarded as an important contributor to phenotypic evolution (23) and a large number of morphological and physiological traits are controlled by multigene families (24, 25). Gene family contraction through gene loss is also a common phenomenon with significant consequences for phenotypic evolution (24–26). To examine whether the evolution of morphological and physiological traits is impacted differentially by gene family expansion/contraction, we calculated an index Dfam for each morphogene and physiogene. Dfam is defined by 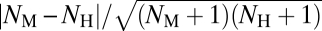 , where NM and NH are the numbers of paralogs that a mouse gene and its human ortholog have in their respective genomes. Dfam is a positive number, with higher values indicating higher rates of gene gain/loss in the family where the focal gene resides. In both mouse and human genomes, physiogenes on average have more paralogs than morphogenes do (mean NM, 5.31 vs. 3.65, P = 1.90E-5, U test; mean NH, 5.18 vs. 3.55, P = 2.95E-5, U test). More importantly, physiogenes have significantly higher Dfam (mean = 0.072) than morphogenes (0.046) (P = 1.03E-3, U test). Using
, where NM and NH are the numbers of paralogs that a mouse gene and its human ortholog have in their respective genomes. Dfam is a positive number, with higher values indicating higher rates of gene gain/loss in the family where the focal gene resides. In both mouse and human genomes, physiogenes on average have more paralogs than morphogenes do (mean NM, 5.31 vs. 3.65, P = 1.90E-5, U test; mean NH, 5.18 vs. 3.55, P = 2.95E-5, U test). More importantly, physiogenes have significantly higher Dfam (mean = 0.072) than morphogenes (0.046) (P = 1.03E-3, U test). Using 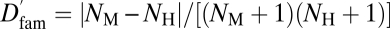 , which ranges between 0 and 1, gave similar results (physiogenes, mean = 0.032 ± 0.0036; morphogenes, 0.022 ± 0.0028). We confirmed these results by comparing Dfam for morphogenes and physiogenes of the same GO categories (Table S4).
, which ranges between 0 and 1, gave similar results (physiogenes, mean = 0.032 ± 0.0036; morphogenes, 0.022 ± 0.0028). We confirmed these results by comparing Dfam for morphogenes and physiogenes of the same GO categories (Table S4).
Hahn and colleagues (27) developed a statistical method to test whether evolutionary expansions and contractions of a gene family follow a random birth-and-death process with constant rates of gain and loss across lineages. We mapped the mouse morphogenes to 630 gene families after exclusion of families that also contain physiogenes. Similarly, we mapped the mouse physiogenes to 667 gene families after exclusion of families that also contain morphogenes. Using gene family composition data from eight mammalian species (Materials and Methods), we found that the random birth-and-death model is rejected slightly more often for physiogene families (41/667 = 6.2%) than for morphogene families (34/630 = 5.4%), although the difference is not statistically significant (P = 0.566, χ2 test). Together with the Dfam result, our analyses show that, whereas physiogene families expand/contract faster than morphogene families, the rate of expansion/contraction is relatively constant across lineages for a given family.
Discussion
In this work, we identified mouse genes that exclusively affect morphological or physiological traits and show that they have distinct properties and modes of evolution. Our study is not completely immune to all ascertainment biases, because the mouse mutants were constructed and the phenotypic data were collected by individual investigators with specific and different research goals. For example, the difference in molecular functions of morphogenes and physiogenes (Fig. 1) could be an artifact if morphological traits tend to be examined in mice lacking transcriptional regulators, whereas physiological traits tend to be examined in mice lacking transporters, channels, receptors, and enzymes. Although such biased and limited phenotyping is possible, we believe that it is relatively uncommon, in part because of the long time and high cost of making mutant mice, compared to phenotyping. In fact, the majority of mutant mice (2,855/4,588 = 62.2%) in our dataset are reported to have both morphological and physiological defects, indicating that they were subject to both morphological and physiological examinations. Furthermore, because mutant mice were almost exclusively generated by biomedical scientists rather than evolutionary biologists, potential ascertainment biases should have minimal impacts on our results of evolutionary properties, because these properties do not differentially influence the design of experiments involving morphogenes versus physiogenes. Note that even if some GO differences between morphogenes and physiogenes are in part caused by ascertainment biases, our subsequent analyses are not affected after the control for the GO differences. The international genetics community has initiated the Knockout Mouse Project (KOMP) to individually knock out every gene in the mouse genome and acquire phenotypic data (28). It will be important to verify our results when such comprehensive data become available.
Because the traits examined in each mutant mouse are somewhat arbitrary, we examined the robustness of our results by random removal of 30% of traits for each gene (Materials and Methods). We then reidentified 887 morphogenes and 834 physiogenes from the reduced data (Materials and Methods). All previously observed differences between morphogenes and physiogenes remain, albeit with weaker statistical support (Figs. S3 and S4), suggesting that the differences would increase with additional phenotyping. Strictly speaking, morphological and physiological traits are not absolutely distinct from each other (7, 8, 24), and their separation was thought to be unnecessary in the study of phenotypic evolution (8). Indeed, >60% of the mouse genes examined affect both morphological and physiological traits. It is possible that no pure morphogene or physiogene would be found if all traits had been examined in all mutant mice. Thus, a more appropriate approach is to compare all genes on the basis of their relative impacts on morphological traits and physiological traits. We classified each gene into one of five groups on the basis of the fraction (fm) of its mutant phenotypes that are morphological (fm = 0, 0 < fm ≤ 1/3, 1/3 < fm ≤ 2/3, 2/3 < fm < 1, and fm = 1). With one exception that is most likely artifactual (Fig. S5 legend), all properties examined show gradual changes from pure physiogenes (fm = 0) to pure morphogenes (fm = 1) whereas the three intermediate groups exhibit intermediate values (Fig. S5). Thus, the differences between morphological and physiological traits are reflected not just in pure morphogenes and physiogenes, but in all genes. Taken together, these analyses show that the separation between morphogenes and physiogenes is both possible and biologically meaningful.
Although our morphogenes and physiogenes are classified largely by the phenotypic effects of deleterious mutations, it is reasonable to assume that most if not all beneficial or neutral mutations in morphogenes affect morphological traits rather than physiological traits and vice versa. Thus, without knowing the genetic basis of phenotypic evolution, which to date is restricted to a limited number of case studies and subject to various biases, we can use properties of morphogenes and physiogenes to make certain predictions about phenotypic evolution. We predict that morphological evolution more often involves transcriptional regulators and gene expression changes whereas physiological evolution more likely involves transporters, channels, receptors, and enzymes and protein sequence changes or gene gains/losses. Interestingly, these predictions are generally consistent with the patterns observed from case studies of a wide variety of natural and domestic organisms, including both animals and plants (4, 7, 9, 29, 30). Thus, our results, although obtained exclusively from two mammals, are probably true in other species as well. Our predictions can guide experimental designs in discerning the genetic mechanisms of phenotypic evolution. The identified differential genetic basis of morphological and physiological defects in mouse may also help identify the molecular underpinnings of human diseases.
One of the most contentious debates on the genetic basis of phenotypic evolution centers on the role of cis-regulatory mutations (4 –9, 19). Although we showed that gene expression clearly evolves faster for morphogenes than for physiogenes (Fig. 3), we did not find predicted cis-regulatory elements or regions to evolve faster in morphogenes (Fig. 4). Rather, almost all of our analyses showed the opposite, with some being statistically significant (Fig. 4). Nevertheless, our analyses have several caveats. First, because genomewide cis-element prediction in mammals is currently based on sequence conservation across multiple species, we can count losses of cis-elements but not gains. The number of gains, however, should approximate the number of losses, if there is no net change of the number of cis-elements in evolution. Thus, it is unlikely, although formally possible, that the rate of cis-element change in morphogenes becomes higher than that in physiogenes when gains of cis-elements are included. Second, cisRED assigns every cis-element to its nearest gene, despite that some cis-elements do not regulate their nearest genes (31). Nevertheless, unless the distributions of the distances between genes and their cis-elements are different between morphogenes and physiogenes, the assignment errors should not bias our comparison, although they reduce the signal/noise ratio and the statistical power. Third, because cis-regulatory elements presumably constitute only a small fraction of noncoding sites and because some cis-elements are distant from the gene being regulated (31), upstream sequence divergence may be generally powerless for comparing the rates of cis-element turnover in morphogenes and physiogenes, although a significant difference was found for the upstream region of 200 nucleotides even after the control of mutation rate (Fig. 4D). These caveats being said, it is interesting to note that our finding of faster expression evolution but slower cis-regulatory evolution of morphogenes than physiogenes is consistent with the observation of poor correlation between cis-element changes and expression divergence in yeasts and vertebrates (32, 33). The relative contributions of cis- and trans-changes to gene expression evolution have been examined in yeast at the genomic scale (34). This approach can be applied to closely related mouse species such that the relative contributions of cis- and trans-changes to morphogene and physiogene expression evolution can be critically evaluated. Furthermore, it is possible that fewer cis-regulatory changes are required in morphogenes than in physiogenes for the same amount of expression evolution, if morphological evolution tends to occur by cooption of preexisting gene networks, whereas physiological evolution tends to occur through one-by-one recruitment of multiple genes (35).
In comparing evolutionary rates, we did not attempt to distinguish between positive selection for advantageous mutations and reduced purifying selection against deleterious mutations, because we are interested in the genetic mechanism rather than the evolutionary force of phenotypic evolution. Whereas some previous studies focused on the genetic basis of adaptive phenotypic evolution (8), we see no reason why the underlying genetic basis should differ between adaptive and neutral phenotypic evolution (24). We expect that our results should apply regardless of the adaptiveness of phenotypic evolution.
Materials and Methods
Morphogenes and Physiogenes.
The data of mouse mutant phenotypes were downloaded from MGI (http://www.informatics.jax.org/) version 4.11. The file MRK_Ensembl_Pheno.rpt contains a list of 5,199 mouse genes with one or more Mammalian Phenotype (MP) IDs showing the phenotypic consequences when the gene is knocked out, knocked down, or mutated by transgenic insertions or point mutations. MP IDs are hierarchically structured. That is, one parent MP ID (e.g., MP:0002102, abnormal ear morphology) represents a phenotype lineage that may include several child MP IDs to describe a more detailed phenotype (e.g., MP:0000026, abnormal inner ear morphology; MP:0002177, abnormal outer ear morphology). In the present study, we used 129 morphological and 183 physiological parent MP IDs (Table S1) to classify the 5,199 mouse genes. The above 183 physiological parent MP IDs include two behavioral parent MP IDs, as the evolutionary patterns of behavioral traits and physiological traits have been proposed to be similar to each other (9). MP IDs that cannot be distinguished as morphological or physiological traits were not used (e.g., MP:0008762, embryonic lethality). If a mouse gene is annotated for a child MP, the parent MP ID that this child MP ID belongs to is used. Here, we defined morphogenes (or physiogenes) as genes exclusively associated with morphological (or physiological) MP IDs. We identified 821 mouse morphogenes and 912 mouse physiogenes for subsequent analysis (Table S2). Our analysis did not include 2,855 mouse genes associated with both morphological and physiological MP IDs. The remaining 611 genes are not associated with any of the morphological or physiological MP IDs. Pleiotropy of each morphogene or physiogene was defined by the number of associated parent MP IDs.
To examine the potential artifact generated by incomplete phenotyping of mouse mutants, we reidentified morphogenes and physiogenes by randomly removing some associated MP IDs of each gene, with every MP ID of every gene having a 30% probability to be removed. The actual removal or retention of a MP ID of a given gene was determined stochastically. The random removal resulted in the loss of some original morphogenes and physiogenes. However, some genes originally associated with both morphological and physiological MP IDs now became morphogenes or physiogenes because of the removal of physiological MP IDs or morphological IDs, respectively. The redefined dataset contained 887 morphogenes and 834 physiogenes.
Genomic Data.
Human genome version NCBI36 and mouse genome version NCBIM36 were used. Chromosomal positions and annotations of human and mouse genes and their orthology relationships based on Ensembl release 45 (http://www.ensembl.org/) were retrieved using BioMart (http://www.biomart.org/). Values of dS and dN between human and mouse orthologs, estimated by the likelihood method, as well as paralog information, were retrieved using BioMart. Gene relocation events resulting in the change of neighboring genes were inferred from the syntenic preservation on the basis of the genomic location of one-to-one human–mouse orthologs. The gene family annotations for the human, chimpanzee, rhesus macaque, mouse, rat, dog, cow, and opossum were from Ensembl release 45. We tested the random birth-and-death model of gene family evolution (27) using CAFÉ 2.0 (http://www.bio.indiana.edu/∼hahnlab/Software.html) with default parameters. This analysis also required the information of the phylogeny and divergence times of the aforementioned eight species, which we obtained from ref. 36. The mouse GO annotations (http://www.geneontology.org/) were downloaded from MGI. Splicing information of mouse genes was retrieved from BioMart. Analysis of gene enrichment in GO categories was performed by FatiGO (http://www.fatigo.org/) (10). Essential genes are those that cause sterility or lethality before puberty when deleted (11). Annotations of predicted cis-regulatory elements of human (cisred_Hsap_9) and mouse (cisred_Mmus_4) genes were obtained from cisRED (http://www.cisred.org/) (20). Human SNP data were based on dbSNP build 129 (ftp://ncbi.nlm.nih.gov/snp/). The upstream sequence divergence was calculated on the basis of the alignment of human–mouse genomes by the University of California, Santa Cruz Genome Browser. Data from human protein–protein interactions were obtained from the Human Protein Reference Database (http://www.hprd.org/).
Microarray Gene Expression Data Analysis.
The GeneAtlas v2 dataset (http://symatlas.gnf.org/) has information about genomewide gene expression levels obtained by hybridizations of RNAs from 73 human nonpathogenic tissues and 61 mouse tissues onto the Affymetrix microarray chips (human, U133A/GNF1H; mouse, GNF1M) (16). The probe sets were assigned to the human and mouse genes following a previous study (37). The expression level detected by each probe set was obtained as the signal intensity (S) computed from the MAS 5.0 algorithm. Tissue specificity of a gene is defined as the heterogeneity of its expression levels across all of the tissues examined and is estimated by 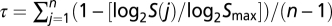 , where n = 61 is the number of mouse tissues examined here and Smax is the highest expression signal of the gene across all tissues (14). Following a previous study (14), we arbitrarily let S(j) be 100 if it is <100. The τ value ranges from 0 to 1, with higher values indicating higher tissue specificity. The GeneAtlas v2 dataset contains 26 common tissues between human and mouse. Expression-profile divergence between a pair of orthologs was measured by 1 − R, where R is Pearson's correlation coefficient between human S and mouse S values across the 26 common tissues. We also computed 1 − R for human–mouse orthologs using the ExonArray dataset, which has more accurate gene expression measures than GeneAtlas v2 (17), although it contains only 6 common tissues between the two species. The expression signals S in the ExonArray data were computed from GeneBASE (http://biogibbs.stanford.edu/∼kkapur/genebase/). The S values were averaged among the three replicated experiments performed for each tissue.
, where n = 61 is the number of mouse tissues examined here and Smax is the highest expression signal of the gene across all tissues (14). Following a previous study (14), we arbitrarily let S(j) be 100 if it is <100. The τ value ranges from 0 to 1, with higher values indicating higher tissue specificity. The GeneAtlas v2 dataset contains 26 common tissues between human and mouse. Expression-profile divergence between a pair of orthologs was measured by 1 − R, where R is Pearson's correlation coefficient between human S and mouse S values across the 26 common tissues. We also computed 1 − R for human–mouse orthologs using the ExonArray dataset, which has more accurate gene expression measures than GeneAtlas v2 (17), although it contains only 6 common tissues between the two species. The expression signals S in the ExonArray data were computed from GeneBASE (http://biogibbs.stanford.edu/∼kkapur/genebase/). The S values were averaged among the three replicated experiments performed for each tissue.
Supplementary Material
Acknowledgments
We thank Meg Bakewell, John Doebley, Masatoshi Nei, Ondrej Podlaha, Wenfeng Qian, David Stern, Zhi Wang, and two anonymous reviewers for valuable comments. This work was supported by National Health Research Institutes intramural funding (to B.Y.L.) and National Institutes of Health research grants (to J.Z.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0910339107/DCSupplemental.
References
- 1.King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- 2.Chimpanzee Sequencing and Analysis Consortium. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature. 2005;437:69–87. doi: 10.1038/nature04072. [DOI] [PubMed] [Google Scholar]
- 3.Glazko G, Veeramachaneni V, Nei M, Makałowski W. Eighty percent of proteins are different between humans and chimpanzees. Gene. 2005;346:215–219. doi: 10.1016/j.gene.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Carroll SB. Evo-devo and an expanding evolutionary synthesis: A genetic theory of morphological evolution. Cell. 2008;134:25–36. doi: 10.1016/j.cell.2008.06.030. [DOI] [PubMed] [Google Scholar]
- 5.Wray GA. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 2007;8:206–216. doi: 10.1038/nrg2063. [DOI] [PubMed] [Google Scholar]
- 6.Stern DL, Orgogozo V. Is genetic evolution predictable? Science. 2009;323:746–751. doi: 10.1126/science.1158997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stern DL, Orgogozo V. The loci of evolution: How predictable is genetic evolution? Evolution. 2008;62:2155–2177. doi: 10.1111/j.1558-5646.2008.00450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoekstra HE, Coyne JA. The locus of evolution: Evo devo and the genetics of adaptation. Evolution. 2007;61:995–1016. doi: 10.1111/j.1558-5646.2007.00105.x. [DOI] [PubMed] [Google Scholar]
- 9.Carroll SB. Evolution at two levels: On genes and form. PLoS Biol. 2005;3:e245. doi: 10.1371/journal.pbio.0030245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Al-Shahrour F, et al. FatiGO +: A functional profiling tool for genomic data. Integration of functional annotation, regulatory motifs and interaction data with microarray experiments. Nucleic Acids Res. 2007;35:W91–W96. doi: 10.1093/nar/gkm260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liao BY, Zhang J. Null mutations in human and mouse orthologs frequently result in different phenotypes. Proc Natl Acad Sci USA. 2008;105:6987–6992. doi: 10.1073/pnas.0800387105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liao BY, Scott NM, Zhang J. Impacts of gene essentiality, expression pattern, and gene compactness on the evolutionary rate of mammalian proteins. Mol Biol Evol. 2006;23:2072–2080. doi: 10.1093/molbev/msl076. [DOI] [PubMed] [Google Scholar]
- 13.He X, Zhang J. Toward a molecular understanding of pleiotropy. Genetics. 2006;173:1885–1891. doi: 10.1534/genetics.106.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao BY, Zhang J. Low rates of expression profile divergence in highly expressed genes and tissue-specific genes during mammalian evolution. Mol Biol Evol. 2006;23:1119–1128. doi: 10.1093/molbev/msj119. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Li WH. Mammalian housekeeping genes evolve more slowly than tissue-specific genes. Mol Biol Evol. 2004;21:236–239. doi: 10.1093/molbev/msh010. [DOI] [PubMed] [Google Scholar]
- 16.Su AI, et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xing Y, Ouyang Z, Kapur K, Scott MP, Wong WH. Assessing the conservation of mammalian gene expression using high-density exon arrays. Mol Biol Evol. 2007;24:1283–1285. doi: 10.1093/molbev/msm061. [DOI] [PubMed] [Google Scholar]
- 18.Tirosh I, Barkai N. Evolution of gene sequence and gene expression are not correlated in yeast. Trends Genet. 2008;24:109–113. doi: 10.1016/j.tig.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 19.Stern DL. Evolutionary developmental biology and the problem of variation. Evolution. 2000;54:1079–1091. doi: 10.1111/j.0014-3820.2000.tb00544.x. [DOI] [PubMed] [Google Scholar]
- 20.Robertson G, et al. cisRED: A database system for genome-scale computational discovery of regulatory elements. Nucleic Acids Res. 2006;34(Database issue):D68–D73. doi: 10.1093/nar/gkj075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao BY, Zhang J. Coexpression of linked genes in mammalian genomes is generally disadvantageous. Mol Biol Evol. 2008;25:1555–1565. doi: 10.1093/molbev/msn101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De S, Teichmann SA, Babu MM. The impact of genomic neighborhood on the evolution of human and chimpanzee transcriptome. Genome Res. 2009;19:785–794. doi: 10.1101/gr.086165.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J. Evolution by gene duplication—an update. Trends Ecol Evol. 2003;18:292–298. [Google Scholar]
- 24.Nei M. The new mutation theory of phenotypic evolution. Proc Natl Acad Sci USA. 2007;104:12235–12242. doi: 10.1073/pnas.0703349104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Demuth JP, Hahn MW. The life and death of gene families. BioEssays. 2009;31:29–39. doi: 10.1002/bies.080085. [DOI] [PubMed] [Google Scholar]
- 26.Wang X, Grus WE, Zhang J. Gene losses during human origins. PLoS Biol. 2006;4:e52. doi: 10.1371/journal.pbio.0040052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hahn MW, De Bie T, Stajich JE, Nguyen C, Cristianini N. Estimating the tempo and mode of gene family evolution from comparative genomic data. Genome Res. 2005;15:1153–1160. doi: 10.1101/gr.3567505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Austin CP, et al. The knockout mouse project. Nat Genet. 2004;36:921–924. doi: 10.1038/ng0904-921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Doebley JF, Gaut BS, Smith BD. The molecular genetics of crop domestication. Cell. 2006;127:1309–1321. doi: 10.1016/j.cell.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 30.Doebley J, Lukens L. Transcriptional regulators and the evolution of plant form. Plant Cell. 1998;10:1075–1082. doi: 10.1105/tpc.10.7.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lettice LA, et al. A long-range Shh enhancer regulates expression in the developing limb and fin and is associated with preaxial polydactyly. Hum Mol Genet. 2003;12:1725–1735. doi: 10.1093/hmg/ddg180. [DOI] [PubMed] [Google Scholar]
- 32.Tirosh I, Weinberger A, Bezalel D, Kaganovich M, Barkai N. On the relation between promoter divergence and gene expression evolution. Mol Syst Biol. 2008;4:159. doi: 10.1038/msb4100198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan ET, et al. Conservation of core gene expression in vertebrate tissues. J Biol. 2009;8:33. doi: 10.1186/jbiol130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tirosh I, Reikhav S, Levy AA, Barkai N. A yeast hybrid provides insight into the evolution of gene expression regulation. Science. 2009;324:659–662. doi: 10.1126/science.1169766. [DOI] [PubMed] [Google Scholar]
- 35.Monteiro A, Podlaha O. Wings, horns, and butterfly eyespots: How do complex traits evolve? PLoS Biol. 2009;7:e37. doi: 10.1371/journal.pbio.1000037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy WJ, Pevzner PA, O’Brien SJ. Mammalian phylogenomics comes of age. Trends Genet. 2004;20:631–639. doi: 10.1016/j.tig.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 37.Liao BY, Zhang J. Evolutionary conservation of expression profiles between human and mouse orthologous genes. Mol Biol Evol. 2006;23:530–540. doi: 10.1093/molbev/msj054. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.