

Abstract
Clostridium difficile has rapidly emerged as the leading cause of antibiotic-associated diarrheal disease, with the transcontinental spread of various PCR ribotypes, including 001, 017, 027 and 078. However, the genetic basis for the emergence of C. difficile as a human pathogen is unclear. Whole genome sequencing was used to analyze genetic variation and virulence of a diverse collection of thirty C. difficile isolates, to determine both macro and microevolution of the species. Horizontal gene transfer and large-scale recombination of core genes has shaped the C. difficile genome over both short and long time scales. Phylogenetic analysis demonstrates C. difficile is a genetically diverse species, which has evolved within the last 1.1–85 million years. By contrast, the disease-causing isolates have arisen from multiple lineages, suggesting that virulence evolved independently in the highly epidemic lineages.
Keywords: homologous recombination, horizontal gene transfer, single nucleotide polymorphism
Clostridium difficile is a Gram-positive, spore-forming anaerobe that has emerged as a major cause of healthcare- and antibiotic-associated diarrhea. After antibiotic therapy, the protective intestinal microbiota is disrupted, whereupon ingested or resident C. difficile hypercolonize the gastrointestinal tract and produce toxins and transmissible spores (1). C. difficile was recognized as a pathogen only three decades ago (2), and a number of emergent PCR ribotypes (a common typing scheme) have been responsible for outbreaks worldwide (3), with different PCR ribotypes dominating both temporally and geographically (4–6). A major outbreak occurred in Canada in 2003 (7, 8), caused by a previously rare PCR ribotype 027. This 027 ribotype has spread globally and now accounts for ∼50% of isolates in United Kingdom and North American hospitals (6, 9, 10). Other recently emerging ribotypes include 001, 017, and 078 (4, 6, 11 –15). The epidemiology of C. difficile is evolving rapidly; yet, despite this continued threat, we have a poor understanding of how or why particular variants emerge.
A comparative genomic hybridization (CGH) approach (16) revealed four major clades, including a hypervirulent clade, which mostly consists of so called B1, NAP1 or 027 strains (based on restriction endonuclease analysis, pulse-field type or ribotype, respectively). Although such typing methods are generally efficient in differentiating isolates, a genome sequence-based approach is more discriminatory and can reveal phylogenetic relationships within specific groups. The C. difficile genome is highly dynamic and readily undergoes genetic exchange of mobile elements (16, 17). However, the impact of homologous recombination between strains is unclear.
High-throughput sequencing technologies have proved to be a powerful approach for studying genetic variation and evolution of monomorphic pathogens such as Salmonella Typhi (18). Here, we generated whole genome sequences from a diverse collection of eight C. difficile human and animal isolates using combined Roche 454 and Sanger sequencing to explore the macroevolution of the C. difficile genome. In addition, we also sequenced 21 isolates representing the hypervirulent clade identified by Stabler et al. (16) with Illumina (Solexa) to investigate the microevolution within this group. We describe a previously unappreciated level of genome variation in terms of both mobile elements and homologous recombination, and confirm the importance of genetic exchange in the evolution of this species. Our results suggest that the core C. difficile genome has been primarily shaped by purifying selection pressure, and that environmental as well as genetic effects may be responsible for its recent expansion as a major pathogen. This study also opens avenues for the development of new epidemiological tools for studying C. difficile transmission routes and for developing interventions to reduce the burden of disease.
Results and Discussion
Macroevolution of the C. difficile Species.
Previous phylogenomic analysis identified four genetically distinct C. difficile clades (16); based on this, we selected six strains representing the broad genetic diversity of C. difficile for whole genome sequencing (Table S1). Genome assemblies were created based on combined 454 (Roche) and Sanger sequencing to increase accuracy and coverage. A consensus whole-genome alignment was used to build a maximum likelihood phylogenetic tree that also includes the published genomes of C. difficile strains 630 (ribotype 012), R20291 and CD196 (both ribotype 027) (Fig. 1A). To minimize the impact of recombined sequences on the tree building, we also used a concatenated alignment of nonrecombining core coding sequences (CDSs) to build a tree of six isolates representing the deep-branching phylogeny, resulting in the same topology (Fig. S1 and SI Text). The resulting phylogeny recapitulates the four major lineages based upon microarray analysis (16) but provides much more depth.
Fig. 1.

Phylogenetic trees of C. difficile based on whole-genome sequences. Arrows and unfilled circles denote insertion and deletion events, respectively. Genomic islands carrying drug resistance genes are shown with asterisks. (A) Deep-branching phylogeny that illustrates the relationships between different lineages/ribotypes are shown by different colors. The four 027 ribotype isolates are collectively represented as node “027s”. Scale bar indicates number of substitutions per site. The root connects to C. bartlettii and C. hiranonis. (B) Split decomposition network indicating microevolution within the hypervirulent lineage. Strain names are colored according to countries of isolation (blue, United States; red, United Kingdom; green, France). Bootstrap values are labeled along branches. The root connects to strain 630.
The phylogenetic tree reveals that the broad genetic diversity of C. difficile is predominantly reflected in the ribotyping scheme. For example, the four 027 ribotype sequences occupy a single lineage (unresolved in Fig. 1A but separated in Fig. 1B). Strains CF5 and M68, which are historic and recent representatives respectively of the 017 ribotype, occupy a distinct lineage. Isolate BI-9, verified as ribotype 001, is more closely related to isolate 630 (ribotype 012), compared with isolate M120 (ribotype 078) that appears to be highly divergent as indicated by its long branch length. The average sequence divergence between M120 and the other isolates is 2.4%, indicative of an old species.
The sequence data were used to estimate the age of the C. difficile species. Dating methods for microbes are imperfect and the subject of controversy, so to find the range of possibilities, we used two independent methods based on different underlying assumptions. One is based on an average synonymous substitution per site (dS) of 0.032 in concatenated nonrecombining core CDSs and a synonymous substitution rate of 2.50 × 10−9 − 1.50 × 10−8 per site per year (Methods). This indicates an age of 1.1–6.4 million years before present. As an alternative, we used BEAST software (19) and specified a calibrated molecular clock rate of 1.15 × 10−10 (Methods). This was calculated based on a sequence divergence of 0.54 between orthologous CDSs of C. difficile and C. tetani, and the hypothesis that the Clostridium lineage diverged 2.34 billion years ago (Ga) (20). Therefore, although C. difficile and C. tetani diverged relatively early in the Clostridium lineage, the divergence time between them should not exceed 2.34 Ga. This analysis resulted in a divergence time of 85 million years. Clearly these methods produce highly divergent estimates, indicating high levels of uncertainly, but which could be viewed as maximum and minimum boundaries.
Our findings have interesting implications on the emergence of C. difficile as a human pathogen. Although the common ancestor of C. difficile dates back millions of years, highly epidemic and disease-causing isolates (017s, 027s, and 078s) are found in all lineages. This suggests there may be certain genetic elements common to all C. difficile strains that underlie virulence. Although C. difficile appears to be an ancient species, it was recognized as a pathogen only three decades ago, indicating that besides genetic modifications, changes in interaction between host and pathogen, as well as other factors such as human activity, hospital design, and antibiotic use, may have contributed to the emergence of C. difficile as a major pathogen.
Microevolution within the Hypervirulent Clade.
To study microevolution within the hypervirulent clade and recent ribotype 027 isolates, a collection of 25 isolates spanning 1985–2007 (Tables S1 and S2) were sequenced using multiplexed Illumina (Solexa) or a combination of 454 (Roche) and Sanger sequencing technologies. SNPs were detected by comparing the sequence of each isolate with the early 027 ribotype isolate CD196 (21). We discovered a total of 1847 SNP differences among 25 isolates; however, 1670 (90.4%) of these SNPs appear in tight clusters and are present only in isolate BI–4 or BI-11 (Fig. 2), indicating that they could have resulted from recent recombination events. We excluded these SNPs from phylogenetic analyses as they could mask the true phylogenetic signal. A split-decomposition network based on remaining SNPs is shown in Fig. 1B. No conflict between placements of branches was identified by split decomposition analysis. A lack of bipartitions in certain parts of the lineage and low bootstrap values can be explained by the scarcity of genetic variation between isolates, and some recombinant sites potentially remaining in the analysis. The placement of the root for this lineage cannot be uniquely determined. This also suggests recombination between isolates sitting at the basal branches of this phylogeny and those outside this group. Interestingly, a Bayesian skyline plot analysis (22) suggests this hypervirulent group has undergone a population expansion around the start of the century (Fig. S2), which coincides with the time when hospital outbreaks caused by this C. difficile ribotype were first reported.
Fig. 2.

SNPs between CD196 and 24 other hypervirulent C. difficile isolates. Outer circle: CDSs of C. difficile CD196 genome, shown on a pair of concentric rings representing both coding strands; two inner circles: G+C% content plot and GC deviation plot (>0% olive, <0% purple); in between: SNPs (blue and red) between CD196 and other isolates, from outer to inner: 2004013, 2004102, 2004118, 2004163, 2006439, 2007140, 2007218, 2007825, 2007833, 2007837, 2007855, BI-1, BI-2, BI-3, BI-4, BI-5, BI-6, BI-6p, BI-7, BI-10, BI-11, BI-13, BI-15, and R20291. The rings representing isolates with large homologous recombination blocks (BI–4 and BI–11) are shown in red.
Extensive Role of Horizontal Gene Transfer in C. difficile Evolution.
The establishment of a rooted phylogeny for C. difficile allowed us to trace various evolutionary events such as genomic insertions and deletions back to where they occurred in the phylogenetic tree. Putative conjugative transposons and bacteriophages account for a large proportion of the mobile elements present within the C. difficile genomes. Many of these mobile elements code for a variety of antibiotic resistance genes (Fig. 1), suggesting a significant role for horizontal gene transfer in resistance acquisition. In isolate 630, CTnCD1, CTnCD3, CTnCD6, and CTnCD7 are closely related to Tn916 in Enterococcus faecalis (17). We found the similarity also extends to CTnCD25 and CTnCD11, but these carry different drug-resistance determinants (CTnCD3, tetracycline; CTnCD25, chloramphenicol; CTnCD11, erythromycin). In both the deep-branching phylogeny and the lineage of hypervirulent isolates, we observed evidence for the same genomic island entering different parts of the phylogenetic tree (SI Text). There are also cases of new insertions occurring within existing genomic islands. For example, copies of the conjugative transposon CTnCD5 found in 2004102, 2006439, 2007855, and BI-13 all contain an extra 7.5-kb cassette (Fig. 1B and Fig. S3). This region contains CDSs encoding a DNA recombinase and aminoglycoside resistance genes aph(2')-Ib and aac(6')-Im. Combining the information from our phylogenetic tree, this suggests that the insertion event within CTnCD5 occurred in the common ancestor of these isolates. M120, which is divergent from the other isolates, harbors a number of unique genomic regions, two of which exhibit >80% sequence similarity to Streptococcus pyogenes and Thermoanaerobacter species, respectively, suggesting gene transfer across very large phylogenetic distances.
Large Chromosomal Region Exchange by Homologous Recombination.
Besides horizontal transfer of mobile elements, bacteria can also evolve through exchange of common chromosomal segments by homologous recombination (23). We examined the distribution of SNPs along the conserved C. difficile genome to test for signatures of recombination. Strikingly, we identified strong evidence for exchange of very large chromosomal regions both within the deep-branching phylogeny and the recent hypervirulent group.
The distribution of SNPs along the genomic backbone of the hypervirulent isolates demonstrates dense SNP clusters in strains BI–4 and BI–11, suggesting imports from different phylogenetic backgrounds into these isolates (Fig. 2). The sizes of these regions range from 9 kb to 170 kb. Further investigation of sequence similarity implies the donors for these recombined regions are not closely related to known C. difficile isolates (SI Text). To identify recombination between ribotypes, SNPs in nonrepetitive regions of the genome were identified between all pairwise combinations of the six isolates (CD196, 630, BI-9, M120, CF5, and M68). Figure 3 shows, as an example, the distribution of SNPs between isolate CF5 and each of the others. A very low level of diversity was observed between isolates CF5 and M68 across the entire chromosome except for several discrete regions, characterized by significantly increased SNP numbers with clear-cut boundaries, which suggests that these regions were acquired through homologous recombination events. The largest of these regions was around 300 kb. The complementary pattern of SNP peaks and valleys between M68 and BI-9, CD196 and 630 indicates a donor similar to BI-9, CD196, and 630 in these chromosomal regions, suggesting this recombination has occurred across a relatively large phylogenetic distance. Similar large blocks of homologous recombination were recently identified in Streptococcus agalactiae, where it was shown that they may arise from Hfr-like conjugation driven by origins of transfer in mobile genomic islands (24). This is also a possible explanation in C. difficile, given the large numbers of mobile elements in the chromosome.
Fig. 3.

Signatures of recombination in the deep-branching phylogeny. The genome-wide distribution of SNPs is shown for each strain against the core genome (excluding repetitive sequences) of strain CF5, which is indicated along the x axis. The y axis gives the number of SNPs in each 500-bp window.
The rate of homologous recombination varies hugely among species (25). To assess the impact of homologous recombination on sequence diversification, we calculated the ratio of recombination/mutation (r/m) within the deep-branching phylogeny, which gives the relative probability that a nucleotide has changed as the result of recombination relative to point mutation (26, 27). The r/m ratio for C. difficile is between 0.63 and 1.13 based on our data. Vos et al. previously compared r/m for different bacteria based on MLST data (28). They calculated this ratio to be 13.6 for Helicobacter pylori, 7.1 for Neisseria meningitidis, and 0.1 for Staphylococcus aureus. Their estimated r/m for C. difficile is 0.2. The difference between their and our estimates may be due to sampling of loci and strains, as the recombination events we have detected seem to be very localized. Despite this, our data suggest that recombination plays a significant role in C. difficile sequence diversification.
Selective Forces Acting upon the C. difficile Genome.
To investigate the selective forces acting on the C. difficile genome, dN/dS, the ratio of nonsynonymous vs. synonymous substitution rate was calculated. A ratio significantly smaller than 1 suggests strong purifying selection, whereas a ratio close to 1 is usually taken as indicating a neutral selection pressure. However, it has been shown that for very closely related genomes, dN/dS can be close to 1 (29), either because time has been too short for selection to act (29) or because nucleotide substitutions within a species may represent segregating polymorphisms rather than fixed differences (30).
dN/dS was calculated for concatenated alignments of CDSs from the nonrepetitive, core genome for each pairwise combination of 9 isolates (630, BI-9, CF5, M68, M120, CD196, BI-1, 2007855, and R20291). It was previously suggested when effective population size is infinite or sufficiently large; the trajectory of 1/(dN/dS) exhibits a linear trend with time, but when population size decreases, the increase of 1/(dN/dS) with time reaches a plateau (29). S (number of synonymous substitutions) or dI (number of intergenic SNPs) have been used as a measure of time since divergence, although synonymous changes and intergenic regions could also be subject to selection forces that deviate from neutral. The 1/(dN/dS) trajectory of C. difficile appears to be nonlinear, regardless of dS or dI being used as the indicator of time (Fig. 4). This pattern is similar to the trajectories reported for Streptococcus pyogenes and the Bacillus cereus+anthracis+thuringiensis complex (29).
Fig. 4.

Trajectory of 1/(dN/dS) within the C. difficile phylogeny over time. The number of intergenic SNPs (A) and synonymous changes (B) serve as measures of time since divergence.
Our data show that between deeply diverging lineages, there is evidence for strong purifying selection (the average dN/dS between strain M120 and the rest is ≈0.08). However, for recently diverged lineages, dN/dS is very close to 1, in agreement with previous analyses (29). This 1/(dN/dS) trajectory suggests nonsynonymous substitutions were purged less efficiently in C. difficile than in E. coli, whose trajectory appears to be linear (29), or that the effects of purifying selection on the C. difficile genome are somewhat delayed. This could be explained by a relatively small effective population size for C. difficile compared with E. coli, which has a broader host-range.
We also investigated genes under positive selection, and identified 12 positively selected CDSs (Table S3), which seems relatively small for such a diverse species. However, among these CDSs are response regulators and surface proteins, including predicted membrane and exported proteins, which are likely candidates for diversifying selection driven by the host immune system. It is very likely that many more variable and positively selected genes exist, but that these are part of the accessory gene pool and therefore not captured in this analysis. Still, our finding does suggest that host immune selection plays a role in C. difficile evolution.
Conclusions
We explored phylogeny, horizontal gene transfer, recombination, and the evolutionary history of C. difficile at the whole genome level and demonstrated significant diversity, with disease-associated isolates emerging from multiple lineages, contradicting the idea that a single lineage evolved to become pathogenic. The level of horizontal gene transfer and recombination confirms that C. difficile has a highly dynamic genome. However, we show that the effect of horizontal exchange extends beyond mobile genetic elements to include large core chromosomal regions transferred over considerable phylogenetic distances. Selection in core CDSs related to surface components was identified, albeit to a limited extent, and investigation of selection pressures acting upon the genome confirms that the majority of the genes are subject to delayed purifying selection.
Methods
Bacterial Isolates.
Isolates were provided by the following individuals: Dale Gerding, Hines VA Hospital, IL (CF5 and BI isolates); Jon Brazier, Anaerobe Reference Laboratory, Cardiff, UK (R20291); Michel Popoff, Institut Pasteur, Paris, France (CD196); Denise Drudy, Centre for Food Safety, University College Dublin, Ireland (M68 and M120); Peter Mullany, Eastman Dental Institute, London, UK (630); and Glenn Songer, Department of Veterinary Science and Microbiology, University of Arizona (all other isolates).
DNA Sequencing and Assembly.
Genomic DNA was prepared according to Wren et al. (31). Isolates were sequenced using 454 Life Sciences GS-20 sequencer (Roche) (R20291), 454 Life Sciences GS-FLX sequencer (Roche) (all other isolates in Table S1), and Illumina (Solexa) Genome Analyzer System (isolates in Table S2) with multiplexed protocol according to the manufacturer's specifications. Paired-end reads were generated for all isolates except CF5, M68, M120, BI-1, 2007855, R20291, and CD196, for which single-end reads were produced. 454 reads were assembled de novo into contigs using newbler (Roche). For isolates with capillary data available, 454 contigs were shredded into reads of comparable length to capillary reads, and assemblies were created using data from both platforms using Phrap. The order of contigs was estimated by comparison with the reference sequence using the MUMmer package (nucmer program) (32), implemented in ABACUS (33). Although some manual error checking was performed, these should still be considered to be draft genomes. Reads from Illumina (Solexa) were directly mapped back to a reference sequence using MAQ version 0.7.1 (34).
Genome Comparison and Identification of Unique Genomic Regions.
Genomic sequences of different isolates were annotated in Artemis (35) and compared with each other using Artemis Comparison Tool (ACT) (36). Orthologs were identified using a FASTA reciprocal-best-hit algorithm, with lower bounds of 30% identity and 80% sequence length. Unique regions were identified using a combination of these approaches.
Phylogenetic Analyses.
The genomic sequences of nine C. difficile isolates (630, BI-9, CF5, M68, M120, CD196, BI-1, 2007855, and R20291) were aligned with ProgressiveMauve (37). Consensus alignments were used to build a maximum likelihood tree with RAxML (38) with 100 resamplings of alignment data. To root the phylogenetic tree, the genomic sequences of Clostridium bartlettii strain DSM 16795 (a close relative of C. difficile) and Clostridium hiranonis DSM 13275 were used. Phylogenetic relationships between hypervirulent isolates were inferred using PHYML (39) and the split decomposition method implemented in SplitsTree4 (40) with 100 bootstraps.
SNPs and Gene Alignments.
Between isolates of different ribotypes, SNP calling was performed using the nucmer program in the MUMmer package (32). All primary SNPs were further checked by FASTA (41) to validate alleles (SI Text). Orthologous CDSs were retrieved from the whole genome alignment of nine C. difficile isolates generated using progressiveMauve (37), with the guide of the fully annotated reference strain 630. We inspected the genome manually and excluded CDSs in bacteriophages, transposons and other mobile elements, as these regions are generally repetitive and can confound SNP finding programs. A multiple sequence alignment for each CDS was obtained by aligning SNP alleles against the sequence of strain 630.
For analysis between hypervirulent isolates, we used MAQ (34) to align Solexa reads to the reference genome CD196 and call SNPs. The identities of hypervirulent isolates were validated by PCR. All SNPs in repetitive regions and mobile elements were excluded. Preliminary SNPs were confirmed for each isolate in all sequencing reads (SI Text). A multiple sequence alignment formed by the concatenated variable positions from all isolates was then used for phylogenetic analyses.
Recombination and Selection Analysis.
We used CLONALFRAME (42) to infer recombination events and calculate r/m ratio within the deep-branching phylogeny. Pairwise dN/dS for concatenated orthologous CDSs was calculated using the method of Nei and Gojobori (43). We used site models M1a and M2a implemented in PAML (44) to identify genes under positive selection, and used individual gene trees built in RAxML (38) to correct for homologous recombination. M1a assumes neutral evolution, and M2a allows positive selection. Likelihoods from the two models were compared by a likelihood ratio test.
Estimates of Age and Population Size.
We used two methods to calculate the age of C. difficile. The first method follows the formula:
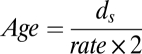 |
where ds is the mean synonymous substitutions per site calculated from concatenated nonrecombining core CDSs after Jukes-Cantor correction (45). The rate represents a synonymous molecular clock rate of 2.50 × 10−9–1.50 × 10−8 per site per year, which is equivalent to a universal mutation rate of 0.0001–0.0002 per genome per generation proposed by Ochman et al. (46). Here we assume 100–300 generations per year (47). As a second measure, we used BEAST (19) to estimate the age of the whole C. difficile collection. To obtain an independent estimate of the molecular clock rate, we identified orthologs between C. difficile and C. tetani, and calculated their sequence divergence following the model of Jukes-Cantor (45). The age of the Clostridium lineage was previously estimated to be 2.34 billion years (20), and we took this to be a maximum divergence time for these two species. This gave rise to a molecular clock rate of 1.15 × 10−10 per site per year. The population history of hypervirulent isolates was inferred using Bayesian skyline plot (22).
Supplementary Material
Acknowledgments
The authors thank Dale Gerding, Jon Brazier, Glenn Songer, and Denise Drudy for providing strains; Sanger Institute sequencing group and pathogen production staff for shotgun and sequence improvement; and Dr. Simon Harris for scientific discussions. We acknowledge Michelle Cairns for PCR-ribotype verification of sequenced isolates. This work was funded by the Wellcome Trust.
Footnotes
The authors declare no conflict of interest.
Data deposition: Draft sequences of complete Clostridium difficile genomes reported in this paper were deposited in the EMBL database (accession codes M68, FN668375; CF5, FN665652; M120, FN665653; BI-9, FN668944; BI-1, FN668941-FN668943; 2007855, FN545816). Solexa reads of hypervirulent isolates were deposited in the EMBL European Short Read Archive with accession numbers ERA000207 and ERA000208.
This article contains supporting information online at www.pnas.org/cgi/content/full/0914322107/DCSupplemental.
References
- 1.Lawley TD, et al. Proteomic and genomic characterization of highly infectious Clostridium difficile 630 spores. J Bacteriol. 2009;191:5377–5386. doi: 10.1128/JB.00597-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartlett JG, Chang TW, Gurwith M, Gorbach SL, Onderdonk AB. Antibiotic-associated pseudomembranous colitis due to toxin-producing clostridia. N Engl J Med. 1978;298:531–534. doi: 10.1056/NEJM197803092981003. [DOI] [PubMed] [Google Scholar]
- 3.Bartlett JG. Narrative review: The new epidemic of Clostridium difficile-associated enteric disease. Ann Intern Med. 2006;145:758–764. doi: 10.7326/0003-4819-145-10-200611210-00008. [DOI] [PubMed] [Google Scholar]
- 4.Cheknis AK, et al. Distribution of Clostridium difficile strains from a North American, European and Australian trial of treatment for C. difficile infections: 2005-2007. Anaerobe. 2009;15:230–233. doi: 10.1016/j.anaerobe.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Brazier JS, Patel B, Pearson A. Distribution of Clostridium difficile PCR ribotype 027 in British hospitals. Eur Surveill. 2007;12 doi: 10.2807/esw.12.17.03182-en. pii=3182. [DOI] [PubMed] [Google Scholar]
- 6.Brazier JS, et al. Distribution and antimicrobial susceptibility patterns of Clostridium difficile PCR ribotypes in English hospitals, 2007-08. Eur Surveill. 2008;13 doi: 10.2807/ese.13.41.19000-en. pii=19000. [DOI] [PubMed] [Google Scholar]
- 7.Loo VG, et al. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med. 2005;353:2442–2449. doi: 10.1056/NEJMoa051639. [DOI] [PubMed] [Google Scholar]
- 8.McDonald LC, et al. An epidemic, toxin gene-variant strain of Clostridium difficile. N Engl J Med. 2005;353:2433–2441. doi: 10.1056/NEJMoa051590. [DOI] [PubMed] [Google Scholar]
- 9.Joseph R, et al. First isolation of Clostridium difficile PCR ribotype 027, toxinotype III in Belgium. Euro Surveill. 2005;10:E051020–E051024. doi: 10.2807/esw.10.42.02815-en. [DOI] [PubMed] [Google Scholar]
- 10.Goorhuis A, et al. Spread and epidemiology of Clostridium difficile polymerase reaction ribotype 027/toxinotype III in The Netherlands. Clin Infect Dis. 2007;45:695–703. doi: 10.1086/520984. [DOI] [PubMed] [Google Scholar]
- 11.Drudy D, Harnedy N, Fanning S, Hannan M, Kyne L. Emergence and control of fluoroquinolone-resistant, toxin A-negative, toxin B-positive Clostridium difficile. Infect Control Hosp Epidemiol. 2007;28:932–940. doi: 10.1086/519181. [DOI] [PubMed] [Google Scholar]
- 12.Kim H, et al. Increasing prevalence of toxin A-negative, toxin B-positive isolates of Clostridium difficile in Korea: Impact on laboratory diagnosis. J Clin Microbiol. 2008;46:1116–1117. doi: 10.1128/JCM.01188-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang H, Fang H, Weintraub A, Nord CE. Distinct ribotypes and rates of antimicrobial drug resistance in Clostridium difficile from Shanghai and Stockholm. Clin Microbiol Infect. 2009;15:1170–1173. doi: 10.1111/j.1469-0691.2009.02992.x. [DOI] [PubMed] [Google Scholar]
- 14.Goorhuis A, et al. Emergence of Clostridium difficile infection due to a new hypervirulent strain, polymerase chain reaction ribotype 078. Clin Infect Dis. 2008;47:1162–1170. doi: 10.1086/592257. [DOI] [PubMed] [Google Scholar]
- 15.Johnson S, et al. Epidemics of diarrhea caused by a clindamycin-resistant strain of Clostridium difficile in four hospitals. N Engl J Med. 1999;341:1645–1651. doi: 10.1056/NEJM199911253412203. [DOI] [PubMed] [Google Scholar]
- 16.Stabler RA, et al. Comparative phylogenomics of Clostridium difficile reveals clade specificity and microevolution of hypervirulent strains. J Bacteriol. 2006;188:7297–7305. doi: 10.1128/JB.00664-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sebaihia M, et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet. 2006;38:779–786. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- 18.Holt KE, et al. High-throughput sequencing provides insights into genome variation and evolution in Salmonella Typhi. Nat Genet. 2008;40:987–993. doi: 10.1038/ng.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheridan PP, Freeman KH, Brenchley JE. Estimated minimal divergence times of the major bacterial and archaeal phyla. Geomicrobiol J. 2003;20:1–14. [Google Scholar]
- 21.Popoff MR, Rubin EJ, Gill DM, Boquet P. Actin-specific ADP-ribosyltransferase produced by a Clostridium difficile strain. Infect Immun. 1988;56:2299–2306. doi: 10.1128/iai.56.9.2299-2306.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 2005;22:1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- 23.Smith JM, Dowson CG, Spratt BG. Localized sex in bacteria. Nature. 1991;349:29–31. doi: 10.1038/349029a0. [DOI] [PubMed] [Google Scholar]
- 24.Brochet M, et al. Shaping a bacterial genome by large chromosomal replacements, the evolutionary history of Streptococcus agalactiae. Proc Natl Acad Sci USA. 2008;105:15961–15966. doi: 10.1073/pnas.0803654105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spratt BG, Maiden MC. Bacterial population genetics, evolution and epidemiology. Philos Trans R Soc Lond B Biol Sci. 1999;354:701–710. doi: 10.1098/rstb.1999.0423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guttman DS, Dykhuizen DE. Clonal divergence in Escherichia coli as a result of recombination, not mutation. Science. 1994;266:1380–1383. doi: 10.1126/science.7973728. [DOI] [PubMed] [Google Scholar]
- 27.Spratt BG, Hanage WP, Feil EJ. The relative contributions of recombination and point mutation to the diversification of bacterial clones. Curr Opin Microbiol. 2001;4:602–606. doi: 10.1016/s1369-5274(00)00257-5. [DOI] [PubMed] [Google Scholar]
- 28.Vos M, Didelot X. A comparison of homologous recombination rates in bacteria and archaea. ISME J. 2009;3:199–208. doi: 10.1038/ismej.2008.93. [DOI] [PubMed] [Google Scholar]
- 29.Rocha EP, et al. Comparisons of dN/dS are time dependent for closely related bacterial genomes. J Theor Biol. 2006;239:226–235. doi: 10.1016/j.jtbi.2005.08.037. [DOI] [PubMed] [Google Scholar]
- 30.Kryazhimskiy S, Plotkin JB. The population genetics of dN/dS. PLoS Genet. 2008;4:e1000304. doi: 10.1371/journal.pgen.1000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wren BW, Tabaqchali S. Restriction endonuclease DNA analysis of Clostridium difficile. J Clin Microbiol. 1987;25:2402–2404. doi: 10.1128/jcm.25.12.2402-2404.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurtz S, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Assefa S, Keane TM, Otto TD, Newbold C, Berriman M. ABACAS: Algorithm-based automatic contiguation of assembled sequences. Bioinformatics. 2009;25:1968–1969. doi: 10.1093/bioinformatics/btp347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–1858. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rutherford K, et al. Artemis: Sequence visualization and annotation. Bioinformatics. 2000;16:944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 36.Carver TJ, et al. ACT: The Artemis Comparison Tool. Bioinformatics. 2005;21:3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
- 37.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stamatakis A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 39.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 40.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 41.Pearson WR, Lipman DJ. Improved tools for biological sequence comparison. Proc Natl Acad Sci USA. 1988;85:2444–2448. doi: 10.1073/pnas.85.8.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Didelot X, Falush D. Inference of bacterial microevolution using multilocus sequence data. Genetics. 2007;175:1251–1266. doi: 10.1534/genetics.106.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 44.Yang Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 45.Jukes TH, Cantor CR. Evolution of protein molecules. Mammalian protein metabolism. 1969;3:21–132. [Google Scholar]
- 46.Ochman H, Elwyn S, Moran NA. Calibrating bacterial evolution. Proc Natl Acad Sci USA. 1999;96:12638–12643. doi: 10.1073/pnas.96.22.12638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gibbons RJ, Kapsimalis B. Estimates of the overall rate of growth of the intestinal microflora of hamsters, guinea pigs, and mice. J Bacteriol. 1967;93:510–512. doi: 10.1128/jb.93.1.510-512.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.