

Abstract
Polyubiquitin chains mediate a variety of biological processes, ranging from proteasomal targeting to inflammatory signaling and DNA repair. Their functional diversity is in part due to their ability to adopt distinct conformations, depending on how the ubiquitin moieties within the chain are linked. We have used the eukaryotic replication clamp PCNA, a natural target of lysine (K)63-linked polyubiquitylation, as a model substrate to directly compare the consequences of modification by different types of polyubiquitin chains. We show here that K63-polyubiquitylated PCNA is not subject to proteasomal degradation. In contrast, linear, noncleavable ubiquitin chains do not promote DNA damage tolerance, but function as general degradation signals. We find that a linear tetraubiquitin chain is sufficient to afford proteasomal targeting through the Cdc48-Npl4-Ufd1 complex without further modification. Although a minimum chain length of four is required for degradation, a longer chain does not further reduce the half-life of the respective substrate protein. Our results suggest that the cellular machinery responsible for recognition of ubiquitylated substrates can make subtle distinctions between highly similar forms of the polyubiquitin signal.
Keywords: DNA damage bypass, polyubiquitin chain linkage, proteasome, protein degradation, UFD pathway
Ubiquitin belongs to a family of posttranslational modifiers that alter the properties of their targets in various ways, usually by affecting their interactions, localization, or stability. Although best known for its role in regulated protein degradation (1), ubiquitin mediates a variety of nonproteolytic functions (2). By means of an enzymatic cascade involving an activating enzyme (E1), a conjugating enzyme (E2), and a ligase (E3) that determines substrate selectivity, ubiquitin is generally attached to its targets through an isopeptide linkage between the modifier’s carboxy (C) terminus and the ϵ-amino group of a lysine (K) residue within the target (1). Its versatility as a signaling molecule is at least in part due to its ability to form polymeric chains. These can adopt a number of different geometries, depending on which of the seven lysines of ubiquitin is used as an acceptor for chain formation (3). Downstream effector proteins that selectively recognize a particular type of chain are believed to mediate the outcome of the modification.
A polyubiquitin chain whose monomers are linked through K48 acts as a signal for degradation by the 26S proteasome (4). K29-linked polyubiquitin chains also mediate degradation, as shown for the ubiquitin fusion degradation (UFD) pathway in yeast, which recognizes a single, noncleavable ubiquitin moiety at the N terminus of a target protein as a substrate for further modification (5). However, the short K29-linked chains assembled by the UFD-specific E3 Ufd4 are relatively inefficient in proteasomal targeting and are therefore extended via K48-linkage by a dedicated enzyme, Ufd2, also called E4 (6, 7). Downstream factors responsible for the recognition of the polyubiquitin chain and the targeting of the modified substrate to the proteasome include the escort factors Cdc48, Npl4, and Ufd1 and ubiquitin adaptors such as Rad23 and/or Dsk2 (8).
K63-linked polyubiquitin chains assembled by the heterodimeric E2 complex of Ubc13 and the E2-like Uev1/Mms2 feature prominently in the NFκB-dependent inflammatory response (9) and also in a system of DNA damage bypass known as the RAD6 pathway (10). Here, the relevant modification target is the eukaryotic sliding clamp PCNA, a processivity factor for replicative DNA polymerases (11). Damage-induced monoubiquitylation promotes the recruitment of damage-tolerant DNA polymerases for a process named translesion synthesis (TLS) (12, 13). In contrast, modification by a K63-linked polyubiquitin chain activates an alternative pathway of damage avoidance that allows cells to overcome replication-blocking lesions in the template strand in an error-free manner, possibly involving a template switch (11, 14). The mechanism by which the K63-linked ubiquitin chains act remains unknown. In the context of NFκB activation, K63-polyubiquitylation is unrelated to proteolysis, as the chains appear to act as scaffolds for the assembly of a signaling complex, but a proteolytic function has not been excluded for the damage tolerance pathway. When linked to a model substrate in vitro, K63-linked polyubiquitin chains in fact trigger proteasomal degradation (15), and recent evidence suggests that they may also function as a degradation signal in vivo (16).
The picture of ubiquitin chain linkage is further complicated by the recent discovery of an E3 in higher eukaryotes, LUBAC, which catalyzes the assembly of linear chains where the ubiquitin moieties are linked in a tandem arrangement via ubiquitin’s amino (N) terminus (17). As the latter is spacially very close to K63, linear chains adopt a conformation almost identical to that of K63-linked chains (18). Yet, although LUBAC was found to be important for NFκB signaling as well (19), the function of the linear chains does not coincide with that of the K63-linked chains, consistent with subtle differences in their recognition by the ubiquitin-binding (UBAN) domain of the Iκ kinase subunit NEMO (18, 20). Moreover, LUBAC can promote the degradation of a model substrate in vivo, indicating a possible function of linear chains in proteasomal targeting (17).
These observations raise the general questions as to what extent linear and K63-linked polyubiquitin chains are interchangeable in their functions, and whether or not they act as degradation signals. PCNA as a natural target of K63-linked polyubiquitylation has provided us with the unique opportunity of directly comparing the effects of linear versus K63-linked polyubiquitylation of a common substrate. Our results indicate that—similar to the NFκB pathway—the system of DNA damage bypass is able to differentiate between linear and K63-linked polyubiquitin chains.
Results
Linear Polyubiquitin Chains Do Not Promote DNA Damage Tolerance.
In order to directly compare the effects of linear versus K63-linked chains on a common substrate, we designed linear fusions of polyubiquitin arrays to the N or C terminus of PCNA (Fig. 1A). We had previously shown that a single ubiquitin fused to PCNA successfully complements a defect in monoubiquitylation at the native site, K164, indicating that the position of ubiquitin on PCNA is not critical for function in TLS (21). In order to allow for some conformational flexibility we designed a series of constructs containing two to four ubiquitin repeats separated by a short linker ( ), and two constructs in which four ubiquitin moieties were joined precisely in a head-to-tail arrangement (
), and two constructs in which four ubiquitin moieties were joined precisely in a head-to-tail arrangement ( and
and  ). In order to prevent further modification, the major acceptor sites for ubiquitin and/or the small ubiquitin-related modifier (SUMO) on PCNA (K164 and K127) and ubiquitin (K29, K48, and K63) were mutated to arginine (indicated by an asterisk in our notation), and disassembly of the chains was prevented by mutation of the C-terminal glycine of each ubiquitin to valine.
). In order to prevent further modification, the major acceptor sites for ubiquitin and/or the small ubiquitin-related modifier (SUMO) on PCNA (K164 and K127) and ubiquitin (K29, K48, and K63) were mutated to arginine (indicated by an asterisk in our notation), and disassembly of the chains was prevented by mutation of the C-terminal glycine of each ubiquitin to valine.
Fig. 1.

Linear noncleavable polyubiquitin chains on PCNA cannot substitute for the K63-linked modification in DNA damage bypass. (A) Schematic view of the linear ubiquitin-PCNA fusion constructs used in this study. Mutations in the open reading frames of ubiquitin and PCNA are indicated only once; the mutant versions are represented as Ub∗ and PCNA∗, respectively. Sequences of linker peptides are shown below the constructs, and symbols correspond to those used in B and C. (B) Linear tetraubiquitin fusions to PCNA rescue the UV sensitivity of rad18 cells to different extents. UV sensitivities were determined for rad18 cells bearing the indicated constructs. (C) The number of ubiquitin units fused to PCNA∗ does not affect the extent of rescue. UV survival assays were carried out as in (B). diamond shape: WT; square shape: rad18; triangle shape: rad18+vector; circle shape: rad18+PCNA∗; square with an “x”inside: rad18 ; solid black triangle: rad18+Ub∗-PCNA∗; solid black circle: rad18
; solid black triangle: rad18+Ub∗-PCNA∗; solid black circle: rad18 ; solid black diamond: rad18+Ub∗-PCNA∗; solid black square: rad18
; solid black diamond: rad18+Ub∗-PCNA∗; solid black square: rad18 ; upside down solid black triangle: rad18
; upside down solid black triangle: rad18 ; *: rad18+UbK63∗-PCNA∗.
; *: rad18+UbK63∗-PCNA∗.
The constructs were expressed from the POL30 promoter in a rad18 strain, which is unable to ubiquitylate endogenous PCNA, and the resulting strains were tested for sensitivity to UV radiation and the alkylating agent methyl methanesulfonate (MMS).  suppressed the damage sensitivity of rad18 cells to some degree (Fig. 1B and Fig. S1A). In contrast, the linkerless versions,
suppressed the damage sensitivity of rad18 cells to some degree (Fig. 1B and Fig. S1A). In contrast, the linkerless versions,  and
and  , did not afford significant rescue beyond the effect of PCNA∗ alone (Fig. 1B and Fig. S1B). Interestingly, all of the linker-bearing fusions,
, did not afford significant rescue beyond the effect of PCNA∗ alone (Fig. 1B and Fig. S1B). Interestingly, all of the linker-bearing fusions,  ,
,  and
and  , conferred damage sensitivities identical to that of the “monoubiquitylated” version, Ub∗-PCNA∗ (Fig. 1C and Fig. S1A). Moreover, rescue of viability by these constructs was completely dependent on the presence of the TLS polymerases, as none of them had any effect on the sensitivity of a rad18 ΔTLS strain, carrying deletions of the genes encoding the budding yeast damage-tolerant polymerases (Fig. S1A). We therefore conclude that the rescue observed with
, conferred damage sensitivities identical to that of the “monoubiquitylated” version, Ub∗-PCNA∗ (Fig. 1C and Fig. S1A). Moreover, rescue of viability by these constructs was completely dependent on the presence of the TLS polymerases, as none of them had any effect on the sensitivity of a rad18 ΔTLS strain, carrying deletions of the genes encoding the budding yeast damage-tolerant polymerases (Fig. S1A). We therefore conclude that the rescue observed with  —as with the monoubiquitin fusion—was due to TLS rather than error-free damage bypass. The failure of the linkerless constructs to support TLS is intriguing, as it might indicate a steric obstruction of the interaction site for the damage-tolerant polymerases on the PCNA-proximal ubiquitin moiety by the head-to-tail linkage.
—as with the monoubiquitin fusion—was due to TLS rather than error-free damage bypass. The failure of the linkerless constructs to support TLS is intriguing, as it might indicate a steric obstruction of the interaction site for the damage-tolerant polymerases on the PCNA-proximal ubiquitin moiety by the head-to-tail linkage.
In order to exclude the possibility that a nonphysiological location of the ubiquitin chain prevented its function in damage bypass, we generated a variant of Ub∗-PCNA∗, named UbK63∗-PCNA∗ (Fig. 1A). In vitro, this arrangement permits polyubiquitin chain formation on K63 of the ubiquitin moiety (22). In a rad18 strain, we observed a suppression of the damage sensitivity that was largely independent of TLS and exceeded the effect of Ub∗-PCNA∗ considerably (Fig. 1C and Fig. S1C), indicating that polyubiquitin-dependent damage bypass was functional, even though the chains were attached to the N terminus of PCNA. Given that none of the linear constructs was able to support error-free damage bypass, these data suggest that linear and K63-linked polyubiquitin chains are functionally distinct.
Linear Polyubiquitin Chains Target PCNA for Proteasomal Degradation.
On Western blots we noticed a dramatic reduction in the abundance of all fusion proteins bearing tetraubiquitin chains when compared with the shorter versions or endogenous PCNA (Fig. 2A and Fig. S2A). The effect was not due to reduced mRNA levels of the corresponding constructs (Fig. S2B). Given the notion that the mimimum length of a K48-polyubiquitin chain for efficient recognition by the 26S proteasome is four ubiquitin moieties (23), we hypothesized that the linear tetrameric chains might act as proteasomal degradation signals. In support of this model, recombinant  , but not native PCNA∗, was degraded by purified 26S proteasome in vitro (Fig. S3A). In vivo we observed increased steady-state levels of the tetraubiquitin fusions in pre1-1 proteasome mutants (Fig. 2B). Moreover, chase experiments with the translation inhibitor cycloheximide indicated that they were degraded in WT cells, but stabilized in the pre1-1 mutant (Fig. 2C). As purified
, but not native PCNA∗, was degraded by purified 26S proteasome in vitro (Fig. S3A). In vivo we observed increased steady-state levels of the tetraubiquitin fusions in pre1-1 proteasome mutants (Fig. 2B). Moreover, chase experiments with the translation inhibitor cycloheximide indicated that they were degraded in WT cells, but stabilized in the pre1-1 mutant (Fig. 2C). As purified 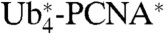 readily formed trimers (Fig. S3B), misfolding was unlikely to be the cause of instability. Hence, these findings suggest that linear polyubiquitin chains of sufficient length on PCNA act as proteasomal degradation signals. Intriguingly, however, the rates of proteolysis varied considerably between the three constructs and were hardly comparable to those of some short-lived endogenous proteins or model substrates (1, 5).
readily formed trimers (Fig. S3B), misfolding was unlikely to be the cause of instability. Hence, these findings suggest that linear polyubiquitin chains of sufficient length on PCNA act as proteasomal degradation signals. Intriguingly, however, the rates of proteolysis varied considerably between the three constructs and were hardly comparable to those of some short-lived endogenous proteins or model substrates (1, 5).
Fig. 2.

Linear noncleavable tetraubiquitin chains target PCNA for degradation by the 26S proteasome. Protein levels of the ubiquitin-PCNA fusion constructs and endogenous PCNA were compared by Western blot with an anti-PCNA antibody. (A) Linear tetraubiquitin chains destabilize the respective fusion proteins. (B) Steady-state protein levels of tetraubiquitin fusions to PCNA are increased in a proteasome mutant. (C) Cycloheximide chase experiments show the degradation of the tetraubiquitin fusion proteins in PRE1 cells and their stabilization in pre1-1. Exponential cultures were treated with 100 μg/mL cycloheximide to inhibit de novo protein synthesis, and samples corresponding to equal culture volumes were processed for Western blot analysis at the indicated time points.
K63-Polyubiquitylation Does Not Target PCNA for Degradation.
Polyubiquitylation of PCNA by K63-linked chains, triggered by conditions of DNA damage, does not result in an obvious decrease in total cellular levels of PCNA (11). However, as the fraction of PCNA ubiquitylated at any given time is exceedingly small, this does not necessarily indicate a nondegradative function of the modification. In fact, proteasome mutants were previously reported to cause DNA damage sensitivity and exhibit an epistatic relationship with mutants in the RAD6 pathway, thus possibly linking proteasome activity to PCNA modification (24). We were therefore interested to directly assess the fate of K63-polyubiquitylated PCNA. As expected, we observed an accumulation of total ubiquitin conjugates in cell extracts of cultures treated with the proteasome inhibitor MG132 or mutated in the genes encoding the proteasome maturation factor Ump1 or the catalytic subunit Pre1 (Fig. 3 A and B). In contrast, the levels of polyubiquitylated PCNA, observable after treatment with the DNA-damaging agent MMS, were not increased, but rather reduced upon attenuation of proteasome activity, and no high molecular weight species accumulated that could have indicated a conversion to longer chains (Fig. 3 C and D). Therefore, it appears that polyubiquitylated PCNA is not normally a substrate of the proteasome. Instead, the reduction in the amount of ubiquitylated PCNA in the presence of MG132 and in the proteasome mutants is likely due to the depletion of free ubiquitin that results from a lack of recycling (25).
Fig. 3.

K63-polyubiquitylated PCNA is not a target of proteasomal degradation. (A) Inhibition of the proteasome by the chemical inhibitor MG132 causes an accumulation of total ubiquitin conjugates. Exponential cultures of HISPOL30 pdr5 cells were treated with 50 μM MG132 for 2 h where indicated, and ubiquitylated species were detected in total extracts by Western blots with an anti-ubiquitin antibody. Detection of phosphoglycerate kinase served as loading control. (B) Mutants with attenuated proteasome activity accumulate total ubiquitin conjugates. Extracts were prepared from the indicated strains and probed as in (A). (C) Damage-induced ubiquitylation of PCNA is reduced upon chemical inhibition of the proteasome. HisPCNA was isolated by denaturing Ni-NTA pull-down from extracts of HISPOL30 pdr5 cells treated with 50 μM MG132 for 2 h and 0.02% MMS for 90 min where indicated, and Western blots were developed with anti-PCNA and anti-ubiquitin antibodies. (D) Damage-induced ubiquitylation of PCNA is reduced in mutants affecting proteasome activity. HisPCNA and its ubiquitylated forms were isolated from the indicated strains and detected as in (C). The high-molecular weight signals in (C) and (D) marked with an asterisk are due to nonspecific isolation of ubiquitin conjugates; they are neither PCNA-reactive nor damage-dependent.
A Linear Polyubiquitin Chain Acts as a General Degradation Signal.
In order to generalize our results and exclude potential PCNA-specific effects on the turnover rate elicited by linear ubiquitin chains, we constructed an analogous fusion of the head-to-tail tetraubiquitin chain to the N terminus of β-galactosidase ( -βGal, Fig. 4A), a model proteasome substrate whose degradation pattern has been studied in detail (26, 5). Whereas the protein by itself (βGal) is stable in yeast cells, fusion of a single, noncleavable ubiquitin moiety to its N terminus (Ub-βGal, Fig. 4A) renders it extremely short-lived (26). Hence, comparing the stability of
-βGal, Fig. 4A), a model proteasome substrate whose degradation pattern has been studied in detail (26, 5). Whereas the protein by itself (βGal) is stable in yeast cells, fusion of a single, noncleavable ubiquitin moiety to its N terminus (Ub-βGal, Fig. 4A) renders it extremely short-lived (26). Hence, comparing the stability of  -βGal with Ub-βGal and βGal should allow an estimation of the efficiency of the linear tetraubiquitin chain as a general degradation signal. Northern blots indicated that the expression levels of all three constructs were comparable (Fig. 4B). However, cycloheximide chase experiments revealed remarkable differences in protein stability (Fig. 4C). Whereas Ub-βGal was degraded within minutes, the levels of
-βGal with Ub-βGal and βGal should allow an estimation of the efficiency of the linear tetraubiquitin chain as a general degradation signal. Northern blots indicated that the expression levels of all three constructs were comparable (Fig. 4B). However, cycloheximide chase experiments revealed remarkable differences in protein stability (Fig. 4C). Whereas Ub-βGal was degraded within minutes, the levels of 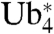 -βGal dropped appreciably over the course of several hours, comparable to the degradation kinetics of the analogous
-βGal dropped appreciably over the course of several hours, comparable to the degradation kinetics of the analogous  . Unmodified βGal remained stable over the course of the entire experiment, and mutation of K29, K48, and K63 in the ubiquitin moiety of Ub-βGal also afforded complete stabilization (Fig. S4). As expected, both Ub-βGal and
. Unmodified βGal remained stable over the course of the entire experiment, and mutation of K29, K48, and K63 in the ubiquitin moiety of Ub-βGal also afforded complete stabilization (Fig. S4). As expected, both Ub-βGal and  -βGal were stabilized in a cim3 mutant, indicating that their degradation is mediated by the proteasome (Fig. 4C). Thus, the linear noncleavable tetraubiquitin chain serves as a general, but relatively inefficient proteasomal degradation signal.
-βGal were stabilized in a cim3 mutant, indicating that their degradation is mediated by the proteasome (Fig. 4C). Thus, the linear noncleavable tetraubiquitin chain serves as a general, but relatively inefficient proteasomal degradation signal.
Fig. 4.

A linear noncleavable tetraubiquitin chain acts as a general, but inefficient degradation signal. (A) Schematic view of the βGal constructs used in this study. The asterisk denotes the ubiquitin mutant (K29/48/63R, G76V). Ub-βGal was originally described as UbV76-V-eΔK-βgal (41). (B) Northern blot analysis indicates similar expression levels of all three βGal constructs upon induction with galactose (GAL). (C)  -βGal and Ub-βGal are degraded by the 26S proteasome with distinct kinetics. After growth in galactose medium for 2 h, a promoter shut-off (by shift to glucose) was combined with a cycloheximide chase (100 μg/mL) to inhibit de novo protein synthesis in the indicated strains, and samples were processed as in Fig. 2C. The βGal construct served as a stable control protein. Lanes labeled “-“ represent samples from cultures grown in glucose medium. Note that degradation of Ub-βGal produces a stable fragment of ca. 90 kDa (26).
-βGal and Ub-βGal are degraded by the 26S proteasome with distinct kinetics. After growth in galactose medium for 2 h, a promoter shut-off (by shift to glucose) was combined with a cycloheximide chase (100 μg/mL) to inhibit de novo protein synthesis in the indicated strains, and samples were processed as in Fig. 2C. The βGal construct served as a stable control protein. Lanes labeled “-“ represent samples from cultures grown in glucose medium. Note that degradation of Ub-βGal produces a stable fragment of ca. 90 kDa (26).
Substrates Marked by Linear Polyubiquitin Chains Are Targeted to the Proteasome by Components of the UFD Pathway.
Given the similarity between the  -βGal construct and a polyubiquitylated UFD substrate, we asked whether degradation of the former would require factors of the UFD pathway. Ufd4, the E3 responsible for the initial modification of Ub-βGal, was found to be dispensable for
-βGal construct and a polyubiquitylated UFD substrate, we asked whether degradation of the former would require factors of the UFD pathway. Ufd4, the E3 responsible for the initial modification of Ub-βGal, was found to be dispensable for  -βGal degradation (Fig. S5A), consistent with the absence of higher modified forms of the fusion protein in WT cells or proteasome mutants (Fig. 4C and Fig. S5). Interestingly, the construct was stabilized in ufd2Δ cells (Fig. S5B). However, a C terminally truncated allele lacking the catalytic U box restored degradation (Fig. S5C), indicating that the requirement for Ufd2 was not due to its E4 activity, but rather to its function in stabilizing the association of the Cdc48-Ufd1-Npl4 complex with the ubiquitin adaptors Rad23 and/or Dsk2 (8). Accumulation of
-βGal degradation (Fig. S5A), consistent with the absence of higher modified forms of the fusion protein in WT cells or proteasome mutants (Fig. 4C and Fig. S5). Interestingly, the construct was stabilized in ufd2Δ cells (Fig. S5B). However, a C terminally truncated allele lacking the catalytic U box restored degradation (Fig. S5C), indicating that the requirement for Ufd2 was not due to its E4 activity, but rather to its function in stabilizing the association of the Cdc48-Ufd1-Npl4 complex with the ubiquitin adaptors Rad23 and/or Dsk2 (8). Accumulation of  -βGal in cdc48-2, npl4-1, and a rad23 dsk2 double mutant indeed suggested that the linear chains are recognized in a manner very similar to K48-linked polyubiquitin chains (Fig. S5 D–F). In summary, it appears that the pathway by which the
-βGal in cdc48-2, npl4-1, and a rad23 dsk2 double mutant indeed suggested that the linear chains are recognized in a manner very similar to K48-linked polyubiquitin chains (Fig. S5 D–F). In summary, it appears that the pathway by which the  -βGal construct is targeted to the proteasome largely overlaps with the UFD pathway, with the notable exception of the initial polyubiquitin chain assembly by E3 and E4. Stabilization of the analogous
-βGal construct is targeted to the proteasome largely overlaps with the UFD pathway, with the notable exception of the initial polyubiquitin chain assembly by E3 and E4. Stabilization of the analogous 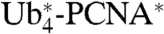 in the npl4-1 mutant indicated processing by the same pathway (Fig. S6).
in the npl4-1 mutant indicated processing by the same pathway (Fig. S6).
Linear Polyubiquitin Chain Length Is Not a Limiting Factor for Degradation.
Our data raised the possibility that the relatively low rate of degradation of our fusion proteins was due to an inefficient recognition of the linear tetraubiquitin chain by the proteasome or its targeting factors. Considering that a K48-linked chain of four ubiquitin units was found to function as the minimal recognition signal for efficient proteasomal degradation (23), we therefore asked whether an increase in chain length could compensate for a poor recognition and thus accelerate the degradation of the model substrate βGal. We duplicated the  module at the N terminus of the protein to generate
module at the N terminus of the protein to generate 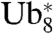 -βGal (Fig. 5A). Degradation of this construct, however, was comparable to that of
-βGal (Fig. 5A). Degradation of this construct, however, was comparable to that of  -βGal (Fig. 5B), indicating that chain length is not a limiting factor for degradation in our system.
-βGal (Fig. 5B), indicating that chain length is not a limiting factor for degradation in our system.
Fig. 5.

Ubiquitin chain length is not a limiting factor in the degradation of linear ubiquitin fusions. (A) Schematic view of a  -βGal construct. Note that each of the
-βGal construct. Note that each of the  modules used to create the octaubiquitin chain is identical to that used in
modules used to create the octaubiquitin chain is identical to that used in  -βGal. (B)
-βGal. (B)  -βGal is degraded at a rate comparable to that of
-βGal is degraded at a rate comparable to that of 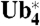 -βGal. Promoter shut-off/cycloheximide chase experiments were performed with the two constructs in a WT strain as described in Fig. 4C.
-βGal. Promoter shut-off/cycloheximide chase experiments were performed with the two constructs in a WT strain as described in Fig. 4C.
Discussion
Why do Linear Chains Not Function in Damage Bypass?
A linear arrangement of ubiquitin molecules adopts an extended conformation identical to that of a K63-linked chain, and many ubiquitin-binding domains make no distinction between linear and K63-linked chains (18). Nevertheless, our fusions of linear tetraubiquitin to PCNA did not rescue a PCNA polyubiquitylation defect in vivo, despite the notion that a K63-chain attached to PCNA’s N terminus is functional. We can envision several reasons for the failure of these constructs to promote damage tolerance. First, it is formally possible that the instability of the fusion proteins prevents efficient error-free damage bypass, although we consider this unlikely because the  construct is active in TLS, and the levels of physiologically K63-modified PCNA are naturally very low. Second, we cannot exclude that the noncleavable nature of the chain interferes with correct function. This may apply if the deubiquitylation step is physiologically relevant for error-free damage bypass—even though removal of monoubiquitin from PCNA is not required for TLS (21). Finally, the linear chains might not be recognized by a K63-selective downstream effector protein that mediates the error-free bypass pathway. On one hand, although the
construct is active in TLS, and the levels of physiologically K63-modified PCNA are naturally very low. Second, we cannot exclude that the noncleavable nature of the chain interferes with correct function. This may apply if the deubiquitylation step is physiologically relevant for error-free damage bypass—even though removal of monoubiquitin from PCNA is not required for TLS (21). Finally, the linear chains might not be recognized by a K63-selective downstream effector protein that mediates the error-free bypass pathway. On one hand, although the  array was bound by the UBAN domain of NEMO (Fig. S7), which is highly selective for linear chains (18, 20), the G76V mutation might interfere with the recognition by a PCNA-specific ubiquitin receptor. On the other hand, there are ubiquitin-binding domains that bind exclusively to K63-linked chains, such as the C-terminal NZF domain of TAB2 (18, 27). At present it is difficult to distinguish between these possibilities, as replacement of our chains with a cleavable version would result in its processing to monoubiquitylated or unmodified PCNA, and characterization of a downstream effector will have to await its identification.
array was bound by the UBAN domain of NEMO (Fig. S7), which is highly selective for linear chains (18, 20), the G76V mutation might interfere with the recognition by a PCNA-specific ubiquitin receptor. On the other hand, there are ubiquitin-binding domains that bind exclusively to K63-linked chains, such as the C-terminal NZF domain of TAB2 (18, 27). At present it is difficult to distinguish between these possibilities, as replacement of our chains with a cleavable version would result in its processing to monoubiquitylated or unmodified PCNA, and characterization of a downstream effector will have to await its identification.
Why Is K63-Polyubiquitylated PCNA Not Degraded?
Genetic data linking DNA damage bypass to proteasome activity have been indirect and ambiguous. While Hofmann and Pickart (15) used a lack of synergism between pre1-1 pre2-2 and rev3 mutants with respect to UV sensitivity as an argument against proteolytic function in error-free damage tolerance, others postulated a role of the proteasome in limiting the mutagenic activity of TLS, based on epistasis and mutation rate analysis (24, 28). We have finally directly assessed the response of polyubiquitylated PCNA to variations in proteasome activity and find no evidence for a degradation of the modified clamp. Our findings instead reflect the global behavior of K63-linked chains, which do not accumulate upon inhibition of the proteasome (29). Hence, the recent finding that proteasome-dependent processing of the transcription factor Mga2 occurs after K63-ubiquitylation by the E3 Rsp5 remains an isolated incident of K63-mediated proteolysis (16). In that study, contributions of K48-linked chains or chain editing by means of Rsp5-associated deubiquitylating activity were not rigorously excluded. Yet, K63-linked chains bind the proteasome with similar efficiency as K48-linked chains (15, 30), and little selectivity was observed in their affinities for the ubiquitin adaptors Rad23 and Dsk2 (31). The most straightforward explanation for the inefficiency of K63-linked chains as a degradation signal on PCNA may therefore be an insufficient chain length. Whereas the minimal number of ubiquitin moieties in a K48-linked chain required for efficient recognition by the proteasome was shown to be four (23), PCNA modifications exceeding the tetraubiquitylated state are undetectable in vivo, and even the latter is much less abundant than the mono- and diubiquitylated forms (32). It remains to be determined how chain length is limited in vivo, but the use of deubiquitylating enzymes such as the mammalian Usp1 (33) may represent an effective strategy for the evasion of degradation.
Linear Polyubiquitin Chains as Degradation Signals.
In vitro, the 26S proteasome is not particularly selective in the recognition of ubiquitylated proteins (15, 34). It is therefore not surprising that linear ubiquitin chains competitively inhibit degradation of K48-polyubiquitylated substrates (23), and a linear noncleavable tetraubiquitin chain fused to a model protein can elicit the degradation of its fusion partner (35). However, little is known about the suitability of linear chains as degradation signals in vivo. Noncleavable tandem arrays of 2–8 ubiquitin units were shown to confer half-lives of less than 10 min to their fusion partners in reticulocyte lysates and cell culture (35, 36). When overexpressed in yeast, they effectively inhibit the proteasomal degradation of short-lived proteins (37). However, extensive modification by further ubiquitylation was noted in these cases, suggesting that the arrays mainly serve as efficient ubiquitin acceptors. In our system, further modification of the ubiquitin moieties, for example via K11, is unlikely, as this should also affect the shorter chains to at least some degree. Yet, instability was observed only for those constructs bearing at least four ubiquitin moieties, and previous reports had demonstrated chain extension via K29 and K48 for UFD substrates (5–7). Thus, a linear tetraubiquitin chain appears to be sufficient to induce proteasomal degradation in vivo.
LUBAC, an E3 that catalyzes linear polyubiquitylation, destabilizes a fusion of ubiquitin to the green fluorescent protein (GFP) in mammalian cell culture when overexpressed (17). At the same time, however, linear chains attached by LUBAC to K285 and/or K309 of NEMO apparently do not promote degradation (19), suggesting that the positioning of the chain on the target may affect its efficiency as a degradation signal. Similarly, we found that turnover rates varied significantly between  and
and  , and degradation of
, and degradation of  -βGal was quite inefficient compared to an analogous UFD substrate. These observations initially suggested that inefficient recognition of the short linear chains by the proteasome might be responsible for the slow turnover. However, extension of the ubiquitin module to eight units did not accelerate degradation, and although linear chains are somewhat less effective at competing for proteasome binding than K48-linked chains (23), association of linear noncleavable tetraubiquitin with the proteasome had been observed in vivo (37). Taken together, these data therefore imply that inefficient processing rather than targeting is responsible for the slow degradation. This scenario is supported by the notion that the proteasome-associated isopeptidase Rpn11 positively contributes to proteolysis in vivo, presumably by removing polyubiquitin chains en bloc from substrates as they enter the channel into the proteasome (38, 39). In our system, the noncleavable tandem ubiquitin array needs to be unfolded and degraded along with the substrate. Considering the tightly folded structure of ubiquitin, this may present a barrier for proteolysis, in particular as substrate unfolding is known to affect degradation rates in vitro (23, 40). Alternatively, prolonged association of the tetraubiquitin module with ubiquitin receptors at the proteasome lid might delay entry of the substrate moiety into the catalytic cavity. In either case, variation of the ubiquitin attachment site might change the way in which the substrate is presented to the proteolytic core, thus ultimately affecting degradation rates.
-βGal was quite inefficient compared to an analogous UFD substrate. These observations initially suggested that inefficient recognition of the short linear chains by the proteasome might be responsible for the slow turnover. However, extension of the ubiquitin module to eight units did not accelerate degradation, and although linear chains are somewhat less effective at competing for proteasome binding than K48-linked chains (23), association of linear noncleavable tetraubiquitin with the proteasome had been observed in vivo (37). Taken together, these data therefore imply that inefficient processing rather than targeting is responsible for the slow degradation. This scenario is supported by the notion that the proteasome-associated isopeptidase Rpn11 positively contributes to proteolysis in vivo, presumably by removing polyubiquitin chains en bloc from substrates as they enter the channel into the proteasome (38, 39). In our system, the noncleavable tandem ubiquitin array needs to be unfolded and degraded along with the substrate. Considering the tightly folded structure of ubiquitin, this may present a barrier for proteolysis, in particular as substrate unfolding is known to affect degradation rates in vitro (23, 40). Alternatively, prolonged association of the tetraubiquitin module with ubiquitin receptors at the proteasome lid might delay entry of the substrate moiety into the catalytic cavity. In either case, variation of the ubiquitin attachment site might change the way in which the substrate is presented to the proteolytic core, thus ultimately affecting degradation rates.
Whether linear polyubiquitin chains naturally act as degradation signals remains an open question. In higher eukaryotes, dedicated ubiquitin binding domains specific for linear chains might shield these from recognition by proteasomal targeting factors, but as linear chains have not been detected in budding yeast (29), this organism might lack relevant receptors, thus resulting in proteasomal targeting through a lack of suitable downstream effectors.
Outlook.
The distinct fates of PCNA modified by linear versus K63-linked polyubiquitin chains highlight the complexity of ubiquitin signaling that has emerged from numerous recent studies. Taken together, they indicate that not only the linkage of a polyubiquitin chain, but also its length, its position on the substrate and the capability to be edited and processed may determine the outcome of the modification. It is likely that the relevant downstream effector proteins responsible for the recognition of a particular chain in the context of its substrate will exhibit distinct interaction properties and will have to be considered individually in order to explain the choice of biological pathway dictated by polyubiquitylation.
Materials and Methods
Yeast Strains and Plasmids.
Standard procedures were followed for the growth and manipulation of Saccharomyces cerevisiae. A list of strains is given in the Supplemental Information (Table S1). Experiments involving the proteasome inhibitor MG132 were carried out in a pdr5 deletion. Temperature-sensitive mutants were pregrown at 25 °C, but experiments addressing protein stability were performed at 30 °C. Plasmids encoding linear fusions of ubiquitin to the pol30(K127/164R) open reading frame were derived from constructs described previously (21), and fusions to βGal from a galactose-inducible construct originally called UbV76-V-eΔK-βgal, obtained from E. Johnson (41). Details about their construction are given in the Supplemental Information. Fusion proteins were detected by Western blot with a polyclonal anti-PCNA (42) or a monoclonal anti-βGal antibody (Promega), respectively.
Detection of Ubiquitin Conjugates.
Total ubiquitin conjugates in denatured cell extracts, prepared as described (43), were detected by Western blots using a monoclonal ubiquitin-specific antibody, P4D1 (Cell Signaling Technologies). Damage-induced ubiquitylation of PCNA was detected by denaturing Ni-NTA affinity chromatography and Western blot analysis as described previously, using PCNA- and ubiquitin-specific antibodies (44). Cells were treated with 0.02% MMS for 90 min to induce the modification. For inhibition of the proteasome, MG132 (50 μM) was added 2 h before inducing DNA damage.
Determination of UV Sensitivities.
UV sensitivities were determined by plating defined numbers of cells from exponential cultures onto YPD medium, irradiation at 254 nm in a UV crosslinker (Stratalinker 2400, Stratagene), incubation in the dark for 3 days, and colony counting. Graphs represent averages and standard deviations of triplicate experiments.
Northern Blot Analysis.
Total RNA was extracted from yeast cultures using an RNeasy mini kit (Qiagen). RNA samples were separated on agarose gels in a buffer containing 30 mM Bis-Tris, 10 mM Pipes, 1 mM EDTA, pH ∼ 6.7, after denaturation by glyoxal. Blots were hybridized with a 464 bp βGal-specific probe generated from a polymerase chain reaction (PCR) product by labeling with Ready-To-Go DNA labeling beads (GE Healthcare). Hybridization was performed in ExpressHyb solution (Clontech) at 68 °C for 1 h.
Determination of Protein Stability by Cycloheximide Chase.
Yeast strains expressing the relevant PCNA∗ constructs were grown in YPD medium at 30 °C to exponential phase and treated with 100 μg/mL cycloheximide to inhibit global protein synthesis. Aliquots were taken at the indicated time points, cell lysates were prepared from equal culture volumes as described (43) and the fusion protein was detected by Western blot with a polyclonal anti-PCNA antibody, along with native PCNA as a loading control. For analysis of the βGal constructs, yeast cultures were grown overnight in uracil-free synthetic complete medium containing 2% lactate as a carbon source, and expression of the constructs was induced by addition of 2% galactose for 2 h. Cells were then shifted to glucose medium containing 100 μg/mL cycloheximide. Aliquots of equal volume were taken at the indicated time points, and the βGal constructs were detected in total extracts by Western blot with a monoclonal anti-βGal antibody (Promega). Detection of phosphoglycerate kinase with a monoclonal antibody (Molecular Probes) served as a loading control.
Supplementary Material
Acknowledgments.
We thank P. Silver for the npl4-1 and cdc48-2 mutants, E. Johnson for Ub-βGal, D. Komander for the UBAN construct, J. Uhler for help with Northern blots and members of the lab for reagents, helpful discussions and critical reading of the manuscript. This work was funded by Cancer Research UK.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.0908764107/-/DCSupplemental.
References
- 1.Ciechanover A, Orian A, Schwartz AL. Ubiquitin-mediated proteolysis: Biological regulation via destruction. Bioessays. 2000;22:442–451. doi: 10.1002/(SICI)1521-1878(200005)22:5<442::AID-BIES6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 2.Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell. 2009;33:275–286. doi: 10.1016/j.molcel.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 3.Ikeda F, Dikic I. Atypical ubiquitin chains: New molecular signals ‘Protein Modifications: Beyond the Usual Suspects’ review series. EMBO Rep. 2008;9:536–542. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chau V, et al. A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science. 1989;243:1576–1583. doi: 10.1126/science.2538923. [DOI] [PubMed] [Google Scholar]
- 5.Johnson ES, Ma PC, Ota IM, Varshavsky A. A proteolytic pathway that recognizes ubiquitin as a degradation signal. J Biol Chem. 1995;270:17442–17456. doi: 10.1074/jbc.270.29.17442. [DOI] [PubMed] [Google Scholar]
- 6.Koegl M, et al. A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell. 1999;96:635–644. doi: 10.1016/s0092-8674(00)80574-7. [DOI] [PubMed] [Google Scholar]
- 7.Saeki Y, Tayama Y, Toh-e A, Yokosawa H. Definitive evidence for Ufd2-catalyzed elongation of the ubiquitin chain through Lys48 linkage. Biochem Biophys Res Commun. 2004;320:840–845. doi: 10.1016/j.bbrc.2004.05.216. [DOI] [PubMed] [Google Scholar]
- 8.Richly H, et al. A series of ubiquitin binding factors connects CDC48/p97 to substrate multiubiquitylation and proteasomal targeting. Cell. 2005;120:73–84. doi: 10.1016/j.cell.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 9.Deng L, et al. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 10.Ulrich HD. Regulating post-translational modifications of the eukaryotic replication clamp PCNA. DNA Repair. 2009;8:461–469. doi: 10.1016/j.dnarep.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 11.Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature. 2002;419:135–141. doi: 10.1038/nature00991. [DOI] [PubMed] [Google Scholar]
- 12.Stelter P, Ulrich HD. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature. 2003;425:188–191. doi: 10.1038/nature01965. [DOI] [PubMed] [Google Scholar]
- 13.Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase η with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol Cell. 2004;14:491–500. doi: 10.1016/s1097-2765(04)00259-x. [DOI] [PubMed] [Google Scholar]
- 14.Zhang H, Lawrence CW. The error-free component of the RAD6/RAD18 DNA damage tolerance pathway of budding yeast employs sister-strand recombination. Proc Natl Acad Sci USA. 2005;102:15954–15959. doi: 10.1073/pnas.0504586102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hofmann RM, Pickart CM. In vitro assembly and recognition of Lys-63 polyubiquitin chains. J Biol Chem. 2001;276:27936–27943. doi: 10.1074/jbc.M103378200. [DOI] [PubMed] [Google Scholar]
- 16.Saeki Y, et al. Lysine 63-linked polyubiquitin chain may serve as a targeting signal for the 26S proteasome. EMBO J. 2009;28:359–371. doi: 10.1038/emboj.2008.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirisako T, et al. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006;25:4877–4887. doi: 10.1038/sj.emboj.7601360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Komander D, et al. Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 2009;10:466–473. doi: 10.1038/embor.2009.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tokunaga F, et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat Cell Biol. 2009;11:123–132. doi: 10.1038/ncb1821. [DOI] [PubMed] [Google Scholar]
- 20.Rahighi S, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell. 2009;136:1098–1109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 21.Parker JL, Bielen AB, Dikic I, Ulrich HD. Contributions of ubiquitin- and PCNA-binding domains to the activity of Polymerase η in Saccharomyces cerevisiae. Nucleic Acids Res. 2007;35:881–889. doi: 10.1093/nar/gkl1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parker JL, Ulrich HD. Mechanistic analysis of PCNA poly-ubiquitylation by the ubiquitin protein ligases Rad18 and Rad5. EMBO J. 2009;28:3657–3666. doi: 10.1038/emboj.2009.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. Recognition of the polyubiquitin proteolytic signal. EMBO J. 2000;19:94–102. doi: 10.1093/emboj/19.1.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Podlaska A, McIntyre J, Skoneczna A, Sledziewska-Gojska E. The link between 20S proteasome activity and post-replication DNA repair in Saccharomyces cerevisiae. Mol Microbiol. 2003;49:1321–1332. doi: 10.1046/j.1365-2958.2003.03635.x. [DOI] [PubMed] [Google Scholar]
- 25.Xu Q, Farah M, Webster JM, Wojcikiewicz RJ. Bortezomib rapidly suppresses ubiquitin thiolesterification to ubiquitin-conjugating enzymes and inhibits ubiquitination of histones and type I inositol 1,4,5-trisphosphate receptor. Mol Cancer Ther. 2004;3:1263–1269. [PubMed] [Google Scholar]
- 26.Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986;234:179–186. doi: 10.1126/science.3018930. [DOI] [PubMed] [Google Scholar]
- 27.Kulathu Y, Akutsu M, Bremm A, Hofmann K, Komander D. Two-sided ubiquitin binding explains specificity of the TAB2 NZF domain. Nat Struct Mol Biol. 2009;16:1328–1330. doi: 10.1038/nsmb.1731. [DOI] [PubMed] [Google Scholar]
- 28.McIntyre J, Podlaska A, Skoneczna A, Halas A, Sledziewska-Gojska E. Analysis of the spontaneous mutator phenotype associated with 20S proteasome deficiency in S. cerevisiae. Mutat Res. 2006;593:153–163. doi: 10.1016/j.mrfmmm.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 29.Xu P, et al. Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell. 2009;137:133–145. doi: 10.1016/j.cell.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haririnia A, D’Onofrio M, Fushman D. Mapping the interactions between Lys48 and Lys63-linked di-ubiquitins and a ubiquitin-interacting motif of S5a. J Mol Biol. 2007;368:753–766. doi: 10.1016/j.jmb.2007.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raasi S, Varadan R, Fushman D, Pickart CM. Diverse polyubiquitin interaction properties of ubiquitin-associated domains. Nat Struct Mol Biol. 2005;12:708–714. doi: 10.1038/nsmb962. [DOI] [PubMed] [Google Scholar]
- 32.Windecker H, Ulrich HD. Architecture and assembly of poly-SUMO chains on PCNA in Saccharomyces cerevisiae. J Mol Biol. 2008;376:221–231. doi: 10.1016/j.jmb.2007.12.008. [DOI] [PubMed] [Google Scholar]
- 33.Huang TT, et al. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat Cell Biol. 2006;8:339–347. doi: 10.1038/ncb1378. [DOI] [PubMed] [Google Scholar]
- 34.Kirkpatrick DS, et al. Quantitative analysis of in vitro ubiquitinated cyclin B1 reveals complex chain topology. Nat Cell Biol. 2006;8:700–710. doi: 10.1038/ncb1436. [DOI] [PubMed] [Google Scholar]
- 35.Prakash S, Inobe T, Hatch AJ, Matouschek A. Substrate selection by the proteasome during degradation of protein complexes. Nat Chem Biol. 2009;5:29–36. doi: 10.1038/nchembio.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stack JH, Whitney M, Rodems SM, Pollok BA. A ubiquitin-based tagging system for controlled modulation of protein stability. Nat Biotechnol. 2000;18:1298–1302. doi: 10.1038/82422. [DOI] [PubMed] [Google Scholar]
- 37.Saeki Y, et al. Intracellularly inducible, ubiquitin hydrolase-insensitive tandem ubiquitins inhibit the 26S proteasome activity and cell division. Genes Genet Syst. 2004;79:77–86. doi: 10.1266/ggs.79.77. [DOI] [PubMed] [Google Scholar]
- 38.Verma R, et al. Role of Rpn11 metalloprotease in deubiquitination and degradation by the 26S proteasome. Science. 2002;298:611–615. doi: 10.1126/science.1075898. [DOI] [PubMed] [Google Scholar]
- 39.Yao T, Cohen RE. A cryptic protease couples deubiquitination and degradation by the proteasome. Nature. 2002;419:403–407. doi: 10.1038/nature01071. [DOI] [PubMed] [Google Scholar]
- 40.Johnston JA, Johnson ES, Waller PR, Varshavsky A. Methotrexate inhibits proteolysis of dihydrofolate reductase by the N-end rule pathway. J Biol Chem. 1995;270:8172–8178. doi: 10.1074/jbc.270.14.8172. [DOI] [PubMed] [Google Scholar]
- 41.Johnson ES, Bartel B, Seufert W, Varshavsky A. Ubiquitin as a degradation signal. EMBO J. 1992;11:497–505. doi: 10.1002/j.1460-2075.1992.tb05080.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Papouli E, et al. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell. 2005;19:123–133. doi: 10.1016/j.molcel.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 43.Silver PA, Chiang A, Sadler I. Mutations that alter both localization and production of a yeast nuclear protein. Genes Dev. 1988;2:707–717. doi: 10.1101/gad.2.6.707. [DOI] [PubMed] [Google Scholar]
- 44.Davies AA, Huttner D, Daigaku Y, Chen S, Ulrich HD. Activation of ubiquitin-dependent DNA damage bypass is mediated by Replication Protein A. Mol Cell. 2008;29:625–636. doi: 10.1016/j.molcel.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.