

Abstract
Deformation of lipid membranes into curved structures such as buds and tubules is essential to many cellular structures including endocytic pits and filopodia. Binding of specific proteins to lipid membranes has been shown to promote membrane bending during endocytosis and transport vesicle formation. Additionally, specific lipid species are found to colocalize with many curved membrane structures, inspiring ongoing exploration of a variety of roles for lipid domains in membrane bending. However, the specific mechanisms by which lipids and proteins collaborate to induce curvature remain unknown. Here we demonstrate a new mechanism for induction and amplification of lipid membrane curvature that relies on steric confinement of protein binding on membrane surfaces. Using giant lipid vesicles that contain domains with high affinity for his-tagged proteins, we show that protein crowding on lipid domain surfaces creates a protein layer that buckles outward, spontaneously bending the domain into stable buds and tubules. In contrast to previously described bending mechanisms relying on local steric interactions between proteins and lipids (i.e. helix insertion into membranes), this mechanism produces tubules whose dimensions are defined by global parameters: domain size and membrane tension. Our results suggest the intriguing possibility that confining structures, such as lipid domains and protein lattices, can amplify membrane bending by concentrating the steric interactions between bound proteins. This observation highlights a fundamental physical mechanism for initiation and control of membrane bending that may help explain how lipids and proteins collaborate to create the highly curved structures observed in vivo.
Keywords: membrane curvature, giant vesicle, lipid domain, protein recognition
Lipid membranes provide a dynamic substrate for biomolecular interactions that underlies environmental response and compartmentalized function in cells. Highly curved membrane structures are critical for a variety of cellular processes including endocytosis, cytoskeletal protrusion, organelle synthesis, and cell division (1). Curved membrane assemblies such as lipid tubules and buds have also been of interest as controllable nanomaterials such as scaffolds for biological–synthetic hybrid materials (2 and 3), and conduits to move species within nano-fluidic networks (4). However, a well-controlled method for the self-assembly of complex membrane structures and networks has yet to emerge.
Cells use a variety of mechanisms to induce curvature in their membranes including cytoskeletal pushing and pulling, binding of curved proteins to membranes, scaffolding of the membrane by curved protein lattices, insertion of amphipathic peptide helices into a single leaflet of the membrane, and asymmetric changes in the membrane lipid composition (1 and 5). Cells orchestrate the size, location, number, and lifetime of a variety of diverse membrane protrusions. Greater understanding of how this extraordinary coordination is accomplished is needed not only to understand cellular processes that depend on protrusion but also to identify principles that could be used to assemble programmable soft-material structures and networks.
Some of the most studied membrane protrusions are endocytic structures such as clathrin-coated pits and caveolae. The curvature of these structures is thought to arise from protein-induced membrane bending, where concentrated application of steric pressure within structures plays an important role. During clathrin-mediated endocytosis it is believed that epsin family proteins insert amphipathic helices in the cytoplasmic membrane leaflet to induce curvature. The structured clathrin coat concentrates epsins leading to membrane budding (6). In the formation of caveolae, caveolins are known to oligomerize (7) and have been hypothesized to deform the bilayer through application of steric pressure (8).
In addition to protein binding, lipid membrane organization may also play a role in membrane bending during endocytosis. Specific membrane species such as cholesterol and sphingomylin are known to colocalize with or enable highly curved membrane structures including clathrin-coated pits (9), caveolae (10), and synaptic vesicles (11), possibly forming domains rich in cholesterol and sphingolipids. A variety of roles for lipid domains relevant to the formation of protrusions have been explored previously (12). Examples include concentration of signaling receptors (10), sorting of membrane proteins (13 and 14), and modification of bilayer mechanical properties (5 and 15). However, the specific mechanisms by which lipid organization collaborates with protein binding to induce membrane deformation remain unknown.
We explore possible collaboration between proteins and lipid domains in membrane protrusion by constructing a simplified synthetic model system using giant unilamellar vesicles (GUVs). These vesicles each contain a lipid domain that strongly binds poly-histidine-tagged proteins (16). Using this synthetic model system we have demonstrated that domains are capable of tightly concentrating protein binding interactions. Remarkably, this concentration leads to lateral steric crowding of proteins within the domain that bends the membrane, inducing spontaneous formation of lipid buds and tubules. Tubule formation relies upon the presence of fluid-phase lipids in the domain and requires a high density of protein attachment that is facilitated by proteins of smaller molecular weight. Most domains yield a single tubule, and tubules frequently consume the entire protein-coated domain such that the domain size tightly defines the tubule surface area. Further, tubule length and diameter were found to vary linearly with vesicle diameter. A simple physical analysis shows that this coupling is consistent with a globally limited membrane tension defined by protein-lipid binding energy. While this synthetic model system is highly simplified, it demonstrates that lipid domains and other confining structures such as protein lattices could aid in the formation of protrusions and define protrusion length scales by concentrating the steric interactions between the lipid bilayer and proteins. Further, these findings suggest an approach for polarizing and ordering lipid-based materials by self-assembly.
Results
Lipid Domains Can Confine Protein Binding on Vesicle Surfaces.
To study the effects of confining protein binding to a specific region on a lipid membrane, we formed GUVs containing insoluble domains that serve as high affinity sites for his-tagged proteins. These vesicles consisted of the lipid DSIDA (distearylglycero triethyleneglycyl iminodiacetic acid) (17) in a 1∶9 molar ratio with matrix lipids (e.g., POPC, DPhPC). When bound to Cu2+, DSIDA has a phase transition temperature of approximately 73 °C. Therefore, when DSIDA is mixed with fluid-phase matrix lipids at room temperature, insoluble domains enriched in DSIDA form on the surfaces of supported lipid bilayers (SLBs) (16) and GUVs. The fluorescent probe, BODIPY, partitions preferentially to fluid-phase regions of the membrane, revealing dark, gel-phase domains, rich in DSIDA. For comparison, we prepared GUVs with the same mole fraction of protein affinity sites uniformly distributed over the entire vesicles surface by mixing the fluid-phase lipid DOIDA (dioleylglycero triethyleneglycyl iminodiacetic acid) (18) at a 1∶9 molar ratio with matrix lipids.
The Cu2+-IDA complex of the DSIDA and DOIDA lipids acts as a high affinity site for histidine and his-tagged proteins. When GUVs containing DOIDA were exposed to his-tagged GFP (his-GFP), an evenly distributed coverage of protein on the vesicle surfaces was observed (Fig. 1A). In contrast, when GUVs containing DSIDA were exposed to his-GFP, protein binding only occurred in a well-defined region of the vesicle surface that strongly colocalized with the DSIDA-rich dark domain (Fig. 1B center, right). Therefore, lipid domains composed of DSIDA confined and concentrated protein binding interactions with the vesicle surface.
Fig. 1.

Lipid domains can confine protein binding leading to localized membrane deformation and tubule formation. (A) GUVs containing 10% DOIDA have a uniform distribution of the membrane dye and surface protein binding (Confocal fluorescence images—Left: BODIPY, Center: GFP, Right: merge). (B) GUVs containing 10% DSIDA form domains that exclude the membrane dye and concentrate protein binding (Confocal fluorescence images—Left: BODIPY, Center: GFP, Right: merge). (C) Protein binding to the domain frequently leads to domain puckering (merged fluorescence confocal image), (D) bulging (wide field epifluorescence merged image), and (E) lipid tubule formation (wide field epifluorescence merged image). (F) Lipid tubules protruding from one vesicle often encounter other vesicles and form stable connections (merged confocal fluorescence image). All vesicles contain 0.3% BODIPY. (Scale bars, 2 μm).
Protein Binding Can Deform Domains into Buds and Tubules.
Concentration of protein binding interactions on domain surfaces led to remarkable changes in their shape. Originally flat domain surfaces were rapidly bent into puckered surfaces (Fig. 1C) and bulges (Fig. 1D) upon protein binding. The most dramatic shape change was the frequent formation of long, thin tubules from domains upon protein binding (Fig. 1E). Membrane deformations and tubule formation were observed within minutes after protein addition (Fig. S1).
From confocal fluorescent images of tubules extending from vesicle surfaces (Fig. 2 A, B) we estimate that 80% of the vesicles forming tubules have a single tubule. Further, many tubules take up the entire domain area (Fig. 1E). These observations suggest that initiation of a tubule by forming an initial high curvature bud presents a higher energetic barrier than extension of an existing bud into a tubule. Membrane shape changes and formation of tubules were not found for DOIDA-containing vesicles, where the protein binding sites were distributed evenly over GUV surfaces, demonstrating that concentration of protein binding at a well-defined, high affinity region of the vesicle surface is required for shape change.
Fig. 2.

Tubule formation is promoted by solubility of fluid-phase lipids in domains. GUVS containing (A)—(10% DSIDA / 89.7% POPC / 0.3% BODIPY ) and (B)—( 10% DSIDA / 89.7% DPhPC / 0.3% BODIPY ) with 2 μM his-GFP. SLBs containing (C)—(10% DSIDA / 89.97% POPC / 0.03% BODIPY) and (D)—(10% DSIDA / 89.7% DPhPC / 0.03% BODIPY) form insoluble domains. White circles contain DSIDA-rich insoluble domains. (E) FCS data (normalized correlation amplitude vs. time) for BODIPY fluorescence shows that fluorophores outside DSIDA domains in a POPC matrix diffuse at about 0.04 μm2/s (light gray), and fluorophores inside these domains diffuse at about 4.7 μm2/s (black). For bilayers composed of DSIDA and DPhPC, domains were too transient to allow separate measurements inside and outside domains. Transient FCS data (dark gray points) show an intermediate slope and permit extraction of two diffusion constants (0.07 μm2/s and 1.7 μm2/s). (F) The frequency of tubule formation for GUVs with various matrix lipids (DPhPC, POPC, and POPC with 0.1% Soy PE). (Scale bar (C, D), 2 μm). (Scale bar (A, B), 5 μm).
Affect of Lipid Composition on Tubule Formation.
We examined the impact of lipid composition on the deformation of lipid domains by protein binding and found that the frequency of tubule formation varied considerably with the lipid composition (Fig. 2). We initially examined the frequency of tubule formation using two different matrix lipids, POPC and DPhPC. We found that tubules frequently formed (0.42 ± 0.07 tubes/vesicle) using 10% DSIDA / 89.7% DPhPC / 0.3% BODIPY while they formed much more rarely (0.02 ± 0.01 tubes/vesicle) using 10% DSIDA / 89.7% POPC / 0.3% BODIPY, Fig. 2 A, B, and F. Additionally, two other unsaturated phosphocholine matrix lipids were tried, DLPC and DOPC. Despite having slightly lower bending rigidities than POPC (∼3.9 × 10-20 J (19)), neither DLPC (∼3.4 × 10-20 J, (20)) nor DOPC (∼1.9 × 10-20 J (19) produced more tubules than POPC (Compare Fig. 2A with Fig. S2). Although the concentration of dye was low in all experiments (0.3 mol %), we cannot rule out its effect on these membrane curvature results.
We hypothesized that tubule formation might depend on solubility of fluid-phase matrix lipids in domains. Enhanced solubility could fluidize domains, reducing curvature energy and facilitating protein packing on domain surfaces. We performed fluorescence correlation spectroscopy (FCS) measurements on supported lipid bilayers to compare the diffusivity of dye molecules within domains, a measure of fluidity. Using 10% DSIDA in a DPhPC matrix, DSIDA-rich domains were observed (Fig. 2D), but were unstable, forming and dissolving multiple times per minute. FCS measurements showed both fast and slow components with diffusion constants characteristic of fluid and gel-phase membranes, respectively, on the minute time scale of the FCS measurements (Fig. 2E). This behavior contrasts with the stable DSIDA-rich domains formed using 10% DSIDA in the POPC matrix (Fig. 2C), which were found to have only a single very slow diffusion component (16).
Domain fluctuations similar to those we observe in DSIDA / DPhPC membranes occur in binary lipid mixtures just above solid-liquid miscibility phase transitions temperatures and have been characterized using FCS (21). Such fluctuations have also been observed just above liquid-liquid phase transition temperatures (22 and 23). Based on these results, we hypothesized that the transient nature of DSIDA / DPhPC domains may indicate that the system is near a miscibility phase transition when observed at room temperature. While these domains appear stable when observed on GUVs (Fig. 1B), substrate-membrane interactions have been known to shift phase transition temperatures of SLBs (24). Therefore, DSIDA / DPhPC mixtures on GUVs may be somewhat below a miscibility phase transition at room temperature, but very near the phase transition on SLBs. The difference in domain stability between SLBs of DSIDA / DPhPC and DSIDA / POPC may reflect a difference in the phase-stability of the two mixtures, which impacts the domain composition of GUVs. To evaluate this possibility we investigated the solubility of DSIDA in both DPhPC and POPC at room temperature. We formed GUVs with decreasing DSIDA content from 7.5 mol % to 3 mol % and measured the area of resulting DSIDA-rich domains (Table S1). Vesicles of DSIDA / POPC contained domains that decreased in area approximately linearly with decreasing DSIDA content. In contrast, vesicles of DSIDA / DPhPC displayed a sharp decrease in domain area between 7.5 mol % and 6 mol % and no micron-scale domains below 6 mol %. These results suggest that at room temperature, the DSIDA / DPhPC mixture is nearer to a miscibility phase transition than is the DSIDA / POPC mixture. Increasing proximity to a phase transition implies that the compositions of the two phases are increasingly equivalent. Therefore DSIDA-rich domains formed in a DPhPC matrix likely contain a greater fraction of the matrix lipid than those formed in a POPC matrix. Increased solubility of the matrix lipid in DSIDA-rich domains could increase domain fluidity, reducing the energetic barrier to tubule formation. This effect could explain the increased formation of lipid tubules with DPhPC in comparison to POPC.
Finally, we hypothesized that DPhPC might aid the formation of tubules because of its negative spontaneous curvature in comparison to other PC lipids tested (25). Lipids of high negative spontaneous curvature might assist tubule formation by populating the negative curvature region at the tubule base or the tubule inner membrane leaflet. For example, phosphatidyl ethanolamine (PE) lipids, which have negative spontaneous curvature, are known to partition to regions of high curvature in cells, often distributing asymmetrically between lipid leaflets to promote curvature (26). We formed GUVs that contained small amounts (0.1–1%) of Soy PE in the DSIDA/POPC bilayers. We had previously observed that Soy PE was highly soluble in domains as evidenced by an increase in size of the domains when larger fractions of Soy PE were included (10–40% Soy PE, Fig. S3). Therefore, we estimate that Soy PE partitions to the domains such that the percentage of Soy PE was likely 1–10% within them. We found that addition of this small fraction of Soy PE produced about 6–7 times more tubules than POPC alone (0.13 ± 0/03 tubes/vesicle). Inclusion of Soy PE in the domain likely increases domain fluidity and encourages negative spontaneous curvature, both of which should reduce the energetic barrier to tubule formation.
Affect of Protein Binding on Tubule Formation.
In addition to lipid composition, variations in the his-tagged protein composition and concentration may impact membrane deformation. We varied the concentration of his-GFP in solution and found a monotonic increase in the frequency of tubule formation of more than fourfold as protein concentration increased from 20 nM to 2 μM (Fig. 3A) (Kd ∼ 2 nM) (16). Below 20 nM, protein bound on the lipid bilayers could not be discerned from background fluorescence. The increase in tubule formation with protein concentrations several orders of magnitude above the dissociation constant suggests that tubule formation relies on a very high fractional occupancy of protein on the domain surface. Additionally, the dissociation constant may increase as protein surface coverage increases.
Fig. 3.

Tubule formation increases with increasing protein concentration and decreases with increasing protein molecular weight, all vesicles 10% DSIDA / 89.7% DPhPC / 0.3% BODIPY. (A) Frequency of tubule formation as a function of protein concentration (his-GFP). (B) Frequency of tubule formation by various his-tagged proteins having different molecular weights (his-GFP-26 kDa, his-mOrange-26 kDa, his-MBP-66 kDa, his-TLR4MD2-125 kDa, all 2 μM).
In Fig. 3B we examine the frequency of tubule formation using four different his-tagged proteins of various molecular weight: his-GFP (26 kDa), his-mOrange (26 kDa), his-MBP (66 kDa), and his-TLR4MD2 (∼125 kDa). We observed that lower molecular weight proteins, which, to a first approximation, will have smaller hydrodynamic radii, produced membrane tubules with significantly higher frequency. GFP and mOrange have the same molecular weight. However, GFP is known to form transdimers, while mOrange has been engineered to prevent dimerization (27). The capacity of GFP to form dimers could increase the amount of protein attached to the domain surface, encouraging crowding and tubule formation. However, the high frequency of tubule formation using mOrange indicates that dimerization is not required for tubule formation. Binding of either TLR4MD2 or MBP protein led to formation of well-defined tubules, though the frequency of tubule formation was much lower in comparison to GFP and mOrange. None of these proteins are known to participate in membrane bending processes in vivo; therefore, the tube formation we observe is likely the result of a general physical mechanism that does not require that attached proteins have a specific protein domain or conformation.
Lipid Tubule Length Is Proportional to Vesicle Diameter.
Since lipid tubules form from domains on the surfaces of vesicles containing a known molar fraction of DSIDA, lipid tubule dimensions (length, diameter) should be related to vesicle dimensions and composition (diameter, domain area). From confocal image scans of the vesicles, we measured the lengths of individual lipid tubules and the diameters of the vesicles they were connected to for varying DSIDA content (7.5, 10, 15 mol %) upon exposure to 2 μM his-GFP. These measurements led to a notable observation—the ratio of the lipid tubule length to the vesicle radius was approximately constant (Fig. 4). Vesicles of increasing DSIDA fraction formed tubules of increasing length relative to vesicle diameter.
Fig. 4.

Lipid tubule length is proportional to vesicle diameter. (A) Lipid tubules formed from GUVs (7.5% DSIDA / 92.2% DPhPC / 0.3% BODIPY). (B) Tubule length as a function of vesicle diameter (7.5%, 10%, and 15% DSIDA in DPHPC with 0.3% BODIPY). (C) Fractional domain surface area, calculated ratio of tubule radius to vesicle radius, and measured ratio of tubule length to vesicle radius. All vesicles exposed to 2 μM his-GFP. (Scale bar, (A) 2 μm).
Since the domain areas were often fully consumed by tubule formation, it was possible to estimate the radius of lipid tubules, RT, from estimates of tubule length, LT, vesicle radius, R, and domain area fraction, AD, by assuming that the domain area is approximately conserved. The average area of DSIDA-rich domains as a percentage of total vesicle surface area was estimated from confocal image stacks (Fig. 4C). Because the fractional domain area and the ratio of tubule length to vesicle diameter are both constant for a given vesicle composition, conservation of domain area implies that tubule diameter is directly proportional to tubule length Eq. 1.
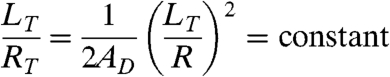 |
[1] |
Discussion
We have shown that protein binding to spatially confined regions of a lipid membrane leads to membrane deformation and tubule formation. Further, the diameter and length of lipid tubules is tightly coupled to size and composition of the vesicles from which they originate, suggesting that global parameters may play an important role as tubules form. Here we examine a possible physical basis for tubule formation and discuss the implications of confined protein binding and associated coupling between tubule and vesicle dimensions.
Lipid Tubule Geometry Can Be Explained by a Global Tension Limit.
Previous studies on the formation of lipid tubules in vitro have primarily focused on mechanical pulling of tubules either by direct application of force or the action of motor proteins (28). In those experiments tubule radius, RT, was found to depend upon the local membrane bending energy, κ, and membrane tension, σ, according to the expression, RT = (2κ/σ)1/2 (29). Here fractional membrane area dilations are small, likely accommodated by vesicle shape fluctuations, such that tension does not increase significantly during pulling of short tubules. Therefore, a constant membrane tension is typically assumed in the analysis, leading to a lipid tubule radius that is invariant with tubule length (28).
In contrast, we observe that the lengths and diameters of lipid tubules formed in our system increase proportionally with the vesicle diameter, suggesting that global parameters such as the total volume, area, and membrane tension could govern the tubule geometry. In particular, our observations raise two questions: (i) why is the ratio of tubule radius to tubule length constant for a given composition and (ii) what limits this ratio?
Distinct from previous studies, we form tubules by a self-assembly process in which proteins crowd onto lipid domains resulting in bending. Protein binding bends the entire domain at once (Fig. S1A), forming lipid tubules that consume as much as the entire domain area (Fig. 1E, Fig 4A). Area dilations of this magnitude are expected to exhaust area available from vesicle shape fluctuations, requiring a decrease in lipid packing density that raises membrane tension substantially (30).
Therefore, it is the boundary condition on membrane tension that primarily differentiates our work from previous tubule pulling experiments and explains our observation of a constant tubule aspect ratio. In previous experiments, small membrane dilations led to approximately constant tension during tubule formation, effectively decoupling tubule pulling from global vesicle dimensions. In our work, transformation of membrane domains into tubules led to large membrane area dilations. Therefore we expect tension to increase, directly coupling tubule formation to vesicle dimensions.
We propose a simple physical analysis based on the principles that protein binding drives membrane deformation, which must conserve domain area and vesicle volume. The stepwise analysis is presented in the SI Text section. Briefly, as protein binding bends the lipid domain, membrane tension increases such that the membrane energy is dominated by the tension term. The energy required to raise the tension balances against the energy available from protein binding to the domain, defining a maximum membrane tension (σmax) that depends on fractional domain area (AD), protein-lipid binding energy (ΔG), and protein binding density ( ), [2].
), [2].
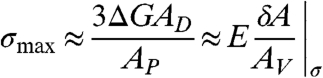 |
[2] |
Since these parameters do not vary with vesicle diameter, the maximum membrane tension is expected to be constant with vesicle diameter. Constant membrane tension implies approximately constant membrane area dilation (δA/AV)|σ across all vesicle diameters, because the majority of area dilation at high tension arises from direct expansion of membrane area, and the area expansion modulus (E) is an intensive property of the membrane. Conserving domain area and vesicle volume, a constant area dilation requires that the ratio of tubule length to tubule diameter be constant for all vesicle diameters. This aspect ratio is limited by the total protein binding energy, a function of fractional domain area, protein-lipid binding constant, and binding density. These predictions are in agreement with our observation of constant tubule aspect ratio and may explain why proteins of smaller molecular weight, which likely increase binding density, form tubules more frequently.
Biophysical Implications of Steric Confinement and Global Coupling.
We have shown that lateral crowding of bound proteins on the surface of a lipid micro-domain causes spontaneous bending of the domain into a stable lipid tubule of well-defined length (Fig. 5A). We first showed that lipid tubules were not observed when protein binding lipids were evenly distributed over GUV surfaces indicating that concentration of protein binding sites, achieved by domain formation, is required for membrane bending. Lipid tubule formation from domains was aided by the presence of fluid-phase lipids of negative spontaneous curvature. These species may aid tubule formation by lowering domain bending energy, fluidizing domains such that dense protein packing is enabled, and facilitating membrane bending by partitioning to the negative curvature regions of the tubule.
Fig. 5.

Lipid tubules form when domains crowd protein binding, likely due to a steric crowding mechanism. (A) Schematic showing how domains could crowd protein binding events, leading to formation of buds and tubules from domains. (B) Comparison of possible mechanisms of domain bending: amplification of bending by helix insertion (C) and direct protein-coat buckling.
Several his-tagged proteins were attached to membrane domains in order to form tubules, none of which have been implicated in membrane bending processes in vivo, suggesting that the mechanism by which tubules are formed does not require a specific protein morphology. The lengths of lipid tubules were observed to vary proportionally with the vesicle diameter, which, according to our analysis, suggests that the system is governed by a global tension limit arising from a balance between protein-lipid binding energy and membrane free energy.
In cellular processes, attachment of specific proteins to lipid bilayers is known to participate in curving membranes as a part of endocytic processes. Several proteins associated with endocytosis have been shown to deform small, originally spherical liposomes into tubules in vitro including amphiphysin (31) and epsin (6), which are required for clathrin-mediated endocytosis, and Sar1p (32), which participates in COPII transport vesicle formation. Several specific mechanisms by which protein attachment bends membranes have been described (1 and 33) including attachment and pushing of the cytoskeleton; insertion of conically shaped transmembrane proteins; amphipathic helix insertion (Fig. 5B); and assembly of curved protein scaffolds.
Our results demonstrate a universal mechanism by which confining structures such as lipid domains, scaffolds, and protein lattices could collaborate with membrane binding proteins to induce membrane curvature. Whenever proteins bind with high affinity to a small region of the membrane that is defined by a sufficiently rigid barrier, lateral protein crowding could cause deformation. As we have demonstrated, this mechanism is sufficient to cause membrane curvature on its own (Fig. 5C) without the requirement to specifically disrupt and deform the membranes via processes, such as amphipathic helix insertion. However, our proposed mechanism could also collaborate with such mechanisms by concentrating their membrane bending effects in a small region (Fig. 5B) or amplifying bending as the density of protein on the surface becomes sufficiently high (Fig. 5C). The prevalence of confining structures in membrane bending events suggests that this mechanism, protein-coat buckling, may play an important role in vivo.
Materials and Methods
Materials.
Lipid molecules including DPhPC (1,2-diphytanoyl-sn-glycero-3-phosphocholine), POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine), and Soy PE (L-α-phosphatidyl ethanolamine) were purchased from Avanti Polar Lipids. DSIDA (Distearylglycero triethyleneglycyl iminodiacetic acid) and DOIDA (Dioleylglycero triethyleneglycyl iminodiacetic acid) were synthesized according to previously reported protocols (17 and 18), Fig. S4. The lipid dye β-BODIPY® 530/550 C5-HPC was purchased from Invitrogen. The proteins his-GFP and his-mOrange each had a molecular weight of approximately 26 kDa and a six histidine tag. His-MBP (Maltose Binding Protein) had a molecular weight of 66 kDa and a six histidine tag. His-TLR4MD2 was purchased from R&D Systems having a molecular weight of approximately 125 kDa with two ten histidine tags. His-MBP and his-TLR4MD2 were fluorescent labeled with Alexa488 probes from Invitrogen. MOPS (3-(N-morpholino) propanesulfonic acid), CuCl2, sucrose and glucose chemicals were purchased from Sigma Aldrich.
Electroformation.
Liposome electroformation was performed according to published protocols (34). We performed electroformation at approximately 70 °C to exceed the highest expected melting temperature of lipid mixtures. Vesicles were electroformed in sucrose solution (∼350 milliosmole(mOsm)).
Online Supporting Methods Include.
GUV slide preparation, microscopy, calculation of tubule formation frequency, formation of supported lipid bilayers, fluorescence correlation spectroscopy, imaging of lipid tubule growth, and measurement of vesicle dimensions.
Supplementary Material
Acknowledgments.
We acknowledge Professor Daniel Fletcher of the University of California, Berkeley, for use of the spinning disc confocal microscope and for helpful discussions on this manuscript. Additionally, we thank Dr. Ross Rounsevell and Dr. Eva Schmid of the Fletcher lab for providing the his-GFP and his-mOrange proteins and for helpful discussions about our results. We thank Dr. Steven Branda of Sandia National Laboratories for cloning of the his-mOrange plasmid DNA. We thank Professor Haw Yang of Princeton University for providing his-MBP protein. We acknowledge Professor Françoise Brochard-Wyart of the Curie Institute, Paris, France and Professor Sarah Keller of the University of Washington for helpful discussions on this work. Membrane synthesis and structural studies were supported by the Division of Materials Science and Engineering for D.Y.S. and J.C.S., and membrane phase and imaging measurements were supported by the Division of Chemical Sciences, Geosciences, and Biosciences for C.C.H. in the Department of Energy’s Office of Basic Energy Sciences, and the Laboratory Directed Research and Development program at Sandia National Laboratories. Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the Department of Energy’s National Nuclear Security Administration under contract DE-AC04-94AL85000.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.0913306107/-/DCSupplemental.
References
- 1.McMahon HT, Gallop JL. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 2005;438:590–596. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- 2.Misra N, et al. Bioelectronic silicon nanowire devices using functional membrane proteins. Proc Nat'l Acad Sci USA. 2009;106:13780–13784. doi: 10.1073/pnas.0904850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou Y. Lipid nanotubes: formation, templating nanostructures and drug nanocarriers. Crit Rev Sol Sciences. 2008;33:183–196. [Google Scholar]
- 4.Karlsson M, et al. Micropipet-assisted formation of microscopic networks of unilamellar lipid bilayer nanotubes and containers. Langmuir. 2001;17:6754–6758. [Google Scholar]
- 5.Huttner WB, Zimmerberg J. Implications of lipid microdomains for membrane curvature, budding, and fission. Curr Opin Cell Biol. 2001;13:478–484. doi: 10.1016/s0955-0674(00)00239-8. [DOI] [PubMed] [Google Scholar]
- 6.Ford MG, et al. Curvature of clathrin-coated pits driven by epsin. Nature. 2002;419:361–366. doi: 10.1038/nature01020. [DOI] [PubMed] [Google Scholar]
- 7.Sargiacomo M, et al. Oligomeric structure of caveolin: implications for caveolae membrane organization. Proc Natl Acad Sci U S A. 1995;92:9407–9411. doi: 10.1073/pnas.92.20.9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sens P, Turner MS. Theoretical model for the formation of caveolae and similar membrane invaginations. Biophys J. 2004;86:2049–2057. doi: 10.1016/S0006-3495(04)74266-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Subtil A, et al. Acute cholesterol depletion inhibits clathrin-coated pit budding. Proc Natl Acad Sci U S A. 1999;96:6775–6780. doi: 10.1073/pnas.96.12.6775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007;8:185–194. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- 11.Thiele C, Hannah MJ, Fahrenholz F, Huttner WB. Cholesterol binds to synaptophysin and is required for biogenesis of synaptic vesicles. Nat Cell Biol. 2000;2:42–49. doi: 10.1038/71366. [DOI] [PubMed] [Google Scholar]
- 12.Groves JT. Bending mechanics and molecular organization in biological membranes. Annu Rev Phys Chem. 2007;58:697–717. doi: 10.1146/annurev.physchem.56.092503.141216. [DOI] [PubMed] [Google Scholar]
- 13.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 14.Baumgart T, et al. Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proc Nat'l Acad Sci USA. 2007;104:3165–3170. doi: 10.1073/pnas.0611357104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parthasarathy R, Yu CH, Groves JT. Curvature-modulated phase separation in lipid bilayer membranes. Langmuir. 2006;22:5095–5099. doi: 10.1021/la060390o. [DOI] [PubMed] [Google Scholar]
- 16.Hayden CC, Hwang JS, Abate EA, Kent MS, Sasaki DY. Directed formation of lipid membrane microdomains as high affinity sites for His-tagged proteins. J Am Chem Soc. 2009;131:8728–8729. doi: 10.1021/ja901157c. [DOI] [PubMed] [Google Scholar]
- 17.Shnek DR, Pack DW, Sasaki DY, Arnold FH. Specific protein attachment to artificial membranes via coordination to lipid-bound copper (II) Langmuir. 1994;10:2382–2388. [Google Scholar]
- 18.Pack DW, Chen GH, Maloney KM, Chen CT, Arnold FH. A metal-chelating lipid for 2D protein crystallization via coordination of surface histidines. J Am Chem Soc. 1997;119:2479–2487. [Google Scholar]
- 19.Niggemann G, Kummrow M, Helfrich W. The bending rigidity of phophatidylcholine bilayers: dependences on experimental method, sample cell sealing and temperature. J Phys II. 1995;5:413–425. [Google Scholar]
- 20.Kummrow M, Helfrich W. Deformation of giant lipid vesicles by electric fields. Phys Rev A. 1991;44:8356–8360. doi: 10.1103/physreva.44.8356. [DOI] [PubMed] [Google Scholar]
- 21.Hac AE, Seeger HM, Fidorra M, Heimburg T. Diffusion in two-component lipid membranes—a fluorescence correlation spectroscopy and monte carlo simulation study. Biophys J. 2005;88:317–333. doi: 10.1529/biophysj.104.040444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korlach J, Baumgart T, Webb WW, Feigenson GW. Detection of motional heterogeneities in lipid bilayer membranes by dual probe fluorescence correlation spectroscopy. Biochim Biophys Acta. 2005;1668:158–163. doi: 10.1016/j.bbamem.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 23.Honerkamp-Smith AR, Veatch SL, Keller SL. An introduction to critical points for biophysicists; observations of compositional heterogeneity in lipid membranes. Biochim Biophys Acta. 2009;1788:53–63. doi: 10.1016/j.bbamem.2008.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaizuka Y, Groves JT. Structure and dynamics of supported intermembrane junctions. Biophys J. 2004;86:905–912. doi: 10.1016/S0006-3495(04)74166-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rand RP, Fuller NL, Gruner SM, Parsegian VA. Membrane curvature, lipid segregation, and structural transitions for phospholipids under dual-solvent stress. Biochemistry. 1990;29:76–87. doi: 10.1021/bi00453a010. [DOI] [PubMed] [Google Scholar]
- 26.Emoto K, et al. Redistribution of phosphatidylethanolamine at the cleavage furrow of dividing cells during cytokinesis. Proc Natl Acad Sci U S A. 1996;93:12867–12872. doi: 10.1073/pnas.93.23.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaner NC, et al. Improved monomeric red, orange, and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 28.Derenyi I, et al. Controlled Nanoscale Motion. Vol 711. Berlin, Heidelberg: Springer; 2007. pp. 141–159. [Google Scholar]
- 29.Derenyi I, Julicher F, Prost J. Formation and interaction of membrane tubes. Phys Rev Lett. 2002;88:238101-1–238101-4. doi: 10.1103/PhysRevLett.88.238101. [DOI] [PubMed] [Google Scholar]
- 30.Evans E, Rawicz W. Entropy-driven tension and bending elasticity in condensed-fluid membranes. Phys Rev Lett. 1990;64:2094–2097. doi: 10.1103/PhysRevLett.64.2094. [DOI] [PubMed] [Google Scholar]
- 31.Takei K, Slepnev VI, Haucke V, De Camilli P. Functional partnership between amphiphysin and dynamin in clathrin-mediated endocytosis. Nat Cell Biol. 1999;1:33–39. doi: 10.1038/9004. [DOI] [PubMed] [Google Scholar]
- 32.Lee MC, et al. Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell. 2005;122:605–617. doi: 10.1016/j.cell.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 33.Hanzal-Bayer MF, Hancock JF. Lipid rafts and membrane traffic. FEBS Lett. 2007;581:2098–2104. doi: 10.1016/j.febslet.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 34.Angelova M, Dimitrov D. Liposome electroformation. Faraday Discuss. 1986;81:303–311. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.