

Abstract
G protein-coupled receptors for dopamine and serotonin control signaling pathways targeted by many psychoactive drugs. A puzzle is how receptors with similar functions and nearly identical binding site structures, such as D2 dopamine receptors and 5-HT2A serotonin receptors, could evolve a mechanism that discriminates stringently in their cellular responses between endogenous neurotransmitters. We used the Difference Evolutionary Trace (Difference-ET) and residue-swapping to uncover two distinct sets of specificity-determining sequence positions. One at the ligand-binding pocket determines the relative affinities for these two ligands, and a distinct, surprising set of positions outside the binding site determines whether a bound ligand can trigger the conformational rearrangement leading to G protein activation. Thus one site specifies affinity while the other encodes a filter for efficacy. These findings demonstrate that allosteric pathways linking distant interactions via alternate conformational states enforce specificity independently of the ligand-binding site, such that either one may be rationally rekeyed to different ligands. The conversion of a dopamine receptor effectively into a serotonin receptor illustrates the plasticity of GPCR signaling during evolution, or in pathological states, and suggests new approaches to drug discovery, targeting both classes of sites.
Keywords: allostery, catecholamines, G protein-coupled receptors
Specificity of ligand recognition by G protein-coupled receptors (GPCR) is critical both for their biological functions and for the effects of therapeutic drugs that target them (1, 2). The receptors for the neurotransmitters dopamine and serotonin mediate very different physiological responses to their respective ligands in regions of the brain where both neurotransmitters are present. Numerous psychoactive drugs target either the transporters for these ligands (3) or the receptors themselves (4), but all display limited efficacy and multiple side effects. Remarkably, the receptors for these functionally distinct neurotransmitters are predicted, based on 44% sequence identity, to have very similar structures in their transmembrane (TM) domain, where ligand binding causes conformational changes that trigger G protein activation. Biochemical studies of D2 dopamine receptors (5–8), D2R, and comparisons to crystal structures for the β-adrenergic receptor (9, 10), adenosine receptor (11) , and rhodopsin (12) delineate a set of residues forming the dopamine binding site, all but three of which are identical in the closely related 5-HT2A serotonin receptor, 5-HT2AR. Thus it seems unlikely that ligand-contacting residues alone are sufficient to confer specificity. A full understanding of the mechanism for ligand discrimination is of intense interest given that 7TMRs make up about 30% of all therapeutic drugs (13). Currently, most therapeutic drugs are thought to bind at the orthosteric site, although many compounds have been shown to bind elsewhere, at allosteric sites, and to impact ligand binding or receptor efficacy (14).
Here, on the assumption that discrimination between serotonin and dopamine has been important for survival throughout evolution of metazoan organisms, we used the Evolutionary Trace (ET) (15) to rank sequence positions in the transmembrane domain according to their evolutionary importance and identified a core of residues of high evolutionary importance, including those specific for the bioamine family, that are in structural contact with the ligand-binding site through no more than two intervening residues. Those whose identities differ between D2R and 5-HT2AR were swapped by site-directed mutagenesis so that each ET residue in the 5-HT2AR sequence replaced the corresponding residue in the D2R. The residue-swapped D2R constructs, mostly single point mutants, were then tested for their abilities to activate G proteins in response to dopamine or serotonin by using a cell-based high-throughput fluorescence assay. Ligand-binding affinities were also assessed using radioligand competition assays, and cell surface expression levels were quantified as well, to ensure that each construct was not grossly misfolded and that similar numbers were available for binding externally supplied ligands.
Results
Difference Evolutionary Trace Analysis Identified Positions Important For Bioamine Receptor Activation Mechanism.
Initially, ET identified one set of residue positions important in all class A GPCR (15) and, likewise, another set that was important specifically in all bioamine receptors. The difference between them, as determined by difference-ET (16, 17) analysis (Fig. 1 and Fig. S1), identified a tight cluster of residues located in the immediate neighborhood of the ligand, as expected (Fig. 1C) (16). Each residue in the cluster is in contact with at least one other, and, remarkably, the cluster surrounds the ligand-binding site, whose position was not considered in the analysis. Because all of these residues were invariant between D2R and 5-HT2AR, this bioamine core was then expanded structurally by up to two shells of evolutionarily important contact residues and up to the top 40% of important residues within either the bioamine family (Fig. 1D) or all of Class A GPCR (Fig. 1E). Of those, a total of 15 residues vary between D2R and 5-HT2AR [Fig. 1G; BW numbering (18) and ET ranks are given in Table S1]. Functional assays verified the hypothesized importance of all of these residues, as every residue-swapped version of D2R displayed significantly altered function in one or more assays.
Fig. 1.

Identification of candidates for specificity-determining residues. (A) The Cα atoms, in blue, mapped onto the β2-adrenergic structure (2RH1) of all positions in the top 20th percentile rank following ET analysis of the transmembrane domains of 402 bioamine receptor sequences. (B) Cα atoms, in cyan, of top 20th percentile positions after ET analysis of all 2512 class A GPCR sequences. (C) Bioamine-specific determinants suggested by subtracting set B (the Class A trace) from set A (the bioamine trace). The residues that cluster in C define a bioamine-specific core that was further extended to include more bioamine-specific neighbors (D) or more residues important in all class A receptors (E) at ranks up to the 40th percentile (see Fig. S1 and Table S1 for details). This set represents 48 candidates for functionally important positions in bioamine receptors (F), of which 15 have different residues in D2R and 5-HT2AR (G).
Activation of Gα16 in response to dopamine was diminished for six of the residue swaps: C118S, S193G, T205M, L379F, V381L, and N418S. Dose-response curves revealed that both the potency and maximal response to dopamine were diminished for C118S and S193G, while L379F showed almost no response to dopamine (Fig. 2C). Binding affinity for dopamine, as monitored by competition for binding of radiolabeled spiperone (19), an antagonist that binds with high affinity to all the D2 variants studied except for V91S, was significantly diminished for four of the residue swaps with diminished activation, C118S, S193G, L379F, N418S, and for an additional four residues, V83L, M117F, Y199F, and L414I (Table S2). Thus 10 of the 15 residue swaps lead to impaired interactions of D2R with dopamine, indicating that the corresponding residues in D2R contribute to dopamine recognition. Because all positions mutated are located within the transmembrane domains and at least two turns from the cytoplasmic face, they are not expected to act through direct alterations in G protein contacts.
Fig. 2.

Mutation of ET positions in D2R alters dopamine response. (A) Typical Ca2+ transients elicited in response to 10 μM dopamine, as measured by fluorescence intensity (relative fluorescence units, RFU) changes in a microplate reader. HEK 293-derived cells stably expressing Gα16 were transiently transfected with plasmids expressing D2R WT or indicated mutants, loaded with dye, and treated with dopamine at t = 10 s. (B) Dopamine maximal responses normalized by receptor surface expression. D2R WT response was defined as 100% (Dashed Line). Eight mutations (Red) reduced receptor responsiveness to dopamine. Data represent mean ± S.E.M. (*, P < 0.05; **, P < 0.001). (C) Dopamine-dose-response curves for D2RWT (▴), I48T (•), N124H (▾) and S193G (♦). Curves are four parameter fits as described in SI Text. (D) Positions that reduced dopamine response (α C) mapped onto the β2-adrenergic receptor structure.
The striking observation that all of the residues with diminished dopamine binding still bind with high affinity to spiperone (Table S2) provides further support for the specificity of our evolution-based approach. Both D2R and 5-HT2AR bind spiperone with high affinity, so swapping residues selected for discrimination between dopamine and serotonin would not be expected to exclude a common ligand.
Positions Outside the Orthosteric Binding Site Control Specificity For D2R Activation Without Altering Ligand Affinities.
ET-identified residues also contribute to discrimination against serotonin responses. The swaps I48T, M117F, N124H, and T205M all resulted in significantly enhanced responses to serotonin (Fig. 3). Three different swaps, L41T, F110W, and S193G, all led to substantial (greater than twofold) increases in affinity for serotonin, and three others, M117F, C118S, and T205M, increased serotonin affinity by at least 40%. One swap, V91S had its most dramatic effect when paired with I48T; as described below, this double mutation yielded dramatic enhancement of serotonin responsiveness above that displayed by I48T alone. Thus all 15 residue swaps enhanced interactions with serotonin, or diminished interactions with dopamine, or both (SI Text and Table S3).
Fig. 3.

Enhanced serotonin responses in D2R with ET residue swaps. (A) Typical Ca2+ transients, recorded as in Fig. 2, in response to 10 μM 5-HT. (B) Dose dependence of responses of I48T and D2RWT to serotonin. (C) Maximal 5-HT responses normalized by surface expression (mean ± SEM) with WT D2R response defined as 100% (Dashed Line). Four mutations (Red) yielded significant enhancement (p < 0.01). (D) Mutations with enhanced 5-HT responses (Red) and inactivating position L379 (Blue) are mapped onto the β2-adrenergic structure. One mutation, S193G, lowered dopamine affinity (E) and enhanced serotonin affinity (F). Specifically bound [3H] binding was measured in the presence of the indicated concentrations of competing ligands (mean ± SEM). g–j. Receptor internalization was determined by loss of surface expression using immunofluorescence in HEK WT (G and H) or cells stably expressing Gα16 (I and J), that were cotransfected with receptor and β-arrestin-EGFP cDNAs. Cells were treated with either 10μM dopamine (G and I) or 10 μM serotonin (H and J).
In sharp contrast to the results obtained with ET residues ranked as highly important is the lack of effect of swapping less important residues. From among 111 transmembrane residues that are different between D2R and 5-HT2AR, we selected five residues that met our distance criteria but ranked in the lowest 20th percentile of evolutionary importance and made the corresponding sequence swaps: I105K, A188N, V191L, I195K, and S409N. Despite their nonconservative character, all of them yielded dopamine responses similar to those of wild-type D2R, and none of them yielded enhanced serotonin responsiveness (Fig. S2). Thus ET rank is a strong predictor of the likelihood that a residue contributes substantially to ligand specificity.
Because the I48T substitution results in enhanced serotonin responsiveness without diminished dopamine responsiveness, we tested the possibility that this substitution simply renders D2R more promiscuous with respect to ligand discrimination using natural and synthetic ligands. No effect of this mutation was observed for responses to histamine, isoproterenol (β-adrenergic agonist), quinpirole (D2R agonist), or DOI (2,5-dimethoxy-4-iodoamphetamine, a partial agonist for 5-HT2AR), supporting the idea that the evolutionary selection of this residue was associated with discrimination between dopamine and serotonin (Fig. S3).
To test whether the enhanced serotonin responsiveness is selective for G protein activation or applies to other downstream signaling pathways, we measured β-arrestin-dependent agonist-induced receptor internalization in cells overexpressing a β-arrestin-EGFP fusion protein. The results revealed that the I48T mutation, which yielded dramatic enhancement of G protein activation by serotonin, gave rise to almost no effect on this alternate pathway (Fig. 3 G–J). Thus its “filtering” function is specific for those conformational changes required for G protein coupling but not those required for phosphorylation and arrestin binding. In contrast, the L379F mutation rendered D2R refractory to internalization by either dopamine or serotonin. Although this mutation reduces binding affinity for both dopamine and serotonin, the concentrations of these used in the internalization assays were high enough to be saturating. Thus this residue allosterically controls conformational switches required for both G protein activation and internalization.
Our working hypothesis is that the 15 ET residues targeted in this study work together to confer responsiveness to dopamine while minimizing responsiveness to serotonin. Unfortunately, a version of D2R with residue swaps at all positions appears to be improperly folded, as judged by a complete lack of response to either dopamine or serotonin. It seems likely that additional residues, perhaps further still from the ligand-binding site, may need to be swapped to allow proper folding in the context of all 15 residue swaps. However, we did find that combining ligand swaps can lead to additive or possibly synergistic effects (Fig. 4). The S193G mutation confers diminished dopamine affinity and enhanced serotonin affinity, without much effect on maximal responses, whereas the N124H and I48T mutations diminish dopamine responsiveness and enhance serotonin responses without much effect on affinities. Combining S193G with either N124H or I48T yields a receptor that is more like a serotonin receptor and less like a dopamine receptor in both its affinities and responsiveness. Moreover, combining N124H and I48T swaps yields higher serotonin responsiveness than either swap by itself. The most dramatic effect was observed when V91S was combined with I48T to yield a receptor whose serotonin responsiveness exceeds the response of WT D2R to saturating dopamine.
Fig. 4.

Enhanced effects of combined mutations. Activation and binding data for dopamine (A and B) or 5-HT (C and D) for D2R WT, I48T, S193G, and the indicated double mutants are shown. Activation data are representative traces for 10 μM dopamine (A) or 5-HT (C). Specific binding (B and D) was determined and curves calculated as in Fig. 3.
Discussion
The most striking conclusions from these results are that (i) evolutionary importance is a strong predictor of determinants of ligand specificity; (ii) ligand-specific responsiveness, or efficacy (20) can be separated from ligand-binding specificity, or affinity, at the structural level; and (iii) specificity of responsiveness is determined by residues that almost certainly do not contact the ligand, in contrast to binding affinity, which is largely determined by ligand-contacting residues. The first conclusion derives from the observations that all highly ranked ET residues tested significantly affected ligand binding or responsiveness, whereas poorly ranked residues had little effect. The second and third conclusions can be drawn partly from the observation that there were only three residue swaps, F110W, C118S, and S193G, that resulted in both significantly lower dopamine affinity and significantly higher serotonin affinity, and these are the only three found at positions whose corresponding residues are less than 4 Å from the ligand in structures of rhodopsin (12), β2-adrenergic receptor, and β1-adrenergic receptor (9, 10). These residues have been previously proposed to be at the surface of the orthosteric binding site (6, 21, 22). Strikingly, of the four residue swaps with the largest enhancement of serotonin responsiveness, I48T, M117F, N124H, and T205M, none is in a ligand-contacting position, and except for M117F, all are at positions at least 10 Å from the ligands in the β-adrenergic receptor structures. T205M paradoxically displays enhanced dopamine binding but reduced dopamine responsiveness. Thus a set of proximal residues largely determine binding affinities, whereas more distant residues constitute a specificity-encoding conformational filter needed to transduce ligand binding into G protein-activating conformational changes at the cytoplasmic face. Of great interest is the observation that the filtering function may or may not be specific for G protein activation. Agonist binding also induces activation of an alternate signaling pathway via GPCR kinases (GRKs), β-arrestin binding, and internalization (23). The observation that one residue, L379, is essential for the conformational changes directing both pathways, whereas another, I48, is selective for G protein activation suggests the intriguing possibility that manipulating specific ET residues by mutagenesis (24) or targeting them with allosteric ligands could be used to bias ligand activation toward specific effectors and downstream pathways, i.e., to enhance functional selectivity or “biased agonism”(25, 26) for experimental or therapeutic purposes.
These conclusions have important implications for our understanding of how allosteric coupling of agonist binding to distant G protein activation has evolved to discriminate between related ligands and for improving rational approaches to drug design. Considerable prior evidence has pointed to the importance of structural features distinct from the binding site that are important for allosteric coupling of agonist binding and G protein activation. These include the DRY motif near the cytoplasmic end of TM3 (27–29) and a glutamate residue (E368 in D2R) in TM6 that forms a salt bridge (“ionic lock”) to the arginine in DRY, as well as tryptophan and proline residues involved in a “rotamer toggle” in TM6 (30, 31), and the NPXXY motif in TM7. Unlike the residues identified in this study, however, these conformational switches are largely invariant, suggesting they mediate universal mechanisms within Class A GPCR that are unlikely to contribute to ligand specificity. By contrast, the present results highlight a previously undescribed set of more variable residues that communicate ligand binding to these generic switch motifs in a ligand-specific way. TM3 residues C118 and M117 are in direct contact with the tryptophan of the rotamer toggle switch, and TM2 residue V83L may communicate with this switch through intervening water molecules that occupy a cavity in the structure (Fig. S4). Likewise TM6 residues L379 and V381, TM7 residue N418, and TM1 residue I48 are positioned so as to influence the NPXXY motif residues through intervening waters. Several of the 15 residues are located on TM3 or TM6, helices that undergo relative motion upon activation (32, 33). These results not only reveal the existence of residues with the unique function of conferring ligand specificity through conformational filtering but also demonstrate that they can be identified computationally and mutated rationally to reprogram efficacy in an allosteric pathway to respond to alternate bound ligands, much as the tumblers of a lock may be adjusted to different keys. The spatial separation of the residues identified here emphasizes the cooperative nature of the allosteric switching induced by ligand-binding in GPCR, with a requirement for concerted movements by multiple structural elements within the transmembrane domain.
From a therapeutic perspective, our results also suggest a paradigm shift in rational, structure-based drug design. Currently such efforts are largely focused on optimizing interactions between ligand functional groups and contact residues in the binding pocket. The results presented here imply that only limited improvement in drug selectivity is likely to be obtained by focusing on such interactions, but that drugs directed to sites outside the orthosteric binding pocket and formed by residues selected during evolution to maximize response specificity might yield dramatic improvements in specificity and/or efficacy either alone or in combination with conventional binding-pocket-directed drugs. Allosteric modulators of this type have already been identified for metabotropic glutamate receptors (34) and in a cannabinoid receptor (35). The sites defined by the residues identified in this study represent potential targets for rational drug design.
In addition to the ligand-specificity determinants identified here in D2R, ET has previously successfully identified residues critical for function in rhodopsin and related Class A GPCR (16) as well as residues that discriminate between G protein signaling and arrestin signaling in the β2-adrenergic receptor (24). Combining the evolutionary trace with crystal structures or homology models of target receptors will be an important part of this approach to drug discovery.
The essential features of the approach are as follows. ET ranks the importance of every residue based on whether its sequence variations correlate mostly with major or with minor evolutionary divergences (15). This is the computational equivalent of laboratory experiments that measure residues importance by how much their mutations perturb the functional response of an assay. In practice, starting with a protein family, its aligned sequences, and the associated divergence tree, ET identifies and ranks best alignment positions that vary between major branches but that have low variation entropy within each one of these branches. Conversely, it will rank poorly positions that vary even between closely related species (36). Important properties of top-ranked ET residues are that they cluster spatially within the structure of a representative family member (37), that these clusters identify functional sites (38), and that they efficiently guide experiments to functional determinants (24, 39–42). Although individual case studies cannot guarantee generality, structural motifs of just a few top-ranked ET residues also prove sufficient to predict protein function on a structural proteomic scale, which is consistent with accurate and large-scale identification of key functional determinants (43–45).
Although there are several useful approaches for identifying functionally important sites in GPCR, the efficiency of the approach described here for finding specificity determinants rests on the likelihood that virtually every amino acid substitution at every position has been tested in the course of evolution so that the evolutionary record of variation and of divergence provides orders of magnitude more information than any strictly experimental approach such as random or targeted mutagenesis or accessibility to chemical modification (46). In particular, these approaches are unlikely to predict residues critical for ligand-selective allosteric conformational coupling and have tended to yield information primarily about the orthosteric binding site.
Beyond its practical implications, the recoding of efficacy by individual mutations casts a unique light on GPCR evolutionary mechanisms. Our observations that single residue variations trigger diverse, sometimes opposite, efficacy changes in distinct ligands implies that, during evolution, single point variations can perturb simultaneously ligand affinities, efficacies, and potentially bias toward downstream signaling pathways, as suggested by the successful use of ET-guided mutations to reprogram effector bias (24). Over evolutionary time, variations would probe a large array of functional changes that reprogram the type, distribution, and strengths of couplings between extracellular upstream ligands and intracellular downstream responses, thus providing, from a network perspective, an evolutionary switchboard mechanism to constantly reconfigure the inputs and outputs at GPCR nodes.
Material and Methods
Evolutionary Trace Analysis.
Evolutionary trace analyses were performed with the real-value ET (38) in bioamine and class A GPCRs using 402 and 2512 sequences, respectively. These sequences were gathered from GPCRDB and aligned separately using ClustalW. As before, alignments were performed on 195 gapless seven transmembrane helix residues, because the loops are highly divergent (16). The residues ranked in the top 20th percentile of importance in the bioamine specific and Class A traces were mapped onto the β2-adrenergic receptor (PDB 2RH1, Fig. S1) and clustered significantly: with z scores of 12.7 and 12.5, respectively (47). The difference between them revealed a significant cluster (z score = 5.4) of 10 amino acids traced in bioamine receptors and not in Class A overall (Fig. S1C). Eight of these bioamine-specific positions clustered within 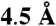 of each other and thus formed a structurally determined core of residues (z score 7.1, Fig. S1C) that should be especially important to bioamine receptor specific functions. An additional nine residues that are important at the 20th percentile for class A GPCRs as a whole, are within
of each other and thus formed a structurally determined core of residues (z score 7.1, Fig. S1C) that should be especially important to bioamine receptor specific functions. An additional nine residues that are important at the 20th percentile for class A GPCRs as a whole, are within 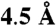 of this initial set (Fig. S1F). Sequence comparison between D2R and 5-HT2AR reveals that all but one of these 17 positions are identical. Extending the criteria to include both Class A and bioamine-specific ET residues to the 40th percentile and within
of this initial set (Fig. S1F). Sequence comparison between D2R and 5-HT2AR reveals that all but one of these 17 positions are identical. Extending the criteria to include both Class A and bioamine-specific ET residues to the 40th percentile and within 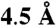 any of the above 17 positions expanded the target residues to 48 (Fig. S1 J and N). Of these, 15 vary between D2 and 5-HT2A receptors and were used for functional-swapping experiments (Fig. S1O). The sequences and alignments inputs, as well as the trace rank file outputs used for these analyses, may be found and reproduced on our GPCR difference-ET server at http://mammoth.bcm.tmc.edu/gpcr/diff_GPCRpaper.html. (The visual tool available in beta version.)
any of the above 17 positions expanded the target residues to 48 (Fig. S1 J and N). Of these, 15 vary between D2 and 5-HT2A receptors and were used for functional-swapping experiments (Fig. S1O). The sequences and alignments inputs, as well as the trace rank file outputs used for these analyses, may be found and reproduced on our GPCR difference-ET server at http://mammoth.bcm.tmc.edu/gpcr/diff_GPCRpaper.html. (The visual tool available in beta version.)
Mutagenesis and Cell Transfection.
The cDNA clone for human dopamine D2Rlong receptor was obtained from the Missouri S&T cDNA Resource Center (www.cdna.org). D2R mutants were generated using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA) and confirmed by DNA sequencing (Lone Star Labs, Houston). All mutants were generated in the human D2Rlong tagged in the N-terminal with the HA epitope.
HEK cells (American Type Culture Collection, Manassas, VA) were stably transfected with the Gα16 cDNA (a gift from M. Simon, Caltech) using lipofectamine 2000 (Invitrogen, Carlsbad, CA) as described by the manufacturer. Cells were maintained in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum supplemented with penicillin, streptomycin, L-glutamine, and G418 (250 μg/l). Cells were grown in a 37 °C humidified environment with 5% CO2. For receptor activation assays, HEK-Gα16 cells were reverse transfected in poly-D-lysine-coated 96-well plates and assays performed 48 hrs post transfection.
Calcium Release Assays.
At the time of assay, cells were washed once with Krebs/Ringer/Hepes (KRH) buffer (120 mM NaCl, 4.7 mM KCl, 2.2 mM CaCl2, 10 mM Hepes, 1.2 mM KH2PO4, 1.2 mM MgSO4, pH 7.4) supplemented with 1.8 g/L glucose. Cells were incubated with 2.5 μM Fluo-4 AM (Invitrogen) for 1 hr at room temperature with pluronic acid F-127 (final conc. 0.02% v/v), and maintained in 1 mM probenecid to inhibit dye efflux. Ca2+-enhanced fluorescence was detected using a Synergy HT plate reader (Biotek, Winooski, VT). Fluorescence was measured for 10 s to establish baseline, and then at t = 10 seconds, drugs (diluted in KRH) were injected into the well and fluorescence measured every half second for 1 min. On the same plate, receptor surface expression was detected using immunofluorescence. Briefly, cells were fixed with 4% paraformaldehyde for 1hr at RT. Cells were incubated with Phosphate Saline buffer (PBS) supplemented with 5% donkey serum (Jackson ImmunoResearch, West Grove, PA) followed by incubations with a mouse anti-HA epitope (Santa Cruz Biotechnologies, Santa Cruz, CA) and Alexa Fluor 488-conjugated donkey antimouse antibody (Invitrogen). Receptor activation data were normalized by the receptor surface expression and traces plotted using GraphPad Prism. Baseline was subtracted to all traces. In some experiments immunolabeling was also carried out in the presence of detergent (0.1% Triton X in TBST with 5% Donkey serum) to check for any major differences in the ratios of surface receptor to total receptor.
Internalization Assays.
HEK WT or HEK-Gα16 cells were transfected, fixed, and protein expression (total and surface) determined as described above. Cells were cotransfected either with receptor-expressing plasmids alone, or with equal amount of receptor and β-arrestin-EGFP cDNAs. Forty eight h after transfection, either HEK WT or HEK-Gα16 were treated at 37 °C with 10 μM dopamine or 5-HT at different time points. Immunodetection was performed by incubating the cells with mouse anti-HA antibody followed by incubations with an Alexa Fluor 594-conjugated donkey antimouse antibody. Endpoint fluorescence values were detected using a Synergy HT plate reader. Cells transfected with pcDNA 3.1(+) instead of receptor cDNA were used to subtract nonspecific fluorescence.
[3H] Spiperone Binding.
Membranes were prepared from transiently transfected HEK-Gα16 cells. 48 h after transfection, cells were scrapped and centrifuged at 1000 × g for 5 min at 4 °C, and the pellets were resuspended in 1 ml of hypotonic buffer (10 mM Tris, 3 mM MgCl2, 2 mM EDTA, pH 7.4) supplemented with protease inhibitors. The homogenates were centrifuged at 13,500 × g for 10 min at 4 °C, and the membrane pellets (from a confluent 100-mm dish) were resuspended by homogenization in 1 ml of binding buffer (10 mM Tris, 150 mM NaCl, pH 7.4). Aliquots of membrane suspension were incubated in binding buffer either with different concentrations of the antagonist [3H] spiperone (in saturation binding experiments) or with increasing concentrations of agonists or antagonists in the presence of 0.6 nM [3H] spiperone (105 Ci/mmol) (in competition binding experiments). Samples were incubated for 90 min at RT prior to filtration through Whatman GF/C filters using a 96-well Inotech filtration apparatus and counted using a Beckman LS1701 scintillation counter. Specific [3H] spiperone binding was defined as total binding less nonspecific binding in the presence of 1 µM sulpiride (Sigma Aldrich, St. Louis). Saturation and competition binding data were analyzed by nonlinear regression analysis using GraphPad Prism 3.0 (GraphPad Software, San Diego), and the equation, B = BmaxL/(L + Kd), where B is bound spiperone, and L is the spiperone concentration. IC50 values were obtained by fitting the data from the competition studies to a one-site competition model. Ki values were determined using the equation Ki = IC50/(1 + L/KD), where L is the concentration of radioligand, and KD is the spiperone concentration for half-maximal binding of [3H] spiperone in the absence of competitor.
Supplementary Material
Acknowledgments.
We thank Monica Galaz Montoya for performing some assays and Rhonald Lua for help with the difference-ET server. This work was supported by grants from the National Institutes of Health (GM066099 and GM079656 to O.L.) and by the Welch Foundation (Q0035 to T.G.W.) and NIH training fellowships (T90 DK070109 and T15 LM007093 to G.J.R.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.0914877107/-/DCSupplemental.
References
- 1.Huber T, Menon S, Sakmar TP. Structural basis for ligand binding and specificity in adrenergic receptors: Implications for GPCR-targeted drug discovery. Biochemistry. 2008;47:11013–11023. doi: 10.1021/bi800891r. [DOI] [PubMed] [Google Scholar]
- 2.Gether U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocr Rev. 2000;21:90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- 3.Rothman RB, Baumann MH. Therapeutic potential of monoamine transporter substrates. Curr Top Med Chem. 2006;6:1845–1859. doi: 10.2174/156802606778249766. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez-Maeso J, Sealfon SC. Agonist-trafficking and hallucinogens. Curr Med Chem. 2009;16:1017–1027. doi: 10.2174/092986709787581851. [DOI] [PubMed] [Google Scholar]
- 5.Javitch JA. Mapping the binding-site crevice of the D2 receptor. Adv Pharmacol. 1998;42:412–415. doi: 10.1016/s1054-3589(08)60776-0. [DOI] [PubMed] [Google Scholar]
- 6.Simpson MM, et al. Dopamine D4/D2 receptor selectivity is determined by a divergent aromatic microdomain contained within the second, third, and seventh membrane-spanning segments. Mol Pharmacol. 1999;56:1116–1126. doi: 10.1124/mol.56.6.1116. [DOI] [PubMed] [Google Scholar]
- 7.Kalani MY, et al. The predicted 3D structure of the human D2 dopamine receptor and the binding site and binding affinities for agonists and antagonists. Proc Natl Acad Sci USA. 2004;101:3815–3820. doi: 10.1073/pnas.0400100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lan H, Durand CJ, Teeter MM, Neve KA. Structural determinants of pharmacological specificity between D(1) and D(2) dopamine receptors. Mol Pharmacol. 2006;69:185–194. doi: 10.1124/mol.105.017244. [DOI] [PubMed] [Google Scholar]
- 9.Cherezov V, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Warne T, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaakola VP, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Palczewski K, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 13.Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- 14.Leach K, Sexton PM, Christopoulos A. Allosteric GPCR modulators: Taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28:382–389. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- 16.Madabushi S, et al. Evolutionary trace of G protein-coupled receptors reveals clusters of residues that determine global and class-specific functions. J Biol Chem. 2004;279:8126–8132. doi: 10.1074/jbc.M312671200. [DOI] [PubMed] [Google Scholar]
- 17.Raviscioni M, He Q, Salicru EM, Smith CL, Lichtarge O. Evolutionary identification of a subtype specific functional site in the ligand binding domain of steroid receptors. Proteins. 2006;64:1046–1057. doi: 10.1002/prot.21074. [DOI] [PubMed] [Google Scholar]
- 18.Ballesteros J, Weinstein H. Integrated methods for modeling G-protein coupled receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 19.Al-Fulaij MA, Ren Y, Beinborn M, Kopin AS. Identification of amino acid determinants of dopamine 2 receptor synthetic agonist function. J Pharmacol Exp Ther. 2007;321:298–307. doi: 10.1124/jpet.106.116384. [DOI] [PubMed] [Google Scholar]
- 20.Kenakin T. Efficacy at G-protein-coupled receptors. Nat Rev Drug Discov. 2002;1:103–110. doi: 10.1038/nrd722. [DOI] [PubMed] [Google Scholar]
- 21.Wiens BL, Nelson CS, Neve KA. Contribution of serine residues to constitutive and agonist-induced signaling via the D2S dopamine receptor: Evidence for multiple, agonist-specific active conformations. Mol Pharmacol. 1998;54:435–444. doi: 10.1124/mol.54.2.435. [DOI] [PubMed] [Google Scholar]
- 22.Javitch JA, Li X, Kaback J, Karlin A. A cysteine residue in the third membrane-spanning segment of the human D2 dopamine receptor is exposed in the binding-site crevice. Proc Natl Acad Sci USA. 1994;91:10355–10359. doi: 10.1073/pnas.91.22.10355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeWire SM, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol. 2007;69:483–510. doi: 10.1146/annurev.physiol.69.022405.154749. [DOI] [PubMed] [Google Scholar]
- 24.Shenoy SK, et al. beta-arrestin-dependent, G protein-independent ERK1/2 activation by the beta2 adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 25.Urban JD, et al. Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 26.Violin JD, Lefkowitz RJ. Beta-arrestin-biased ligands at seven-transmembrane receptors. Trends Pharmacol Sci. 2007;28:416–422. doi: 10.1016/j.tips.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 27.Kim JH, Cho EY, Min C, Park JH, Kim KM. Characterization of functional roles of DRY motif in the 2nd intracellular loop of dopamine D2 and D3 receptors. Arch Pharm Res. 2008;31:474–481. doi: 10.1007/s12272-001-1181-x. [DOI] [PubMed] [Google Scholar]
- 28.Rovati GE, Capra V, Neubig RR. The highly conserved DRY motif of class A G protein-coupled receptors: Beyond the ground state. Mol Pharmacol. 2007;71:959–964. doi: 10.1124/mol.106.029470. [DOI] [PubMed] [Google Scholar]
- 29.Bhattacharya S, Hall SE, Li H, Vaidehi N. Ligand-stabilized conformational states of human beta(2) adrenergic receptor: Insight into G-protein-coupled receptor activation. Biophys J . 2008;94:2027–2042. doi: 10.1529/biophysj.107.117648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi L, et al. Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem. 2002;277:40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- 31.Swaminath G, et al. Probing the beta2 adrenoceptor binding site with catechol reveals differences in binding and activation by agonists and partial agonists. J Biol Chem. 2005;280:22165–22171. doi: 10.1074/jbc.M502352200. [DOI] [PubMed] [Google Scholar]
- 32.Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- 33.Sheikh SP, Zvyaga TA, Lichtarge O, Sakmar TP, Bourne HR. Rhodopsin activation blocked by metal-ion-binding sites linking transmembrane helices C and F. Nature. 1996;383:347–350. doi: 10.1038/383347a0. [DOI] [PubMed] [Google Scholar]
- 34.Kew JN. Positive and negative allosteric modulation of metabotropic glutamate receptors: Emerging therapeutic potential. Pharmacol Ther. 2004;104:233–244. doi: 10.1016/j.pharmthera.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 35.Price MR, et al. Allosteric modulation of the cannabinoid CB1 receptor. Mol Pharmacol. 2005;68:1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- 36.Mihalek I, Res I, Lichtarge O. A family of evolution-entropy hybrid methods for ranking protein residues by importance. J Mol Biol. 2004;336:1265–1282. doi: 10.1016/j.jmb.2003.12.078. [DOI] [PubMed] [Google Scholar]
- 37.Madabushi S, et al. Structural clusters of evolutionary trace residues are statistically significant and common in proteins. J Mol Biol. 2002;316:139–154. doi: 10.1006/jmbi.2001.5327. [DOI] [PubMed] [Google Scholar]
- 38.Yao H, et al. An accurate, sensitive, and scalable method to identify functional sites in protein structures. J Mol Biol. 2003;326:255–261. doi: 10.1016/s0022-2836(02)01336-0. [DOI] [PubMed] [Google Scholar]
- 39.Sowa ME, et al. Prediction and confirmation of a site critical for effector regulation of RGS domain activity. Nat Struct Biol. 2001;8:234–237. doi: 10.1038/84974. [DOI] [PubMed] [Google Scholar]
- 40.Ribes-Zamora A, Mihalek I, Lichtarge O, Bertuch AA. Distinct faces of the Ku heterodimer mediate DNA repair and telomeric functions. Nat Struct Mol Biol. 2007;14:301–307. doi: 10.1038/nsmb1214. [DOI] [PubMed] [Google Scholar]
- 41.Kobayashi H, Ogawa K, Yao R, Lichtarge O, Bouvier M. Functional rescue of beta-adrenoceptor dimerization and trafficking by pharmacological chaperones. Traffic. 2009;10:1019–1033. doi: 10.1111/j.1600-0854.2009.00932.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baameur F, et al. Role for the regulator of G-protein signaling homology domain of G protein-coupled receptor kinases 5 and 6 in beta 2-adrenergic receptor and rhodopsin phosphorylation. Mol Pharmacol. 2010;77:405–415. doi: 10.1124/mol.109.058115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ward RM, et al. Evolutionary Trace Annotation Server: automated enzyme function prediction in protein structures using 3D templates. Bioinformatics. 2009;25:1426–1427. doi: 10.1093/bioinformatics/btp160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ward RM, et al. De-orphaning the structural proteome through reciprocal comparison of evolutionarily important structural features. PLoS ONE. 2008;3:e2136. doi: 10.1371/journal.pone.0002136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Erdin S, Ward RM, Venner E, Lichtarge O. Evolutionary trace annotation of protein function in the structural proteome. J Mol Biol. 2010;396:1451–1473. doi: 10.1016/j.jmb.2009.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Javitch JA, et al. The fourth transmembrane segment of the dopamine D2 receptor: Accessibility in the binding-site crevice and position in the transmembrane bundle. Biochemistry. 2000;39:12190–12199. doi: 10.1021/bi001069m. [DOI] [PubMed] [Google Scholar]
- 47.Mihalek I, Res I, Yao H, Lichtarge O. Combining inference from evolution and geometric probability in protein structure evaluation. J Mol Biol. 2003;331:263–279. doi: 10.1016/s0022-2836(03)00663-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.