

Abstract
The earliest stages of animal development occur without the benefit of zygotic transcription. The absence of transcription necessitates that all changes in the levels of specific proteins must be controlled by post-transcriptional mechanisms, such as the regulated translation of stored maternal mRNAs. One of the major challenges to investigating translational mechanisms is the availability of reliable methods for assaying the translational state of specific mRNAs. The most definitive assay of an mRNA's translational state is polyribosome association; mRNAs actively translated are engaged with polyribosomes while mRNAs translationally repressed are not. While linear gradient centrifugation is commonly used to purify polyribosomes from a wide variety of cell types in different organisms, the isolation of polyribosomes from Xenopus oocytes, eggs and embryos presents some unique challenges. Here we detail the methodology for the isolation and analysis of polyribosomes from Xenopus oocytes, eggs and embryos using step gradient centrifugation. We present detailed protocols, describe the critical controls and provide several examples to guide the interpretation of experimental results regarding the translational state of specific mRNAs.
Keywords: translational control, repression, polyribosome, Xenopus development
1. Introduction
The earliest stages of animal development occur in the absence of zygotic transcription (1-4). These circumstances necessitate that all changes in the levels of specific proteins must be controlled by post-transcriptional mechanisms, such as the regulated translation of stored maternal mRNAs. For example, in the oocytes of frogs, mice and marine invertebrates, mRNAs encoding the c-mos and cyclin proteins are translationally repressed. During oocyte maturation the translation of these mRNAs is activated and the synthesis of the c-mos and cyclin proteins drives the completion of meiotic maturation (5-8).
Many cell fate decisions in vertebrate embryos are initiated during the post-transcriptional phase of development and are subject to translational control (1, 9). Embryonic cells with different fates accumulate unique maternal proteins, referred to as maternal determinants. These determinants activate specific signaling pathways that in turn ultimately activate specific sets of zygotic genes. For example, translation of the maternal mRNA encoding the Xenopus Wnt11 protein is activated in the embryonic cells that will direct the formation of Spemann's organizer. Blocking the translation of the Wnt11 mRNA disrupts Wnt signaling and embryos develop abnormally due to defects in organizer formation and function (10-12). Together these and other examples demonstrate that the regulated translation of maternal mRNAs is important for promoting oocyte maturation, driving the embryonic cell divisions and modulating cell fate decisions in all metazoan organisms (13-17).
Polyribosome isolation and analysis
One of the major challenges to investigating translational mechanisms is the availability of reliable methods for assaying the translational state of specific mRNAs. The most definitive assay of an mRNA's translational state is polyribosome association; mRNAs actively translated are engaged with polyribosomes while mRNAs translationally repressed are not. The traditional assay used to monitor mRNA translation and polyribosome association involves fractionating cell lysates on a linear sucrose gradient (18-22). The resulting gradient fractions are analyzed to reveal the presence of the characteristic repeating UV absorption peaks corresponding to polyribosomes of varying sizes that migrate at the bottom of the gradient. The individual polyribosome and non-polyribosome fractions are analyzed to determine the presence of specific mRNAs and reveal their translational activity.
Xenopus polyribosomes
While linear gradient centrifugation is commonly used to purify polyribosomes from a wide variety of cell types in different organisms the isolation of polyribosomes from Xenopus oocytes, eggs and embryos presents some unique challenges. First, oocytes, eggs and embryos contain large quantities of carbohydrates, membranes and yolk proteins (23). The presence of these potentially interfering components must be considered when generating and using extracts for polyribosome isolation. For example, the detergents that disrupt membranes are a critical component of the buffers used for the preparation of extracts from Xenopus. Otherwise, if the membranes were left intact they could potentially interfere with gradient fractionation. Second, the polyribosome content of Xenopus oocytes, eggs and embryos is quite low compared to tadpole stage embryos or other cell types (24). Consequently, the fractions from linear gradient analysis of extracts from these sources lack the series of peaks that typically indicate the presence of polyribosomes. This paucity of polyribosomes makes it more practical to use simpler step gradients instead of linear gradients for Xenopus polyribosome isolation (25-27). Third, one reason that polyribosome content of Xenopus oocytes, eggs and embryos contain relatively few polyribosomes is that many mRNAs in these cells are translationally repressed (15). The repressed mRNAs reside in specific large mRNP particles and these particles can migrate in sucrose gradients at the same position as polyribosomes. Such co-migration can make it difficult to determine whether an mRNA resides within a repressive mRNP or is associated with polyribosomes. Therefore, it is imperative that all experiments include appropriate controls to address this issue and distinguish between these possibilities.
Here we detail the methodology for the isolation and analysis of polyribosomes from Xenopus oocytes, eggs and embryos using step gradient centrifugation. We present detailed protocols, describe the critical controls and provide several examples to guide the interpretation of experimental results regarding the translational state of specific mRNAs.
2. Description of method
2.1 Overview
To analyze the translational state of specific mRNAs, polyribosomes are isolated from Xenopus oocytes, eggs and embryos using a simple sucrose step gradient (Figure 1) (27, 28). The resulting material in the pellet fraction contains the polyribosomes and associated mRNAs while the supernatant fraction contains the non-polyribosome associated mRNAs. To ensure that mRNAs are present in the pellet fraction due to their association with polyribosomes each fractionation experiment is repeated with the addition of EDTA to the polyribosome buffers. EDTA chelates Mg2+ ions, causing polyribosomes to dissociate and release bound mRNAs into the supernatant fraction. Therefore the analysis of each sample involves performing two fractionations in parallel; one under standard conditions and one in the presence of EDTA. The quantitative analysis of mRNAs from the polyribosomal and non-polyribosomal fractions can reveal the translational status of specific maternal mRNAs. This general strategy readily lends itself to monitoring how translation of a specific mRNA changes during the course of development or comparing the translation of different mRNAs within a single cell type.
Figure 1. Monitoring the translational behavior of Xenopus mRNAs by polyribosome analysis.
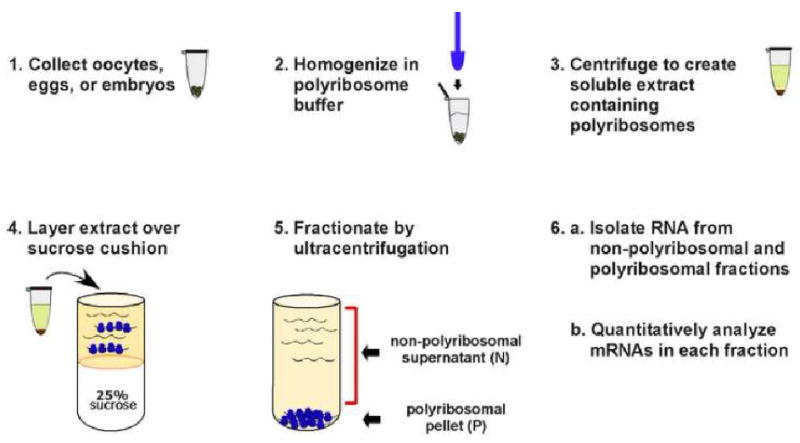
Extacts prepared from oocytes, eggs or are fractionated using sucrose step gradients. During centrifugation the non-polyribosomal mRNAs remain in the supernatant fraction while the polyribosome associated mRNAs partition to the bottom of the tube. RNAs extracted from the non-polyribosomal and polyribosomal fractions are analyzed using quantitative methods to determine whether specific mRNAs are polyribosome associated and therefore translationally active.
2.2 Detailed methods
2.2.1 Collection of oocytes, eggs and embryos
Oocytes were harvested from anesthetized females (0.02% benzocaine) and manually defolliculated with forceps in oocyte ringers (100mM NaCl, 1.8mM KCl, 1mM MgCl2, 2mM CaCl2, 5mM NaHCO3 pH 7.0) (29). Xenopus oocytes, eggs and embryos were obtained as described (29). Oocytes were collected and used immediately or frozen on dry ice and stored at -80°C for later use. Eggs were harvested from females injected with chorionic gonadotropin (Sigma). For fertilization, eggs were collected in a glass dish and mixed with macerated testes. After 10 minutes the dish was filled with 0.25× MMR and embryos were allowed to develop at room temperature. Eggs and embryos were dejellied with 2% cysteine-HCl, pH 8.0 and used immediately or frozen on dry ice and stored at -80°C for later use. We have compared polyribosome analysis from fresh versus frozen oocytes, eggs and embryos and have not observed a significant difference between frozen and non-frozen samples (Fritz and Sheets, unpublished).
2.2.2 Cell lysate preparation and gradient fractionation
Polyribosome buffer (PB-300mM KCl, 2mM MgCl2, 20mM Tris-HCl pH 7.5, 4μg/ml polyvinylsulfate) is prepared on ice (27). Immediately prior to use, polyribosome buffer is supplemented with sodium deoxycholate to 0.5% (from freshly prepared 10% w/v stock), dithiothreitol (DTT) to 4mM and rRNasin (Promega) to 25U/ml. A maximum of twenty, but no fewer than five oocytes, eggs or embryos are homogenized on ice (4°C) in 500μl of polyribosome buffer (PB). An additional 500μl of PB is added to the homogenate and mixed by pipetting several times. Homogenates are spun in a microfuge at 12,000× g, 4°C for 15 minutes. The supernatant from the clearing spin is removed, taking care not to disturb the cell debris pellet, and transferred to a 15ml falcon tube. To each supernatant either 2ml of PB or 2ml of PB plus 15mM EDTA is added. Diluted supernatants are layered over 2.5ml of PB, or PB+EDTA, containing 25% (w/v) sucrose and subjected to ultracentrifugation at 149,000× g, 4°C for 2 hours, in a Beckman SW55Ti rotor.
2.2.3 Fraction collection and RNA extraction
Following ultracentrifugation, the supernatant fractions are recovered (taking care not to disturb the pellet) and transferred to sterile 10ml centrifuge tubes. RNA is isolated from the nonpolyribosomal supernatant fractions with two extractions using an equal volume of phenol/chloroform. The extracted RNA is precipitated overnight with 2.5 volumes of ethanol, 1/10th volume of 5M NaCl, and 50μg of glycogen at -20°C. The RNA is further purified by addition of an equal volume of 8M LiCl and precipitation at -20°C for at least 4 hours. The precipitate is collected by microcentrifugation (10,000×g, 4°C for 30 minutes), washed with 150μl of cold 70% ethanol, resuspended in DEPC-treated water, and stored at -80 °C.
2.2.4 Isolation of RNA from polyribosomal pellet
The polyribosome pellet is resuspended in 800 μl Trizol and incubated for 5 min. After the addition of 160μl of chloroform, the sample is vortexed for 15 seconds and incubated 2 minutes at room temperature. Following microcentrifugation (12,000 × g, 15 minutes, 4°C) the recovered aqueous layer is mixed with an equal volume of phenol/chloroform, vortexed and the mixture spun for 2 minutes at 12,000 × g. The aqueous layer is mixed with 450μl isopropanol, vortexed and incubated 10 minutes at room temperature. After microcentrifugation (10 minutes, 12,000 × g, 4°C) the supernatant is removed and the pellet washed (70% ethanol). The pellet is air dried briefly at room temperature and resuspended in 30 μl DEPC-H2O.
2.2.5 The isolation of RNA from unfractionated Xenopus oocytes, eggs and embryos
Total RNA was isolated from Xenopus oocytes, eggs or embryos by Trizol extraction (Gibco BRL) following the manufacturer's instructions, with the addition of a phenol/chloroform extraction prior to the precipitation of RNA with isopropanol. RNA was resuspended in DEPC-treated water and stored at -80°C.
2.2.6 Quantitative RNA analysis
To reliably monitor the translation of specific mRNAs and compare the results from different samples and experiments requires methods for accurately detecting specific mRNAs in the polyribosome and nonpolyribosome fractions. We rely upon quantitative RNA blot hybridization (5). Quantitative RT-PCR can be used but this method relies upon oligo-dT priming via the mRNA's poly (A) tail for first strand cDNA synthesis. Since polyadenylation of Xenopus maternal mRNAs is highly regulated, RT-PCR may not the ideal strategy for detecting specific mRNAs. This problem with RT-PCR could be potentially avoided by using random hexamers to prime first-strand cDNA synthesis instead of oligo-dT. Random hexamer priming can also be problematic as there is a vast excess of rRNAs and tRNAs relative to mRNAs in Xenopus oocytes, eggs and embryos. The presence of these abundant RNAs makes efficient cDNA synthesis with random priming challenging and thus mRNAs may not be accurately represented in the cDNA.
2.2.7 RNA Blot Hybridization Analysis
RNA for analysis is treated with deionized glyoxal and subjected to electrophoresis through 1% agarose (Seakem LE), 10mM sodium phosphate (pH 7.0) at 50 volts for 3-4 hours with recirculation (5). RNA is transferred to Pall Biodyne A nylon membranes via capillary action with 20× SSC for 16-24 hours. The transferred RNA is fixed to the membrane by UV crosslinking and baking at 80°C for two hours. Glyoxal adducts are removed by boiling the filters in 20mM Tris, pH 8.0 for 3 minutes. To detect specific mRNAs, single-stranded antisense DNA probes are generated with asymmetric PCR using either phage promoter primers or template - specific primers (100pmol), 1μg of template DNA, 0.2mM dTTP, dGTP and dATP, 0.02mM dCTP and 50-150μCi of 32P-dCTP per 50μl reaction. Probes are purified using G-50 columns as recommended by the manufacturer (General Electric). Filters are prehybridized at least 1 hour at 65°C in hybridization solution (1% BSA, 7% SDS, 0.25mM NaPhosphate, pH 7.2 and 1mM EDTA). Following replacement of the hybridization solution, filters are hybridized overnight at 65°C with 1.0×106 to 3.5×106 cpm/ml of probe. Hybridized filters are washed 3 times for 30 minutes in 250ml wash solution (5% SDS, 40mM NaPhosphate, pH 7.2 and 1mM EDTA) at 65°C. Longer wash times (up to 16 hours) are necessary when filters were simultaneously hybridized with multiple probes. Hybridization signals are quantified using a phosphorimager (Molecular Dynamics).
3. Example of polyribosome analysis
To illustrate how polyribosome analysis can be used to investigate the translational activity of Xenopus maternal mRNAs we will use the results from our previous study of mRNAs that encode proteins of the BMP signaling pathway, Fritz and Sheets 2001 (27) (Figures 2A and B).
Figure 2. The polyribosome association of maternal mRNAs encoding proteins of the BMP signaling pathway is regulated during oocyte maturation and embryogenesis.
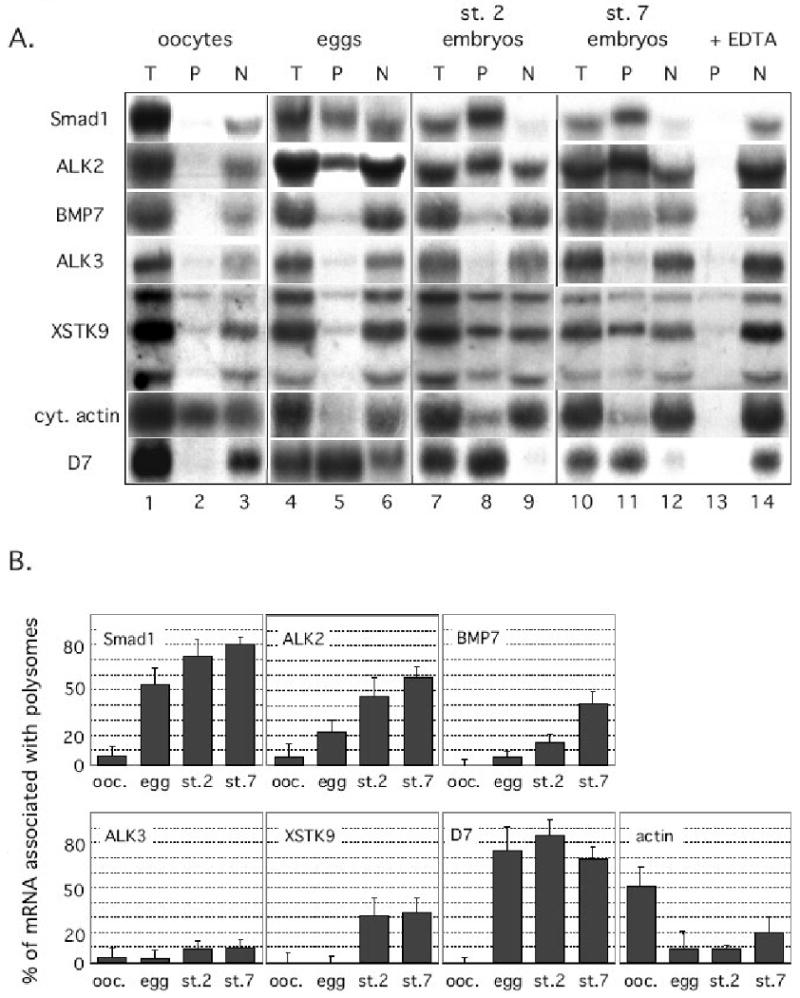
[A] Polyribosomal (P) and non-polysomal (N) fractions were prepared from Xenopus laevis oocytes (lanes 1-3), eggs (lanes 4-6), 2-cell stage embryos (lanes 7-9) and stage 7 blastula embryos (lanes 10-14). Total RNA was isolated from unfractionated samples (T), the polyribosomal (P) and non-polyribosomal supernatant (N) fractions. The RNAs were analyzed by blot hybridization. Filters were hybridized with probes to detect Xenopus mRNAs encoding the ALK2 type I receptor, the ALK3 type I receptor, the BMP7 ligand, the Smad1 transcription factor, the XSTK9 type II receptor, the D7 and cytoskeletal actin proteins. Shown is a representative RNA blot analyzing each mRNA at each stage of development. The EDTA control experiment is only shown for the st.7 polyribosome fractionation (lanes 13 and 14). mRNAs that are genuinely associated with polyribosomes will fractionate with the non-polyribosomal supernatant in the presence of EDTA. [B] Quantitative analysis of polyribosome association. The fraction of each mRNA population associated with polyribosomes at different stages of Xenopus development was quantitated using a phosphorimager (Molecular Dynamics). The RNA blots from three independent experiments were analyzed and the average polyribosome association was calculated for each mRNA and plotted for each stage of development. Polyribosome association was defined as the amount of mRNA in the pellet fraction minus the amount of RNA in the pellet that forms in the presence of EDTA. Reprinted from Developmental Biology, 2001. 236(1): p. 230-243. Regulation of the mRNAs encoding proteins of the BMP signaling pathway during the maternal stages of Xenopus development. Fritz, B.R. and M.D. Sheets Developmental Biology, 2001. 236(1): p. 230-243 with permission from Elsevier.
Lysates from Xenopus oocytes, unfertilized eggs and embryos were separated into polyribosomal (P) and nonpolyribosomal (N) fractions. Each fraction was analyzed using quantitative RNA blot hybridization to detect the presence of mRNAs encoding the BMP7 ligand, two closely related putative BMP type I receptors (ALK2 and ALK3), a BMP type II receptor (XSTK9) and a BMP-specific transcription factor (Smad1) (30-37). A sample of these results is shown in Figure 2A and a quantitative summary of three independent experiments is shown in Figure 2B.
Analyzing the polyribosome association of mRNAs that encode proteins of the BMP pathway at different stages of development indicate that the translation of these mRNAs is highly regulated. All mRNAs analyzed were inefficiently associated with polyribosomes and found predominantly in the nonpolyribosomal (N) fraction from oocytes (Fig. 2A, compare lanes 2 and 3). The small amount of the mRNA encoding the XSTK9 type II BMP receptor present in the polyribosomal fraction from both oocytes and eggs (Fig. 2A, lanes 2 and 5) was not due to a bona fide ribosome association, since these interactions were not disrupted by EDTA (not shown). During oocyte maturation, a significant fraction of the ALK2 and Smad1 mRNA populations load onto polyribosomes (Fig. 2A, compare lanes 2 and 5; Fig. 2B: 22% of the ALK2 mRNA and 53% of the Smad1 mRNA was associated with polyribosomes in eggs). In contrast, the ALK3 and XSTK9 mRNAs remained predominantly in the non-polyribosomal fraction from eggs (Fig. 2A, compare lanes 2 and 5).
Additional translational regulation was observed when polyribosome analysis was performed using Xenopus embryos. The amounts of the BMP7 and XSTK9 mRNAs associated with polyribosomes increased significantly compared to unfertilized eggs by the early blastula stages (Fig. 2A, st.7 embryos, compare lanes 8 and 11, Fig. 2B). In contrast, the association of ALK3 mRNA with polyribosomes was not efficient at any stage examined (Fig. 2A, lanes 2, 5, 8 and 11, Fig. 2B). Thus, the BMP7 and XSTK9 mRNAs are efficiently recruited to polyribosomes during Xenopus embryogenesis, while the ALK3 mRNA was not associated with polyribosomes in oocytes, eggs and maternal embryonic stages.
Activation of the Xenopusbone morphogenetic protein (BMP) pathway during early development requires maternal signaling proteins, but that requirement is not understood. Our polyribosome analysis indicates that the translation of several different mRNAs encoding proteins of the BMP pathway is regulated during Xenopus development. These mRNAs were either not associated or inefficiently associated with polyribosomes in oocytes, and each was recruited to polyribosomes at different stages of developmental. Such temporal regulation may be one mechanism to ensure the appropriate assembly and activation of the BMP pathway.
Important controls
There are three important controls that need to be performed for each sample analyzed. First, the amount of each mRNA in the combined polyribosome and non-polyribosome fractions is compared to the amount of each mRNA present in the equivalent amount of unfractionated material (total RNA) for each sample. Only samples from experiments where the amount of each message in the combined polyribosomal pellet and nonpolyribosomal supernatant is at least 75% of the unfractionated starting material are used for analysis. This ensures that substantial amounts of material are not lost during fractionation. Second, the polyribosome association of mRNAs whose translational behaviors have been well characterized must be analyzed for each sample. For these controls we use the D7 and cytoskeletal actin mRNAs. Previous studies demonstrated that the D7 mRNA is not loaded onto polyribosomes in oocytes, but translated in eggs and embryos (Fig. 2A lanes 2 and 3) (38). The cytoskeletal actin mRNA exhibits the opposite behavior; it is translationally active in oocytes, but not in eggs or embryos (Fig. 2A lanes 2 and 3)(39, 40). The analysis of these well-characterized mRNAs in fractions from all samples ensures that the polyribosome fractionation accurately reflects the translation activity of the mRNAs being analyzed. Only samples in which the control mRNAs fractionate correctly should be considered for further analysis. Third, to ensure that an mRNA's presence in the polyribosome fraction is due to its association with polyribosomes each fractionation experiment is repeated with the addition of EDTA to the polyribosome buffers. The chelation of Mg2+ ions by EDTA causes polyribosomes to dissociate and release any bound mRNAs into the supernatant fraction. Therefore, the analysis of each sample requires fractionating samples and comparing the fractionation that occurs in the presence of EDTA. mRNAs are considered to be associated with polyribosomes only if they are released in the supernatant fraction in the presence of EDTA.
Future directions
The methodology outlined here allows investigators to evaluate the translational states of different mRNAs at a single stage of development and monitor how these states change over time. The major limitation of this strategy as presented is that only a modest number of mRNAs can be analyzed when RNA blot hybridization is used for assaying the presence of specific mRNAs. In other systems it has been possible to couple polyribosome isolation with microarray technologies to comprehensively define the translational state of all mRNAs present (20-22). In principle this same approach could be applied to Xenopus development. However, there are certain problems that remain to be addressed for this approach to be successful. In particular, to create labeled probes from mRNAs for microarray analysis requires the production of cDNA. This cDNA is frequently generated by oligo-dT priming using the poly (A) tails present on mRNAs. As pointed out above the poly (A) tails of maternal mRNAs are highly regulated and the presence or absence of poly (A) heavily influences translation. Therefore, to thoroughly analyze translational changes that occur during Xenopus development a different strategy should be considered for labeling polyribosome associated mRNAs. Recent developments in RNA-seq methods may provide an alternative for monitoring mRNA expression that circumvents these and other potential problems of microarray analysis (41).
Recently a new method termed ribosome profiling was developed for high-resolution analysis of ribosome mRNA interactions (42). Ribosome profiling defines precisely what regions of a message are being occupied by translating ribosomes. The application of this methodology to Xenopus mRNAs from different stages of development and mRNAs from different tissues could define and provide new insights into the precise ribosome-mRNA interactions that regulate translation. As these new tools become available it should ultimately be feasible to comprehensively define all the translational changes that occur during development and define translational events that are confined to specific embryonic cells.
Acknowledgments
This work was supported by grants from the NIH HD43996 (M.S), the Beckman Foundation, the Pew Scholars Program and the James D. Shaw and Dorothy Shaw Fund of the Greater Milwaukee Foundation and the University of Wisconsin Graduate School. RSH was funded by a grant from the National Cancer Institute-National Institutes of Health (R01CA095898).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Heasman J. Semin Cell Dev Biol. 2006;17:93–8. doi: 10.1016/j.semcdb.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 2.Vardy L, Orr-Weaver TL. Trends Cell Biol. 2007;17:547–54. doi: 10.1016/j.tcb.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 3.Farley BM, Ryder SP. Crit Rev Biochem Mol Biol. 2008;43:135–62. doi: 10.1080/10409230801921338. [DOI] [PubMed] [Google Scholar]
- 4.Tadros W, Lipshitz HD. Development. 2009;136:3033–42. doi: 10.1242/dev.033183. [DOI] [PubMed] [Google Scholar]
- 5.Sheets MD, Fox CA, Hunt T, Vande Woude G, Wickens M. Genes & Development. 1994;8:926–38. doi: 10.1101/gad.8.8.926. [DOI] [PubMed] [Google Scholar]
- 6.Sheets MD, Wu M, Wickens M. Nature. 1995;374:511–6. doi: 10.1038/374511a0. [DOI] [PubMed] [Google Scholar]
- 7.Stebbins-Boaz B, Hake LE, Richter JD. EMBO Journal. 1996;15:2582–92. [PMC free article] [PubMed] [Google Scholar]
- 8.Gebauer F, Xu W, Cooper GM, Richter JD. Embo J. 1994;13:5712. doi: 10.1002/j.1460-2075.1994.tb06909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.White JA, Heasman J. J Exp Zool B Mol Dev Evol. 2008;310:73–84. doi: 10.1002/jez.b.21153. [DOI] [PubMed] [Google Scholar]
- 10.Kofron M, Birsoy B, Houston D, Tao Q, Wylie C, Heasman J. Development. 2007;134:503–13. doi: 10.1242/dev.02739. [DOI] [PubMed] [Google Scholar]
- 11.Tao Q, Yokota C, Puck H, Kofron M, Birsoy B, Yan D, Asashima M, Wylie CC, Lin X, Heasman J. Cell. 2005;120:857–71. doi: 10.1016/j.cell.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 12.Schroeder KE, Condic NL, Eisenberg LM, Yost HJ. Developmental Biology. 1999;214:288–97. doi: 10.1006/dbio.1999.9426. [DOI] [PubMed] [Google Scholar]
- 13.Audic Y, Garbrecht M, Fritz B, Sheets MD, Hartley RS. Developmental Dynamics. 2002;225:511–21. doi: 10.1002/dvdy.10191. [DOI] [PubMed] [Google Scholar]
- 14.Vasudevan S, Seli E, Steitz JA. Genes Dev. 2006;20:138–46. doi: 10.1101/gad.1398906. [DOI] [PubMed] [Google Scholar]
- 15.Colegrove-Otero LJ, Minshall N, Standart N. Critical Reviews in Biochemistry & Molecular Biology. 2005;40:21–73. doi: 10.1080/10409230590918612. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Forinash KD, McGivern J, Fritz B, Dorey K, Sheets MD. Mol Cell Biol. 2009;29:3791–802. doi: 10.1128/MCB.01865-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lund E, Liu M, Hartley RS, Sheets MD, Dahlberg JE. RNA. 2009;15:2351–63. doi: 10.1261/rna.1882009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rubin GM. Methods Cell Biol. 1975;12:45–64. doi: 10.1016/s0091-679x(08)60951-6. [DOI] [PubMed] [Google Scholar]
- 19.Sagliocco FA, Moore PA, Brown AJ. Methods Mol Biol. 1996;53:297–311. doi: 10.1385/0-89603-319-8:297. [DOI] [PubMed] [Google Scholar]
- 20.Beilharz TH, Preiss T. Brief Funct Genomic Proteomic. 2004;3:103–11. doi: 10.1093/bfgp/3.2.103. [DOI] [PubMed] [Google Scholar]
- 21.Arava Y. Methods Mol Biol. 2003;224:79–87. doi: 10.1385/1-59259-364-X:79. [DOI] [PubMed] [Google Scholar]
- 22.Arava Y, Wang Y, Storey JD, Liu CL, Brown PO, Herschlag D. Proc Natl Acad Sci U S A. 2003;100:3889–94. doi: 10.1073/pnas.0635171100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gurdon JB, Wickens MP. Methods Enzymol. 1983;101:370–86. doi: 10.1016/0076-6879(83)01028-9. [DOI] [PubMed] [Google Scholar]
- 24.Woodland HR. Dev Biol. 1974;40:90–101. doi: 10.1016/0012-1606(74)90111-0. [DOI] [PubMed] [Google Scholar]
- 25.Baum EZ, Wormington WM. Dev Biol. 1985;111:488–98. doi: 10.1016/0012-1606(85)90500-7. [DOI] [PubMed] [Google Scholar]
- 26.Hyman LE, Wormington WM. Genes Dev. 1988;2:598–605. doi: 10.1101/gad.2.5.598. [DOI] [PubMed] [Google Scholar]
- 27.Fritz BR, Sheets MD. Developmental Biology. 2001;236:230–43. doi: 10.1006/dbio.2001.0324. [DOI] [PubMed] [Google Scholar]
- 28.Wormington M. Methods in Cell Biology. 1991;36:167–83. doi: 10.1016/s0091-679x(08)60277-0. [DOI] [PubMed] [Google Scholar]
- 29.Methods Cell Biol. 1991;36:367–87. doi: 10.1016/s0091-679x(08)60288-5. [DOI] [PubMed] [Google Scholar]
- 30.Nishimatsu S, Iwao M, Nagai T, Oda S, Suzuki A, Asashima M, Murakami K, Ueno N. Febs Lett. 1992;312:169–73. doi: 10.1016/0014-5793(92)80928-a. [DOI] [PubMed] [Google Scholar]
- 31.Nishimatsu S, Suzuki A, Shoda A, Murakami K, Ueno N. Biochemical & Biophysical Research Communications. 1992;186:1487–95. doi: 10.1016/s0006-291x(05)81574-8. [DOI] [PubMed] [Google Scholar]
- 32.Nishimatsu S, Oda S, Murakami K, Ueno N. FEBS Letters. 1992;303:81–4. doi: 10.1016/0014-5793(92)80482-v. [DOI] [PubMed] [Google Scholar]
- 33.Graff JM, Thies RS, Song JJ, Celeste AJ, Melton DA. Cell. 1994;79:169–79. doi: 10.1016/0092-8674(94)90409-x. [DOI] [PubMed] [Google Scholar]
- 34.Graff JM, Bansal A, Melton DA. Cell. 1996;85:479–87. doi: 10.1016/s0092-8674(00)81249-0. [DOI] [PubMed] [Google Scholar]
- 35.Hawley SH, Wunnenberg-Stapleton K, Hashimoto C, Laurent MN, Watabe T, Blumberg BW, Cho KW. Genes & Development. 1995;9:2923–35. doi: 10.1101/gad.9.23.2923. [DOI] [PubMed] [Google Scholar]
- 36.Thomsen GH. Development. 1996;122:2359–66. doi: 10.1242/dev.122.8.2359. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki A, Kaneko E, Ueno N, Hemmati-Brivanlou A. Developmental Biology (Orlando) 1997;189:112–22. doi: 10.1006/dbio.1997.8652. [DOI] [PubMed] [Google Scholar]
- 38.Dworkin MB, Shrutkowski A, Dworkin-Rastl E. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:7636–40. doi: 10.1073/pnas.82.22.7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ballantine JE, Woodland HR, Sturgess EA. J Embryol Exp Morphol. 1979;51:137–53. [PubMed] [Google Scholar]
- 40.Sturgess EA, Ballantine JE, Woodland HR, Mohun PR, Lane CD, Dimitriadis GJ. J Embryol Exp Morphol. 1980;58:303–20. [PubMed] [Google Scholar]
- 41.Akkers RC, van Heeringen SJ, Jacobi UG, Janssen-Megens EM, Francoijs KJ, Stunnenberg HG, Veenstra GJ. Dev Cell. 2009;17:425–34. doi: 10.1016/j.devcel.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Science. 2009;324:218–23. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]