

Abstract
Approximately 7 days after HSV-1 corneal infection, BALB/c mice develop tissue-destructive inflammation in the cornea termed herpes stromal keratitis (HSK), as well as periocular skin lesions that are characterized by vesicles, edema, and fur loss. CD4+ T cells and Th1 cytokines contribute to both the immunopathology in the cornea and the eradication of viral replication in the skin. We demonstrate that disruption of CD40/CD154 signaling does not impact the initial expansion of CD4+ T cells in the draining lymph nodes, but dramatically reduces the persistence and Th1 polarization of these cells. Despite the reduced Th1 response, CD154−/− mice developed HSK and periocular skin disease with similar kinetics and severity (as assessed by clinical examination) as wild-type (WT) mice. However, when the composition of the inflammatory infiltrate was examined by flow cytometric analysis, CD154−/− mice exhibited significantly fewer CD4+ and CD8+ T cells and neutrophils than WT mice at the peak of HSK. Moreover, CD4+ T cells from infected corneas of CD154−/− mice produced significantly less IFN-γ than those of WT mice when stimulated with viral Ags in vitro. The IFN-γ production of cells from infected corneas of WT mice was not affected by addition of anti-CD154 mAb to the stimulation cultures. This suggests that CD154 signaling is required at the inductive phase, but not at the effector phase, of the Th1 response within the infected cornea. We conclude that local disruption of CD40/CD154 signaling is not likely to be a useful therapy for HSK.
Herpes simplex virus type 1 infection of the mouse cornea leads to a transient epithelial lesion caused by HSV-1 replicating in and destroying corneal epithelial cells. This lesion is evident by 2 days after infection and typically requires an additional 2 days to heal (1). Such lesions are not associated with permanent loss of vision. However, by ~7 days after infection, a mild haze develops in the infected mouse cornea stroma, and limbal blood vessels begin to invade this normally avascular tissue. The inflammation is progressive, leading to severe neovascularization and opacity. This secondary inflammation in most mouse models is mediated by CD4+ T cells, in part through the production of the Th1 cytokines IL-2 and IFN-γ (2–5). The majority of infiltrating cells in the inflamed cornea are neutrophils, which appear to be the proximal mediators of corneal tissue destruction (6,7), presumably through the release of molecules that degrade the extracellular matrix of the corneal stroma. A similar inflammatory response, referred to as herpes stromal keratitis (HSK),3 leads to scarring and permanent loss of vision in humans.
The normal mouse cornea is considered to be an immune-privileged tissue and lacks MHC class II-positive dendritic cells (DC). One group reported the presence of a population of MHC class II-negative DC in the central mouse cornea (8,9), but this DC population was not detected in another similar study (10). There clearly are MHC class II-positive DC (many exhibiting Langer hans cell morphology) that are abundantly present at the limbus (region between the cornea and conjunctiva) (10,11). These cells migrate into the cornea after HSV-1 infection and appear to directly or indirectly participate in the activation of HSV-1-specific CD4+ T cells in the draining lymph nodes (DLN) as well as in the restimulation of CD4+ T cells that infiltrate the infected cornea (12).
The ligation of the TCR on naive CD4+ T cells by an immunogenic MHC/peptide complex typically does not provide an adequate signal to induce their proliferation and differentiation. In stead, such interactions usually lead to a state of unresponsiveness, referred to as anergy (13). However, when DC acquire Ags at sites of infection or inflammation, they mature and express costimulatory molecules and cytokines that stimulate the additional intracellular signaling required for CD4+ T cell proliferation and differentiation (14). For instance, CD80 and CD86 that are expressed on mature DC induce signaling through CD28 on naive CD4+ T cells; and IL-12 and IL-18 secreted by mature DC induce signaling through IL-12R and IL-18R on CD4+ T cells. Together this signaling leads to CD4+ T cell proliferation and differentiation into Th1 cells. However, this initial proliferation and Th1 differentiation can only be sustained if DC activation and survival are maintained through signaling pathways that are induced by CD4+ Tcells. Thus, activated CD4+ T cells transiently express a variety of surface molecules, including CD154 (a TNF receptor family member) that binds to CD40 (a TNF family member) on DC and induces a signaling pathway(s) that leads to increased expression of CD80 and CD86 (15,16). CD40/CD154 interactions have also been shown to be important for production of IL-12 and IL-18 (17–19) by APCs. This reciprocal activation produces the robust Th1 CD4+ T cell response that is required for the eradication of many viral and bacterial infections. Signaling induced by CD154 also appears to be essential for the establishment of B cell and CD8+ T cell memory (20,21).
When effector CD4+ T cells infiltrate infected tissue, they again receive TCR signaling through recognition of epitopes on tissue APC. This secondary stimulation can lead to expansion, retention, and cytokine secretion by Ag-specific CD4+ T cells in the infected tissue. The contribution of APC costimulation and cytokine production to the expansion and cytokine pattern of effector CD4+ T cells within infected tissue remains enigmatic. The requirement for APC costimulation and cytokine production will probably vary depending on the epitope density, TCR affinity, and cytokine milieu within a particular infected tissue (22). Clearly the proper regulation of CD4+ T cell function at sites of infection is critical to optimize elimination of the infectious agent with minimal immunopathology. In support of this, we have shown in our model system a requirement for local B7/CD28 interactions in the development of HSK (23). That study clearly demonstrated a need for costimulation within the inflamed tissue and led us to investigate the potential role of CD40/CD154 interaction after ocular HSV-1 infection.
This study tested the involvement of the CD40/CD154 costimulatory interaction in the initial induction of a Th1 response in the DLN and in the subsequent development of HSK after HSV-1 corneal infection of BALB/c mice. Our findings demonstrate that CD40/CD154 interaction is required for an optimal Th1 response to HSV-1 in the DLN, but is not required for CD4+ T cell production of Th1 cytokines within the infected cornea. A reduced Th1 response and milder inflammation were observed in the infected corneas of CD154−/− mice, but this did not alter the clinical manifestations of the disease.
Materials and Methods
Mice and virus infection
A breeding pair of mice with a targeted disruption of the CD154−/− mouse gene (CD154−/−) on the BALB/c background was obtained from Howard Hughes Medical Institute, Section of Immunobiology, Yale University School of Medicine (New Haven, CT), and bred in our animal facility at University of Pittsburgh. Female CD154−/− mice and wild-type (WT) BALB/c mice (Frederick Cancer Research Center, Frederick, MD) were used in experiments at 6 – 8 wk of age. All mice were anesthetized by i.m. injection of 2 mg of ketamine hydrochloride and 0.04 mg of xylazine (Phoenix Scientific, St. Joseph, MO) in 0.2 ml of HBSS (BioWhittaker, Walkersville, MD). Topical corneal infection was then achieved by scarifying the central cornea 15 times with a sterile 30-gauge needle in a criss cross pattern and applying 3 μl of RPMI 1640 (BioWhittaker, Rockland, ME) containing 105 PFU of HSV-1. The RE strain of HSV-1 used in these studies was grown in Vero cells, and intact virions were purified on OptiPrep gradients according to the manufacturer’s instructions (Accurate Chemical & Scientific Corp., Westbury, NY), quantified by virus plaque assay, and stored at –80°C.
Monitoring of HSV-1 corneal and skin disease
Corneal disease was monitored in a masked fashion by slit-lamp examination on alternate days after HSV-1 corneal infection. By 2 days after HSV-1 corneal infection, BALB/c mice uniformly exhibit dendritic-shaped epithelial lesions that are caused by HSV-1 replication in and destruction of corneal epithelial cells. The epithelial lesions typically heal by 4 days after infection. HSK characterized by corneal opacity and neovascularization begins 7 days after HSV-1 corneal infection in 90 –100% of mice. Opacity and neovascularization develop concurrently and are monitored by slit-lamp examination. HSK is scored on the basis of opacity as: 1+, mild corneal haze; 2+, moderate opacity; 3+, severe opacity obscuring the iris; or 4+, corneal perforation. Periocular skin disease was assessed according to severity and area involved: 1+, confined blepharitis; 2+, moderate blepharitis; 3+, regional blepharitis with vesicles; or 4+, regional blepharitis with >2 mm of periocular skin involvement and vesicles.
Preparation of cornea single-cell suspension
At various times after HSV-1 corneal infection, mice were scored for HSK, and corneas were removed and incubated with PBS-EDTA to separate the epithelial layer. Individual corneal stromas were rinsed, cut into quarters, treated with collagenase type I (84 U/cornea; Sigma-Aldrich, St. Louis, MO) for 2 h at 37°C, and triturated until no apparent tissue fragments remained. The single-cell suspension of each cornea was washed and then subjected to flow cytometric analysis or cytokine assay.
Immunomagnetic separation and cytokine assay
The DLN (cervical and submandibular lymph nodes) were excised 7 and 14 days after HSV-1 infection, and single-cell suspensions were prepared in assay medium (RPMI 1640 plus 5% FCS, 10 mM HEPES buffer, and antibiotics). CD4+ T cells and CD11c+ DCs were isolated by two rounds of magnetic separation using MACS mouse CD4 (L3T4) and MACS mouse CD11c (N418) MicroBeads, respectively, according to the manufacturer’s instructions (Miltenyi Biotec, Auburn, CA). In all experiments the positively selected cells were shown by flow cytometry to be >97% marker positive. The purified CD4+ T cells (5 × 105) or cells from individual corneas were cultured in 96-well, round-bottom plates (BD Bio sciences, Franklin Lakes, NJ) with CD11c+ DCs (2.5 × 104). The DCs were pulsed with either UV-inactivated RE strain HSV-1 (UV-HSV, 5 × 107 original PFU/ml) or 100 μl of HSV-1 Ags. The cultures were incubated at 37°C for 72 h, and the cytokine content of the supernatant fluids was measured by ELISA.
The HSV-1 Ags were extracted from Vero cells (3.5 × 106/ml) 10 h after RE HSV-1 infection (multiplicity of infection of 4). The extraction involved three freeze/thaw cycles, two 2-min homogenization cycles using the Retsch MM 300 Mixer Mill homogenizer (F. Kurt Retsch, Haan, Germany), UV inactivation, and clarification by centrifugation at 200 × g.
ELISA
Microtiter plates (Dynex Immulon 4HBX; Thermo Labsystems, Franklin, MA) were coated overnight with primary anti-cytokine capture Ab. The plates were washed and blocked with 1% BSA in PBS. The supernatant fluids and standards were added and incubated at room temperature for 2 h. The plates were washed again, and secondary biotinylated anti-cytokine detection Ab was added and incubated at room temperature for 1 h. The plates were then washed and developed with strepavidin-HRP (BD Pharmingen, San Diego, CA) and its substrate ((3,3′ 5,5′-tetramethylbenzidine base; Life Technologies, Gaithersburg, MD). The colored reaction product was measured with an enzyme immunoassay plate reader at 450 nm. The amount of cytokine in each supernatant was extrapolated from a standard curve. The capture/detection Abs were as follows: IL-4, 11B11/BVD6-24G2 (from American Type Culture Collection, Manassas, VA); and IFN-γ, R4-6A2 (from American Type Culture Collection)/polyclonal goat anti-mouse IFN-γ (R&D Systems, Minneapolis, MN). The detection sen sitivities were 7.8 pg/ml for IL-4 and 15.6 pg/ml for IFN-γ.
Flow cytometry
For flow cytometric analysis, single-cell suspensions of DLN (1 × 106 cells/tube) or corneas (one cornea per tube) were prepared on the designated day after HSV-1 infection and transferred to 5-ml, polystyrene, round-bottom tubes (BD Biosciences). Cells were incubated with anti mouse CD16/CD32 (FcγRIII/II; 2.4G2; BD Pharmingen) to prevent non specific binding of fluoresceinated mAbs, then stained for cell surface markers. The following Abs were purchased from BD Pharmingen: FITC conjugated anti-CD8α (Ly.2; clone 53-6.7), FITC-conjugated anti-CD8β.2 (clone 53-5.8), PE-conjugated anti-CD4 (RM4-5), and PerCP-conjugated anti-CD45 (30-F11). FITC-conjugated anti-CD45R/B220 (RA3-6B2) and allophycocyanin-conjugated anti-GR-1 (RB6-8C5) were purchased from Caltag Laboratories (Burlingame, CA). The cells were then fixed in 1% paraformaldehyde (Electron Microscopy Sciences, Fort Washington, PA) and analyzed on a FACSCalibur (BD Biosciences) using WinMDI data analysis software (J. Totter, The Scripps Clinic, La Jolla, CA). Due to the small sample, cornea cells were not counted before analysis, but the entire sample was analyzed to provide an estimate of the absolute number of each population of cells in the cornea. The DLN cells were counted, and the absolute number of marker-positive cells was determined by multiplying the percentage of marker-positive cells by the total number of cells.
Statistics
The differences in DLN cellularity and cornea day 21 cellular infiltrations between WT and CD154+ mice were analyzed by unpaired t test. The differences in IFN-γ and IL-4 production in in vitro cultures from all groups were analyzed by one-way ANOVA followed by the Tukey post test to assess the significance of differences between individual groups. The differences in HSK and skin disease were assessed by longitudinal analysis with the SAS program using the SAS PROC MIXED procedure with the repeated measures option. This analysis was chosen because the repeated observations were taken over time within the same mouse. It was also chosen for the ability to analyze and account for different covariance patterns of the repeated measurements. Overall group and time effects were then tested for HSK and skin disease outcomes (24,25).
Results
Is CD40/CD154 costimulation required for the expansion of B and T lymphocytes in DLN of HSV-1-infected mice?
HSV-1 corneal infection results in the expansion of CD4+ T cells, CD8+ T cells, and B cells within the DLN. To assess the role of CD40/CD154 interaction in T cell and B cell expansion, the corneas of WT and CD154−/− BALB/c mice were infected with HSV-1. At 7 or 14 days after infection, DLN cells were counted, and the absolute number and percentage of CD4+ T cells, CD8+ T cells, and B cells were determined by flow cytometric analysis. There was a modest, but statistically significant, difference in the absolute numbers of CD4+ T cells between WT and CD154−/− mice 7 days after corneal infection. However, there was no significant difference in the percentage of CD4+ T cells or in the number of CD45+ cells, suggesting that the difference in the absolute number of CD4+ T cells is probably not biologically significant. Statistical differences in the absolute number and percentage of CD8+ T cells between WT and CD154 mice were also observed on day 7 (Fig. 1). In WT mice there was only a modest (17%) reduction in total DLN cells between 7 and 14 days after infection, which resulted primarily from a reduced number of B cells, with the numbers of CD4+ and CD8+ T cells remaining nearly constant (Fig. 1). In contrast, the total number of DLN cells decreased by 60% in CD154−/− mice between 7 and 14 days after infection. Again, the biggest reduction was in the B cell population (76%), but significant reductions were also seen in CD4+ T cells (40%) and CD8+ T cells (50%). Thus, whereas CD40/CD154 interaction is only minimally involved in the initial expansion of CD4+ and CD8+ T cells and B cells in the DLN after HSV-1 corneal infection, all three populations undergo a more rapid contraction in the absence of CD40/CD154 costimulation.
FIGURE 1.

DLN cellularity at 7 (A) and 14 (B) days after HSV-1 corneal infection. At designated times after corneal infection, DLN were excised from five individual WT or CD154−/− mice, and single-cell suspensions were counted and stained with fluorescein-conjugated Abs against CD45, CD4, CD8, or B220. Flow cytometric analysis was performed to determine the percentage and absolute number of the different populations. Data are presented as the mean ± SEM for each cell type. Asterisks indicate a significant (*, p < 0.05; **, p < 0.01; ***, p < 0.001) difference between WT and CD154−/− mice, as assessed by unpaired Student’s t test. The data are pooled from six separate experiments.
Is CD40/CD154 costimulation required for Th1 polarization of CD4+ T cells?
Costimulation through CD40/CD154 reportedly can favor Th1 or Th2 polarization of CD4+ T cells depending on the model under investigation (26,27). To determine whether CD40/CD154 costimulation favors Th1 polarization after HSV-1 corneal infection, the DLN were excised 7 or 14 days after infection, purified CD4+ T cells were stimulated in vitro with UV-HSV-1-pulsed DLN DC for 72 h, and secreted IFN-γ and IL-4 were measured by ELISA. We did not observe a significant difference in the capacity of DC from the DLN of WT or CD154−/− mice to stimulate CD4+ T cells, so all data are presented with WT DC only. Although the DC were purified from the DLN of infected mice, DLN CD4+ T cells failed to produce detectable IFN-γ or IL-4 when the DC were not pulsed with UV-HSV-1 (data not shown). When CD4+ T cells obtained from the DLN 7 or 14 days after HSV-1 corneal infection were stimulated with UV-HSV-1-pulsed DC, the CD154−/− CD4+ T cells produced significantly less IFN-γ and significantly more IL-4 than those from WT mice (Fig. 2). The addition of IL-12 to HSV-1 Ag-stimulated cultures of CD154−/− CD4+ T cells from day 7 DLN significantly augmented IFN-γ production and decreased IL-4 production (Fig. 2). In contrast, exogenous IL-12 only modestly affected the cytokine pattern of CD154−/− CD4+ T cells from day 14 DLN.
FIGURE 2.

DLN CD4+ T cells exhibit reduced Th1 polarization after HSV-1 infection of CD154−/− mice. CD4+ T cells and CD11c+ DC were purified on the designated day from DLN of WT or CD154−/− mice by two rounds of magnetic separation. CD4+ T cells (5 × 105) were stimulated in vitro using UV-HSV-1-pulsed DC (2.5 × 104) in a 96-well plate. Where indicated, cultures received recombinant mouse IL-12 (2 ng/ml). Cytokine concentrations in culture supernatants were measured at 72 h by ELISA. The experiment was repeated twice with similar results. Representative data are recorded as the mean ± SEM concentration of IFN-γ (A) or IL-4 (B) in triplicate wells. In all cases, asterisks refer to the significance (*, p < 0.05; **,p < 0.01; ***, p < 0.001) of differences relative to the column to the immediate left, as assessed by one-way ANOVA with post-tests. No stimulation was observed with a homogenate of uninfected Vero cells.
The above observations suggested that the DLN of CD154−/− mice contained a significant population of naive CD4+ T cells that could be skewed to Th1 polarization by in vitro stimulation in the presence of exogenous IL-12. This population of naive HSV-1-specific CD4+ T cells appeared to be largely absent in the DLN by 14 days after infection. We hypothesized that a similar population of naive HSV-1-specific CD4+ T cells was present in the DLN of WT mice, but their requirement for exogenous IL-12 for Th1 skewing was supplanted by CD40/CD154 costimulation. To test this, CD4+ T cells were purified from the DLN of WT mice 7 and 14 days after HSV-1 corneal infection and were restimulated with UV-HSV-1-pulsed DC in the presence or the absence of anti-CD154 mAb. At the highest dose (100 μg/ml), anti-CD154 reduced the IFN-γ production of CD4+ T cells obtained 7 days after infection by 59% and increased their IL-4 production by [H11022]5-fold (Fig. 3A). In contrast, anti-CD154 only modestly reduced IFN-γ production and did not influence IL-4 production by WT CD4+ Tcells obtained 14 days after corneal infection (Fig. 3B). These data suggest that a majority of CD4+ T cells in the DLN 7 days after HSV-1 corneal infection are not yet polarized to a Th1 cytokine pattern and can be pushed to Th2 polarization by blocking CD40/ CD154 costimulation. In contrast, it appears that the majority of CD4+ T cells in the DLN 14 days after infection are either already polarized to a Th1 cytokine pattern or do not require CD40/CD154 costimulation for Th1 polarization.
FIGURE 3.
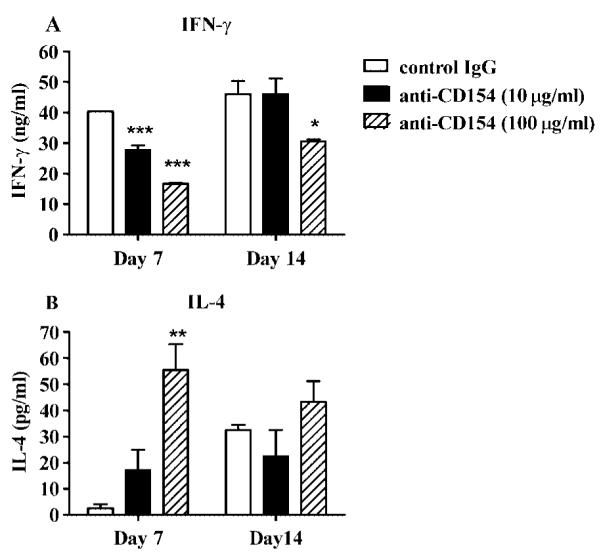
CD154 signaling is required for the in vitro Th1 differentiation of WT DLN CD4+ T cells. On the designated day after HSV-1 corneal infection, CD4+ T cells and CD11c+ DC were purified from WT DLN. CD4+ T cells (5 × 105) were cultured with UV-HSV-1-pulsed DC (2.5 × 104) with control hamster IgG (100 μg/ml) or blocking anti-CD154 mAb. The experiment was repeated twice with similar results. Representative data are recorded as the mean ± SEM concentration of IFN-γ (A) or IL-4 (B) in triplicate wells. Asterisks indicate a statistically significant (*, p < 0.05; **, p *lt; 0.01; ***, p < 0.001) difference between control IgGand anti-CD154 mAb-treated cultures, as assessed by one-way ANOVA and post-tests. Control Ab treatment did not significantly affect IFN-γ production (not shown).
The rapid contraction in the B cell population in the DLN of CD154−/− mice from days 7–14 after HSV-1 corneal infection was associated with failure to develop an IgG anti-HSV-1 Ab response (Fig. 4). IgG anti-HSV-1 Abs were detectable in the serum of WT mice by 7 days after infection and reached maximal levels by 14 days. In contrast, IgG anti-HSV-1 Abs were not detectable at either time in the serum of CD154−/− mice.
FIGURE 4.
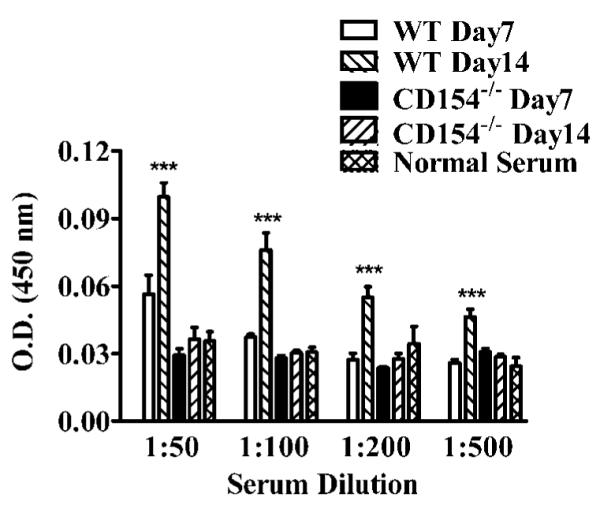
CD154−/− mice do not produce an IgG Ab response to HSV-1. Serum was collected from five WT and five CD154−/− mice at the designated times after HSV-1 corneal infection and tested for IgG anti HSV Ab by ELISA. The experiment was repeated twice with similar results. Representative data are presented as the mean ± SEM. The ODs in wells coated with an uninfected Vero homogenate were not significantly higher than those in uncoated wells (not shown). Asterisks indicate a statistically significant (*, p < 0.05; **, p < 0.01; ***, p < 0.001) difference between serum samples, as assessed by one-way ANOVA and post-tests.
Does disruption of the CD40/CD154 costimulation influence the severity of periocular skin disease and HSK?
Control of HSV-1 replication in the periocular skin and development of inflammation in the cornea are both regulated in part by Th1 cytokines (2). Because the Th1 cytokine response was blunted in the DLN of CD154−/− mice after HSV-1 corneal infection, we anticipated that these mice would display more severe periocular skin disease, but less severe HSK. However, these predictions were not supported by the data. Although the periocular skin disease appeared to be somewhat more severe early (day 7) in CD154−/− mice, their overall disease progression was not significantly different from that in WT mice (Fig. 5). The WT and CD154−/− mice exhibited a 100% incidence of HSK (not shown). Moreover, the kinetics and severity of HSK (assessed clinically) were similar in WT and CD154−/− mice (Fig. 6). Thus, CD154−/− mice appear to reach a threshold level of Th1 cytokine production required to control HSV-1 replication in the skin and regulate clinical manifestations of HSK. Additionally, we determined that there was no difference in initial viral clearance in the corneas of WT and CD154−/− mice by plaque assay (data not shown).
FIGURE 5.
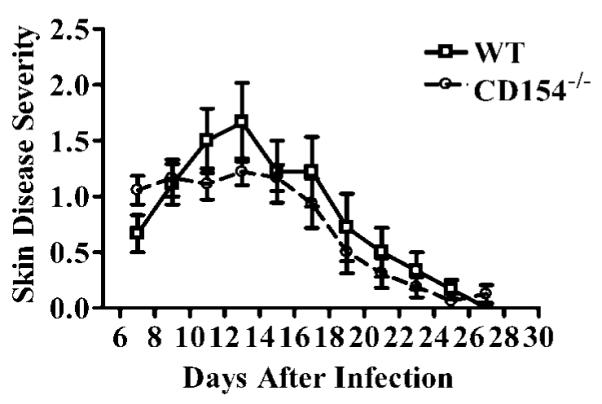
Kinetics of HSV-1 periocular skin disease progression in WT and CD154−/− mice. Skin disease severity was evaluated over time for nine WT and CD154−/− mice in a masked fashion on the designated days after HSV-1 corneal infection. The experiment was repeated twice with similar results. Representative data are recorded as the mean ± SEM. No significant differences were detected between WT and CD154−/− mice by longitudinal analysis with the SAS program.
FIGURE 6.
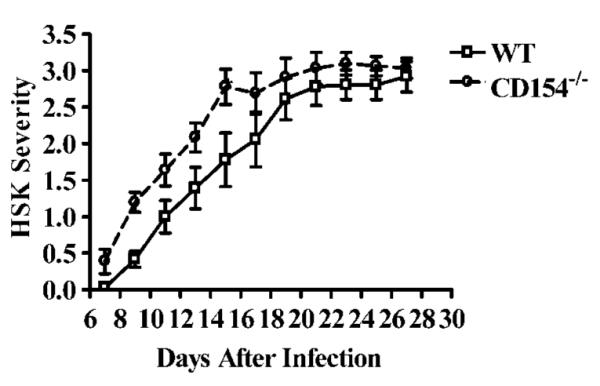
Kinetics of HSK progression in WT and CD154−/− mice. HSK severity was evaluated over time for nine WT and CD154−/− mice in a masked fashion on the designated days after HSV-1 corneal infection. The experiment was repeated twice with similar results. Representative data are recorded as the mean ± SEM. No significant differences were detected between WT and CD154−/− mice by longitudinal analysis with the SAS program.
Clinical evaluation of HSK is based on slit-lamp assessment of corneal opacity, which appears to result from a combination of cellular infiltration, disruption of the collagen matrix, edema, and scarring. To directly measure the degree of inflammatory infiltration, flow cytometric analysis was performed on single-cell sus pensions of corneas obtained from WT and CD154−/− mice 21 days after HSV-1 corneal infection (time of peak HSK). The over all inflammatory infiltrate (CD45+ cells), CD4+ T cells, CD8+ Tcells, and granulocytes (GR1+ cells) were all significantly reduced in the corneas of CD154−/− mice (Fig. 7).
FIGURE 7.
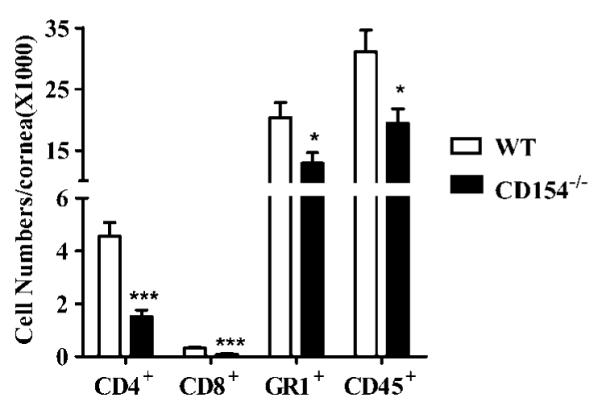
Reduced leukocyte infiltration in CD154−/− mouse corneas with HSK. Six corneas from WT or CD154−/− mice were excised 21 days after HSV-1 corneal infection, and single-cell suspensions from individual corneas were stained for CD4, CD8, GR1, and CD45. The entire sample was analyzed by flow cytometric analysis to determine the absolute number of each cell type per cornea. The experiment was repeated twice with similar results. Representative data are presented as the mean ± SEM. Asterisks indicate a significant (*, p < 0.05; **, p < 0.01; ***, p < 0.001) difference between WT and CD154−/− mice, as assessed by unpaired Student’s t test.
The reduced inflammatory infiltrate in infected corneas of CD154−/− mice is associated with reduced IFN-γ production
Granulocytic infiltration and survival in HSV-1-infected corneas is in part controlled by Th1 cytokines (3, 4, 28). Therefore, we compared IFN-γ production by cells derived 21 days after infection from infected corneas of WT and CD154−/− mice after in vitro stimulation with HSV-1 Ag-pulsed DC (Fig. 8). One corneal equivalent (~5000 CD4+ T cells) of cells derived from WT corneas produced significantly more IFN-γ than those derived from CD154−/− mice after in vitro stimulation with viral Ags. Cultures of corneal cells from WT or CD154−/− mice failed to produce detectable IFN-[H9253] when incubated with DC, but no viral Ags, or with viral Ags, but no DC (not shown). One corneal equivalent of cells from infected WT corneas (200-μl cultures) produced 645 ± 161 pg of IFN-γ (range, 153-1383 pg) when stimulated with HSV-1 Ag-pulsed DC. Corneal cells from CD154−/− mice produced significantly ( p < 0.05) less IFN-γ (96 ± 142 pg/cornea; range, 0 –303 pg).
FIGURE 8.
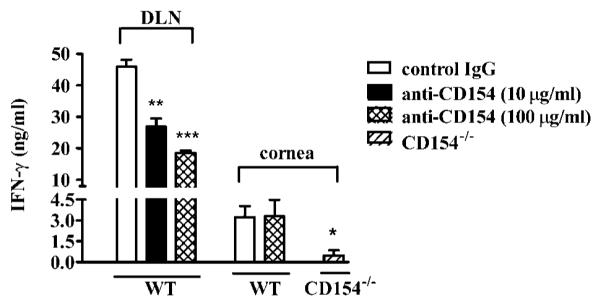
CD154 signaling is not required for in vitro IFN-γ production by cells from HSV-1-infected corneas. Corneas with HSK severity scores of 2.5–3.0 were obtained from four WT or four CD154−/− mice 21 days after HSV-1 corneal infection. CD4+ T cells and CD11c+ DC were purified from DLN obtained 7 days after infection. Single-cell suspensions of individual corneas or DLN CD4+ T cells were stimulated with the same HSV-1 Ag-pulsed DC for 72 h in vitro with control IgG or anti-CD154 mAb. Supernatants were collected and assayed for IFN-γ by ELISA. Control Ab did not influence IFN-γ production (not shown). The experiment was repeated with similar results. Pooled data are recorded as the mean ± SEM. Asterisks indicate a statistically significant (*, p < 0.05; **, p < 0.01; ***, p < 0.001) difference in IFN-γ production between control IgG-treated and anti-CD154 mAb-treated WT DLN cells or between corneal cells from WT or CD154−/− mice. Differences among cultures were evaluated by one-way ANOVA with post-tests
The reduced IFN-γ production by cells from CD154−/− mice could be due to the reduced number of T cells present in their infected corneas. Alternatively, the CD4+ T cells in the infected corneas might require CD40/CD154 costimulation for Th1 cyto kine production in vitro, as seen with CD4+ T cells from day 7 DLN. To address this issue, cells derived from day 21 corneas or purified CD4+ T cells from day 7 DLN of WT mice were stimulated for 72 h in vitro using the same virus-pulsed DC in the presence or the absence of anti-CD154 mAb. IFN-γ production was measured in culture supernatants by ELISA (Fig. 8). The anti-CD154 mAb significantly reduced IFN-γ production by CD4+ T cells from day 7 DLN, but had no effect on IFN-γ production by cells derived from day 21 corneas. It is noteworthy that IL-4 was not detectable in cultures of corneal cells from CD154−/− mice stimulated with HSV-1 Ag-pulsed DC or in cultures of corneal cells from WT mice that were stimulated with HSV-1 Ag-pulsed DC in the presence or the absence of anti-CD154 mAb (data not shown). Together, these findings demonstrate that 1) CD4+ T cells in the infected cornea at the peak of HSK do not require CD40/CD154 costimulation for IFN-γ production under the in vitro conditions used; 2) the reduced IFN-γ production by cells from infected corneas of CD154−/− mice is most likely due to the reduced numbers of HSV-1-specific CD4+ T cells present; 3) CD4+ T cells in the infected corneas of WT mice, unlike those present in day 7 DLN cannot be skewed to IL-4 production by Ag stimulation in the absence of CD40/CD154 costimulation; and 4) the IL-4-producing CD4+ T cells observed in the day 7 DLN of CD154−/− mice are not detectable in their corneas at the peak of HSK.
Discussion
In humans, HSK develops as a result of recurrent reactivation of latent HSV-1 in sensory neurons and shedding of virus at the corneal surface. The resulting corneal lesions trigger recurrent bouts of inflammation and scarring (HSK), leading to progressive corneal stromal opacity and potentially to blindness. In a mouse model of HSK, Th1 cytokines are necessary regulators of corneal inflammation (2). Interestingly, HSK often occurs in the absence of detectable replicating virus in the human cornea and occurs in mice at a time when replicating virus has been cleared from the cornea. These observations suggest that HSK can be triggered by very low levels of virus or viral proteins and perhaps without any virion release. Consistent with the latter possibility is the observation of one group that HSV-1 corneal infection leads to an autoimmune attack on corneal tissue. In that model, an HSV-1 protein, UL6, induced autoimmunity by serving as a molecular mimic of a corneal protein (29–31). However, another group obtained conflicting results (32), and T cells obtained from human corneas with HSK react with HSV-1 Ags, but not with UL6 or corneal extracts (33, 34). Another proposal is that HSK in mice results from bystander activation of T cells of irrelevant specificity (35–38). That observation was made in TCR transgenic SCID mice in a system in which virus replication was not controlled, and mice succumbed to infection during the earliest stages of HSK development. In fact, when HSV-1 replication was controlled normally in the corneas of TCR transgenic SCID mice, bystander activation of CD4+ T cells leading to HSK did not occur (35).
Our studies clearly demonstrate the presence of CD4+ T cells in corneas of mice with HSK, and that these cells produce IFN-γ when stimulated with HSV-1 Ags presented by DCs. IFN-γ was not detectable in cultures of corneal cells that received DCs that were not pulsed with HSV-1 Ags or DCs pulsed with a normal corneal extract (unpublished observation). Moreover, after PMA/ ionomycin stimulation of cells from stromas of corneas with HSK, IFN-γ was produced exclusively by CD4+ T cells (our unpublished observations). Taken together, these data strongly suggest that in normal BALB/c mice, HSV-1-specific CD4+ T cells are the main source of IFN-γ and are thus critical regulators of HSK.
A recent study addressed the role of CD40/CD154 in regulating the T cell response to HSV-1 in C57BL/6 mice, a strain that is more resistant than BALB/c mice to lethal infection and to virus induced inflammation in the cornea (39). This study showed that CD154 deficiency resulted in decreased CD4+ and CD8+ T cell responses to viral Ags and increased mortality after HSV-1 foot pad infection. However, the route of infection and the use of a resistant strain of mice in that study precluded examining the contribution of CD40/CD154 costimulation to virus eradication and immunopathology within a clinically relevant model. In this study we addressed the role of CD40/CD154 interactions in both the inductive and effector phases of the immune response in BALB/c mice, a strain that is susceptible to HSK after corneal infection.
During the first 7 days after HSV-1 corneal infection, the numbers of CD4+ and CD8+ T cells and B cells were dramatically expanded in the DLN of both WT and CD154−/− BALB/c mice. Although CD4+ T cells increase dramatically during the first 7 days after corneal infection, we have not been able to detect IFN-γ-secreting CD4+ T cells in day 7 DLN by intracellular cytokine staining (our unpublished observation). Thus, up to 7 days after infection most CD4+ T cells in the DLN appear to be in an immature state and not yet polarized to a Th1 or Th2 cytokine pattern. Our current findings demonstrate that differentiation of CD4+ T cells from day 7 DLN into Th1 cells in vitro is regulated by CD40/ CD154 signaling. Thus, CD4+ T cells from day 7 DLN of CD154−/− mice produced significantly less IFN-γ and more IL-4 than their WT counterparts when stimulated with HSV-1 Ags in vitro. Moreover, CD4+ T cells from WT mice could be skewed to less IFN-γ and more IL-4 production by adding anti-CD154 mAb to stimulation cultures. Conversely, CD4+ T cells from CD154−/− mice could be skewed to greater production of IFN-γ and reduced production of IL-4 by adding IL-12 to stimulation cultures. Taken together, these findings suggest that CD154-induced signaling is not required for the initial expansion of HSV-1-specific CD4+ T cells in the DLN, but is important for their differentiation into Th1 cells. Exogenous IL-12 can at least partially relieve the requirement for CD154 in the Th1 differentiation of HSV-1-specific CD4+ T cells, consistent with the known capacity of CD40 signaling to induce IL-12 production by DC (17,18).
In WT mice, the number of CD4+ and CD8+ T cells remained surprisingly constant in the DLN from 7–14 days after HSV-1 corneal infection. During the same period, the number of CD4+ and CD8+ T cells declined by ~50% in the DLN of CD154−/− mice. The reduced numbers of CD4+ and CD8+ T cells could reflect reduced proliferation, increased cell death, or an increased rate of egress from the DLN. We cannot now definitively distinguish among these possibilities. The CD4+ T cells that were present in the day 14 DLN of WT and CD154−/− mice appeared to be more polarized in their cytokine pattern than those present in day 7 DLN. The CD4+ T cells in the day 14 DLN of WT mice continued to express a predominantly Th1 cytokine pattern when stimulated with HSV-1 Ags, which was only modestly altered by adding anti-CD154 to the stimulation cultures. The CD4+ T cells in the day 14 DLN of the CD154−/− mice continued to produce a mixed Th1 and Th2 cytokine pattern when stimulated with HSV-1 Ags in vitro, which was only modestly altered by the addition of exogenous IL-12 to the stimulation cultures.
Given the deficient Th1 response in the DLN of CD154−/− mice after HSV-1 corneal infection, we anticipated that these mice would exhibit less severe HSK when examined clinically. In fact, no significant difference was observed in the incidence, kinetics, or average severity of HSK between WT and CD154−/− mice. How ever, our clinical scoring of HSK is based on the degree of corneal opacity, which probably reflects a combination of cellular infiltration, edema, disruption of the normal orientation of collagen fibrils, and scarring. The degree of contribution of each of these factors to the opacity score is unknown.
In humans with HSK, scarring causes permanent loss of vision, and the degree of scarring is probably influenced by the magnitude and duration of leukocytic infiltration. Therefore, it was important to examine the composition of the cellular infiltrate in the infected corneas of WT and CD154−/− mice. Indeed, flow cytometric analysis revealed a significant reduction in leukocytic infiltration in corneas of CD154−/− mice compared with those of WT mice that received the same clinical score for HSK. Corneas of CD154−/− mice contained significantly reduced numbers of CD4+ T cells and produced significantly less IFN-γ when stimulated with HSV-1 Ag-pulsed DCs. The reduced CD4+ T cell infiltrate and IFN-γ production were associated with a 37% reduction in infiltrating granulocytes, consistent with our previous demonstration thatIFN-γ regulates neutrophil extravasation into HSV-1-infected corneas (3). The fact that this dramatic reduction in the inflammatory infiltrate did not translate into reduced opacity suggests that the inflammatory cells themselves do not contribute as much to opacity as other factors. Of the putative factors that contribute to corneal opacity in this model, all are reversible, except scarring. Scarring results from the production of inappropriate extracellular matrix by myofibroblasts attempting to repair the lesion caused by the inflammatory cells in the cornea. It is reasonable to speculate that the degree of scarring is directly related to the magnitude and duration of the inflammatory infiltrate. Thus, the reduced infiltrate in the infected corneas of the CD154−/− mice could ultimately impact long term corneal clarity.
It is noteworthy that the CD4+ T cells in the infected corneas of WT mice at the peak of HSK severity, unlike those in the day 7 DLN, did not require CD40/CD154 interaction for IFN-γ production. It would appear, therefore, that the CD4+ T cells in the inflamed cornea are already polarized to the Th1 cytokine pattern or do not require CD40/CD154 costimulation for differentiation in vitro. The fact that we can detect IFN-γ-producing CD4+ T cells by intracellular cytokine staining directly ex vivo in the cornea, but not in the DLN, favors the former hypothesis (our unpublished observation). Either way, the reduced Th1 response in the infected corneas of CD154−/− mice does not appear to reflect a requirement for CD40/CD154 costimulation within the cornea. Although we cannot rule out a requirement for CD40/CD154 costimulation in the cornea early in the development of HSK, our data do not support the concept of exploiting local blockade of this costimu latory signal as therapy for HSK.
Lesions develop in the periocular skin of mice ~7 days after HSV-1 corneal infection. CD4+ T cells and Th1 cytokines play an important role in controlling HSV-1 skin lesions (2,40). Thus, the cells and cytokines that are responsible for immunopathology in the HSV-1-infected cornea are also important for controlling virus replication in the skin. The challenge in attempting to regulate HSK through inhibition of CD4+ T cell function is to avoid rendering other tissues susceptible to uncontrolled virus replication. Therefore, it was encouraging to note that the reduced Th1 response to HSV-1 in CD154+ mice was not associated with in creased susceptibility to periocular skin disease after HSV-1 corneal infection. It would appear that the CD154−/− mice achieved a threshold level of Th1 cytokine production required to avoid the severe periocular skin disease seen in HSV-1-infected mice when IFN-γ was neutralized (2) or use an IFN-γ-independent mechanism to block viral damage to the periocular skin. Thus, therapeutic approaches that blunt, but do not eliminate, the Th1 response to HSV-1 could provide useful therapy for HSK without rendering mice susceptible to HSV-1 replication in the skin.
Acknowledgments
We thank Teresa Lee for her expert technical assistance, and Regis Rumpf for his assistance with the DLN studies.
Footnotes
- HSK
- herpes stromal keratitis
- DC
- dendritic cell
- DLN
- draining lymph node
- WT
- wild type
This work was supported by Grants EY010359 and EY08098 from the National Eye Institute, National Institutes of Health (Bethesda, MD); an unrestricted research grant from Research to Prevent Blindness (New York, NY); and a grant from the Eye and Ear Foundation (Pittsburgh, PA).
References
- 1.Hendricks RL, Weber PC, Taylor JL, Koumbis A, Tumpey TM, Glorioso JC. Endogenously produced interferon α protects mice from herpes simplex virus type 1 corneal disease. J. Gen. Virol. 1991;72:1601. doi: 10.1099/0022-1317-72-7-1601. [DOI] [PubMed] [Google Scholar]
- 2.Hendricks RL, Tumpey TM, Finnegan A. IFN-γ and IL-2 are protective in the skin but pathologic in the corneas of HSV-1-infected mice. J. Immunol. 1992;149:3023. [PubMed] [Google Scholar]
- 3.Tang Q, Hendricks RL. IFN-γ regulates PECAM-1 expression and neutrophil infiltration into herpes simplex virus-infected mouse corneas. J. Exp. Med. 1996;184:1435. doi: 10.1084/jem.184.4.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang Q, Chen W, Hendricks RL. Proinflammatory functions of IL-2 in herpes simplex virus corneal infection. J. Immunol. 1997;158:1275. [PubMed] [Google Scholar]
- 5.Niemialtowski MG, Rouse BT. Predominance of Th1 cells in ocular tissues during herpetic stromal keratitis. J. Immunol. 1992;149:3035. [PubMed] [Google Scholar]
- 6.Yan X-T, Tumpey TM, Kunkel SL, Oakes JE, Lausch RN. Role of MIP-2 in neutrophil migration and tissue injury in the herpes simplex virus-1-infected cornea. Invest. Ophthalmol. Vis. Sci. 1998;39:1854. [PubMed] [Google Scholar]
- 7.Thomas J, Gangappa S, Kanangat S, Rouse BT. On the essential involvement of neutrophils in the immunopathologic disease: herpetic stromal keratitis. J. Immunol. 1997;158:1383. [PubMed] [Google Scholar]
- 8.Hamrah P, Liu Y, Zhang Q, Dana MR. The corneal stroma is endowed with a significant number of resident dendritic cells. Investigative Ophthalmol. Vis. Sci. 2003;44:581. doi: 10.1167/iovs.02-0838. [DOI] [PubMed] [Google Scholar]
- 9.Hamrah P, Zhang Q, Liu Y, Dana MR. Novel characterization of MHC class II-negative population of resident corneal Langerhans cell-type dendritic cells. Invest. Ophthalmol. Vis. Sci. 2002;43:639. [PubMed] [Google Scholar]
- 10.Brissette-Storkus CS, Reynolds SA, Lepisto AJ, Hendricks RL. Identification of a novel macrophage population in the normal mouse corneal stroma. Invest. Ophthalmol. Vis. Sci. 2002;43:2264. [PMC free article] [PubMed] [Google Scholar]
- 11.Rodrigues M, Rowden G, Hackett J, Bakos I. Langerhans cells in the normal conjunctiva and peripheral cornea of selected species. Invest. Ophthalmol. Vis. Sci. 1997;21:759. [PubMed] [Google Scholar]
- 12.Hendricks RL, Janowicz M, Tumpey TM. Critical role of corneal Langerhans cells in the CD4- but not CD8-mediated immunopathology in herpes simplex virus-1-infected mouse corneas. J. Immunol. 1992;148:2522. [PubMed] [Google Scholar]
- 13.Appleman LJ, Boussiotis VA. T cell anergy and costimulation. Immunol. Rev. 2003;192:161. doi: 10.1034/j.1600-065x.2003.00009.x. [DOI] [PubMed] [Google Scholar]
- 14.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu. Rev. Immunol. 1996;14:233. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 15.Yang Y, Wilson JM. CD40 ligand-dependent T cell activation: requirement of B7-CD28 signaling through CD40. Science. 1996;273:1862. doi: 10.1126/science.273.5283.1862. [DOI] [PubMed] [Google Scholar]
- 16.Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu. Rev. Immunol. 2001;19:225. doi: 10.1146/annurev.immunol.19.1.225. [DOI] [PubMed] [Google Scholar]
- 17.Koch F, Stanzl U, Jennewein P, Janke K, Heufler C, Kämpgen E, Romani N, Schuler G. High level IL-12 production by murine dendritic cells: upregulation via MHC class II and CD40 molecules and downregulation by IL-4 and IL-10. J. Exp. Med. 1996;184:741. doi: 10.1084/jem.184.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cella M, Scheidegger D, Palmer-Lehmann K, Lane P, Lanzavecchia A, Alber G. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T-T help via APC activation. J. Exp. Med. 1996;184:747. doi: 10.1084/jem.184.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karni A, Koldzic DN, Bharanidharan P, Khoury SJ, Weiner HL. IL-18 is linked to raised IFN-γ in multiple sclerosis and is induced by activated CD4+ T cells via CD40-CD40 ligand interactions. J. Neuroimmunol. 2002;125:134. doi: 10.1016/s0165-5728(02)00018-8. [DOI] [PubMed] [Google Scholar]
- 20.Borrow P, Tough DF, Eto D, Tishon A, Grewal IS, Sprent, Flavell RA, Oldstone MB. CD40 ligand-mediated interactions are involved in the generation of memory CD8+ cytotoxic T lymphocytes (CTL) but are not required for the maintenance of CTL memory following virus infection. J. Virol. 1998;72:7440. doi: 10.1128/jvi.72.9.7440-7449.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Arpin C, Dechanet J, Van Kooten C, Merville P, Grouard G, Briere F, Banchereau J, Liu YJ. Generation of memory B cells and plasma cells in vitro. Science. 1995;268:720. doi: 10.1126/science.7537388. [DOI] [PubMed] [Google Scholar]
- 22.Xu M, Lepisto AJ, Hendricks RL. Co-stimulatory requirements of effector T cells at inflammatory sites. DNA Cell Biol. 2002;21:461. doi: 10.1089/10445490260099755. [DOI] [PubMed] [Google Scholar]
- 23.Chen H, Hendricks RL. B7 costimulatory requirements of T cells at an inflammatory site. J. Immunol. 1998;160:5045. [PubMed] [Google Scholar]
- 24.Brown HARP. Applied Mixed Models in Medicine. Wiley; New York: 1999. [Google Scholar]
- 25.Twisk JWR. Applied Longitudinal Data Analysis for Epidemiology. Cambridge University Press; Cambridge: 2003. [Google Scholar]
- 26.Howland KC, Ausubel LJ, London CA, Abbas AK. The roles of CD28 and CD40 ligand in T cell activation and tolerance. J. Immunol. 2000;164:4465. doi: 10.4049/jimmunol.164.9.4465. [DOI] [PubMed] [Google Scholar]
- 27.Whitmire JK, Flavell RA, Grewal IS, Larsen CP, Pearson TC, Ahmed R. CD40-CD40 ligand costimulation is required for generating antiviral CD4 T cell responses but is dispensable for CD8 T cell responses. J. Immunol. 1999;163:3194. [PubMed] [Google Scholar]
- 28.Chen W, Tang Q, Hendricks RL. Ex vivo model of leukocyte migration into herpes simplex virus-infected mouse corneas. J. Leukocyte Biol. 1996;60:167. doi: 10.1002/jlb.60.2.167. [DOI] [PubMed] [Google Scholar]
- 29.Avery AC, Zhao Z-S, Rodriguez A, Bikoff EK, Soheilian M, Foster CS, Cantor H. Resistance to herpes stromal keratitis conferred by an IgG2a-derived peptide. Nature. 1995;376:431. doi: 10.1038/376431a0. [DOI] [PubMed] [Google Scholar]
- 30.Panoutsakopoulou V, Sanchirico ME, Huster KM, Jansson M, Granucci F, Shim DJ, Wucherpfennig KW, Cantor H. Analysis of the relationship between viral infection and autoimmune disease. Immunity. 2001;15:137. doi: 10.1016/s1074-7613(01)00172-8. [DOI] [PubMed] [Google Scholar]
- 31.Zhao Z-S, Granucci F, Yeh L, Schaffer PA, Cantor H. Molecular mimicry by herpes simplex virus-type 1: autoimmune disease after viral infection. Science. 1998;279:1344. doi: 10.1126/science.279.5355.1344. [DOI] [PubMed] [Google Scholar]
- 32.Deshpande SP, Lee S, Zheng M, Song B, Knipe D, Kapp JA, Rouse BT. Herpes simplex virus-induced keratitis: evaluation of the role of molecular mimicry in lesion pathogenesis. J. Virol. 2001;75:3077. doi: 10.1128/JVI.75.7.3077-3088.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Verjans GM, Feron EJ, Dings ME, Cornelissen JG, Van Der LA, Baarsma GS, Osterhaus AD. T cells specific for the triggering virus infiltrate the eye in patients with herpes simplex virus-mediated acute retinal necrosis. J. Infect. Dis. 1998;178:27. doi: 10.1086/515586. [DOI] [PubMed] [Google Scholar]
- 34.Verjans GM, Remeijer L, van Binnendijk RS, Cornelissen JG, Volker-Dieben HJ, Baarsma SG, Osterhaus AD. Identification and characterization of herpes simplex virus-specific CD4+ T cells in corneas of herpetic stromal keratitis patients. J. Infect. Dis. 1998;177:484. doi: 10.1086/517382. [DOI] [PubMed] [Google Scholar]
- 35.Deshpande S, Zheng M, Lee S, Banerjee K, Gangappa S, Kumaraguru U, Rouse BT. Bystander activation involving T lymphocytes in herpetic stromal keratitis. J. Immunol. 2001;167:2902. doi: 10.4049/jimmunol.167.5.2902. [DOI] [PubMed] [Google Scholar]
- 36.Gangappa S, Babu JS, Thomas J, Daheshia M, Rouse BT. Virus-induced immunoinflammatory lesions in the absence of viral antigen recognition. J. Immunol. 1998;161:4289. [PubMed] [Google Scholar]
- 37.Gangappa S, Deshpande SP, Rouse BT. Bystander activation of CD4+ T cells accounts for herpetic ocular lesions. Invest. Ophthalmol. Vis. Sci. 2000;41:453. [PubMed] [Google Scholar]
- 38.Gangappa S, Deshpande SP, Rouse BT. Bystander activation of CD4+ T cells can represent an exclusive means of immunopathology in a virus infection. Eur.J. Immunol. 1999;29:3674. doi: 10.1002/(SICI)1521-4141(199911)29:11<3674::AID-IMMU3674>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 39.Edelmann KH, Wilson CB. Role of CD28/CD80 – 86 and CD40/ CD154 costimulatory interactions in host defense to primary herpes simplex virus infection. J. Virol. 2001;75:612. doi: 10.1128/JVI.75.2.612-621.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hendricks RL, Tumpey TM. Concurrent regeneration of T lymphocytes and susceptibility to HSV-1 corneal stromal disease. Curr. Eye Res. 1991;10:47. doi: 10.3109/02713689109020357. [DOI] [PubMed] [Google Scholar]