

Abstract
Protein crystallographers are often confronted with recalcitrant proteins not readily crystallizable, or which crystallize in problematic forms. A variety of techniques have been used to surmount such obstacles: crystallization using carrier proteins or antibody complexes, chemical modification, surface entropy reduction, proteolytic digestion, and additive screening. Here we present a synergistic approach for successful crystallization of proteins that do not form diffraction quality crystals using conventional methods. This approach combines favorable aspects of carrier-driven crystallization with surface entropy reduction. We have generated a series of maltose binding protein (MBP) fusion constructs containing different surface mutations designed to reduce surface entropy and encourage crystal lattice formation. The MBP advantageously increases protein expression and solubility, and provides a streamlined purification protocol. Using this technique, we have successfully solved the structures of three unrelated proteins that were previously unattainable. This crystallization technique represents a valuable rescue strategy for protein structure solution when conventional methods fail.
Keywords: protein crystallography, surface entropy reduction, carrier-driven crystallization, MBP, fixed-arm, rescue strategy
Introduction
Solving the structure of a protein using X-ray crystallography is accomplished via a multi-stage process which requires successful completion of each stage before proceeding to the next. One of the largest obstacles to obtaining crystal structures is the need for milligram quantities of highly purified, soluble protein. Once soluble protein is obtained and purified, it must be crystallized in a form that yields diffraction quality crystals, which can also represent a serious hurdle. According to recent statistics from the Joint Center for Structural Genomics (http://www.jcsg.org), only 9.8% of expressed proteins generate crystals of sufficient size and quality for X-ray screening. Of these crystals, 74% demonstrate adequate diffraction for data collection. Despite having diffraction-quality crystals, only 50% of these are solved.
Historically, when faced with a problem along the crystallography pipeline, many crystallographers have taken advantage of specific rescue strategies. Several methods of chemical modification have been developed, including reductive lysine methylation1–3 and carboxymethylation.4,5 In addition, crystallization of orthologous and/or isomeric proteins is also a respectable alternative. An in situ proteolysis method utilizing batch protease digestion has also been established.6,7 However, if these strategies are insufficient to produce diffraction quality crystals, what other options are available? In recent years, several different rescue techniques for managing intractable proteins have been utilized.
One such approach involves crystallization of the target protein in complex with its endogenous binding partners. However, expression and purification of such partners are not always possible, nor do all target proteins have known binding partners. An alternative to crystallization of endogenous multi-protein assemblies is complexation to an antibody. Many proteins (membrane proteins and viral capsid proteins in particular) have been crystallized in complex with high affinity antibody fragments8–17 (Supporting Information Table 1). Antibody-mediated crystallization14,18 is thought to increase the likelihood of crystallization by providing a large, hydrophilic interaction surface for initiating crystal lattice contacts and by effectively limiting the conformational flexibility of solvent exposed loop regions.14,19,20 There are some drawbacks that also must be considered. Generating the antibody fragments has become a routine process, but is still costly, time-consuming and labor-intensive.19,21 To create an antibody complex suitable for crystallization, large quantities of soluble protein must be generated for both the antibody fragment and the target protein, which can be problematic in many cases. The antibodies must display high affinity binding to the native conformation of the target protein, and must be soluble/stable under the same conditions.21,22 A major consideration for this technique is that soluble, stable, and specific antibodies must be produced individually for each target protein to be studied.
Another rescue strategy uses crystallization of a large carrier protein fused to the protein of interest.23 Many different carrier fusion proteins have been used in this manner (Table I), including maltose binding protein (MBP),27,30,36,38 glutathione-S-transferase (GST),46–49 thioredoxin (TRX),54,55 and lysozyme.50,52,53,56 In many cases, the presence of the carrier increases the expression level and solubility of the protein of interest.57–59 Positioning the carrier on the N-terminus of the polypeptide can have a “chaperone-like” effect, aiding in protein folding and increasing the yield of active product.60,61 Many such carrier proteins are affinity tags, which also streamlines the purification protocol, making this method highly adaptable for high-throughput screening.62,63
Table I.
Successfully Solved Structures Using Carrier-Driven Crystallizationa
| Carrier Protein | Target Protein | Size (AA) | Resolution (Å) | Linker sequence |
|---|---|---|---|---|
| MBP (∼366AA) | Hepatitis B surface antigen24 | 14 | 2.7-2.9 | GS |
| IAPP25 | 22 | 1.75–1.86 | AAA | |
| MutS (C-term)26 | 34 | 2.0 | HM | |
| MATa127 | 50 | 2.1–2.3 | AAAAA | |
| Huntingtin (N-term)28 | 66 | 3.5–3.7 | AAA | |
| TIM40/MIA4029 | 82 | 3.0 | SSSVPGRGSIEGRPEF | |
| gp2130 | 88 | 2.5 | AAA | |
| Monobody YSX131 | 91 | 2.0 | GSSGSS | |
| Monobody YS131 | 93 | 1.8 | GSS | |
| IPS1 CARD domain32 | 93 | 2.1 | SAMA | |
| CRFR1 ECD33 | 96 | 1.96–3.4 | AAAEF | |
| ZP3 (ZP-N domain)34 | 102 | 2.3–3.1 | AAA | |
| L3035 | 103 | 2.31 | SSSVPGRGSIEGRA | |
| gp2136 | 108 | — | AAA | |
| designed helical protein37 | 108 | 1.9 | SSSNNNNNNNNNN | |
| SarR38 | 115 | 2.3 | AAAEF | |
| Argonaute2 PAZ domain39 | 136 | 2.8 | AAAEF | |
| PTH1R ECD40 | 174 | 1.95 | AAAEF | |
| Der p 7 Mueller et al (in press) | 198 | 2.35 | AAA | |
| CD3841 | 256 | 2.4 | N/Ab | |
| 2OST42 | 298 | 2.65 | AAA | |
| RACK1A43 | 324 | 2.40 | AAA | |
| NEDD844 | 431 | 2.8 | AAA | |
| GST (∼217AA) | GP41 neutralizing antigen45 | 6 | 2.5 | SDLVPRGSM |
| C-terminal Fibrinogen gamma chain46 | 14 | 1.8 | SDP | |
| alpha-Na/K ATPase (ankyrin binding domain)47 | 25 | 2.6 | SDLVPRGS | |
| AML-1 NMTS48 | 31 | 2.7 | SDLVPRGSRRASVGS | |
| DREF49 | 53 | 2.5 | SDLVPRGS | |
| Lysozyme (∼163AA) | TELSAMc + E80-TELSAM50 | 80 | 2.4–2.6 | AGP |
| β2ARd,51 | 282 | 2.8 | — | |
| β2ARd,52 | 282 | 2.4 | — | |
| Adenosine A2A receptore,53 | 308 | 2.6 | — | |
| Thioredoxin (∼109AA) | Puf60 UHM54 | 100 | 2.2 | GSAM |
| VanHf,55 | 322 | 3.0 | N/Ab |
All target proteins listed in this table are fused C-terminal to the carrier protein, unless otherwise stated.
Information not available.
TELSAM was fused N-terminal to the lysozyme carrier protein.
Residues 231–262 of the β2AR have been replaced by residues 2-161 of T4 lysozyme.
Adenosine A2A receptor residues 209–221 have been replaced by residues 2-161 of T4 lysozyme.
VanH was fused N-terminal to the thioredoxin carrier protein. The structure of this fusion protein has not yet been reported.
From a crystallographic standpoint, addition of the carrier protein to the crystallizable unit can be both advantageous and detrimental. Because the structures of the carrier proteins used in this process have been solved to high resolution, they can be used as search models to solve the phase problem via molecular replacement. In addition, the carrier proteins used in these studies often crystallize readily, which suggests that they can provide molecular surfaces that are conducive for crystal lattice formation, similar to the addition of an antibody. However, the unavoidable linker region connecting the carrier to the protein of interest becomes a possible source for structural heterogeneity, a less pertinent issue in antibody-mediated crystallization. In the hopes of reducing conformational flexibility, minimizing the length of the linker region appears to be of paramount importance.23
In some cases, the increased expression and solubility of a carrier protein alone is not sufficient to produce diffraction quality crystals. There are several reported instances where expression of a recalcitrant target protein fused downstream of MBP has produced a high yield of soluble, active protein in adequate quantities for crystallization,64–67 yet diffraction quality crystals were not obtained.
Another rescue strategy now used with problematic systems is the powerful surface entropy reduction (SER) technique.68–70 Protein crystallization is an entropy-driven process—a delicate thermodynamic interplay between the ordering of protein molecules within a crystal lattice and the release of water molecules ordinarily bound to the surface of those protein molecules.71,72 SER attempts to compensate for the entropic behavior of large hydrophilic side chains on the protein surface.73 Random motions by these side chains can subtly increase the entropy of the system to the point that forming positive lattice contacts is inhibited.69 Therefore, by mutating patches of surface-exposed large, charged residues to small nonpolar amino acids, the surface entropy is reduced, allowing formation of crystal contacts featuring backbone amide and carbonyl groups.68
Though the advantages of SER lie in crystal lattice formation, there are some drawbacks to this technique. Altering the electrostatic properties of a protein can alter its biochemical behavior, and its interaction with other protein binding partners. If the structure of the protein target is unknown, determining the appropriate amino acid sequence to mutate—without disrupting the protein structure or physiology—can be difficult. In addition, replacing solvent-exposed hydrophilic residues with nonpolar amino acids can decrease the solubility and stability of the protein.68,74 Therefore, relatively unstable or less soluble proteins are not optimal candidates for SER.
To increase the probability of obtaining diffraction-quality crystals for problematic proteins, we have devised a flexible technique that harnesses the advantages of carrier-driven crystallization and surface entropy reduction. This approach works synergistically to increase the likelihood of crystallization. We have selected MBP for use in this system due to its high solubility, published success with this affinity tag, and its streamlined purification protocol. MBP is fused to the N-terminus of the target protein. The linker joining the two proteins has been truncated as published in Center et al. (1998) to create as little conformational heterogeneity as possible. To expand the capabilities of this system and improve the probability of crystallization, we have generated entropy reducing mutations on solvent exposed loops on the surface of the MBP based on the known crystal structures. These mutations maximize the potential for crystallization of the resulting fusion protein. Five easily interchangeable “cassettes,” carrying different combinations of the MBP/SER mutations, were created for use in this system (Table II).
Table II.
SER Mutations Present in the MBP-SER Cassettes
| Vector | SER mutations |
|---|---|
| pMALX(A) | D82A/K83A |
| pMALX (B) | E172A/N173A |
| pMALX(C) | D82A/K83A/K239A |
| pMALX(D) | E172A/N173A/K239A |
| pMALX(E) | D82A/K83A/E172A/N173A/K239A |
By producing a set of vectors with different SER mutations on the MBP rather than on the target protein, we negate the need to create SER mutations on each target, providing for a straightforward and efficient cloning protocol requiring minimal cost and effort. Using the SER technique in conjunction with the MBP carrier protein also addresses the issue of decreased protein solubility and stability. This method minimizes the risk of inadvertent mutagenesis of biologically important residues on the target protein—a potential hazard of the SER technique. Here, we present three case studies of unrelated target proteins (ranging in size from 198 to 324 amino acids) whose structures were determined using this approach.
Discussion
To date, in our laboratory, we used the tandem fixed-arm MBP/SER mutation system toward the crystallization of seven unrelated proteins whose structures could not be solved by conventional means. We have successfully solved the structures of three of these proteins—2OST (Table I),42 RACK1A,43 and Der p 7 (Mueller et al (doi:10.1016)). We have also obtained crystals for two (TargetA and TargetB) of the remaining four proteins (Table III). Co-crystals of TargetA (431 amino acids) in complex with DNA diffract to 2.3 Å resolution. However, these crystals are badly twinned, and simultaneously, have problems with pseudo-symmetry. Efforts are currently underway to optimize the crystal growth conditions for TargetB (292 amino acids). Thus far, the other two proteins (Targets C and D) have not yielded diffraction-quality crystals.
Table III.
Proteins for Which the MBP(SER) System has been Thus Far Unsuccessfula
| Target protein | Biological role | Size | Crystals? | Solved? |
|---|---|---|---|---|
| TargetA + DNA | Y-family polymerase | 431 amino acids | Yes | No (perfect hemihedral twinning) |
| TargetB | Protein sulfotransferase | 292 amino acids | Yes | No (insufficient diffraction quality) |
| TargetC + DNA | X-family polymerase | 357 amino acids | No | No |
| TargetD | Transcriptional regulator | 242 amino acids | No | No |
Before transferring these proteins to the MBP(SER) system, we first attempted to crystallize them alone or in the presence of ligands. Where appropriate, different species of proteins were used, utilizing multiple construct lengths in each case. These particular target proteins displayed low solubility in the absence of a fusion partner, so SER mutations were not used. In multiple cases, chemical modification was also used. However, these proteins generated no usable crystals.
Taken together, in a field where less than 10% of expressed proteins are solved and deposited (http://www.jcsg.org), we have increased our success rate to over 40%, with proteins that previously have been difficult to solve. Because of the small sample size (n = 7), these values may not be statistically significant, but are definitely encouraging.
Though the MBP/SER system can increase the likelihood of successfully crystallizing difficult proteins, the approach is not infallible. There are specific issues to consider. While N-terminal fusion of the MBP to the target protein can be beneficial for protein expression and purification, the significant size of the MBP may serve to limit the size of the target protein that can then be expressed in E. coli. Historically, MBP fusion proteins with smaller targets, relative to the size of the MBP, have yielded more structures (Table I), though a few targets larger than 150 residues have been determined. The synergistic nature of the additional SER mutations may increase the likelihood that a larger target protein could be crystallized. Placement of the MBP at the N-terminus of the fusion protein is generally considered to be advantageous due to its chaperone-like behavior. However, in cases where the N-terminus of the target protein is crucial for functionality or for protein-protein interaction, N-terminal MBP could be disruptive. On the other hand, it should be noted that MBP fusion proteins have been used to solve structures of large multi-protein complexes.44
In short, the tandem fixed-arm MBP/SER system represents a simple, cost-effective method for crystallization of problematic proteins that encompasses the advantageous properties of carrier-driven crystallization and surface entropy reduction. This technique represents another tool that can be used as a viable rescue strategy for problematic protein crystallography.
Experimental Methods
Vector design
The pMAL-c2x (New England Biolabs) vector was used as the backbone to create the pMALX vector, based on the reported vector,30,36 with a few alterations. The MBP amino acid sequence was originally modified to include mutations E359A/K362A/D363A and was truncated after Asn367, removing the Factor Xa protease cleavage site. The linker region encodes three alanine residues, as in the original vector,36 but was created by insertion of NotI and NheI restriction sites upstream of the EcoRI site in the original pMAL-c2x multicloning region [Fig. 1(A)].
Figure 1.
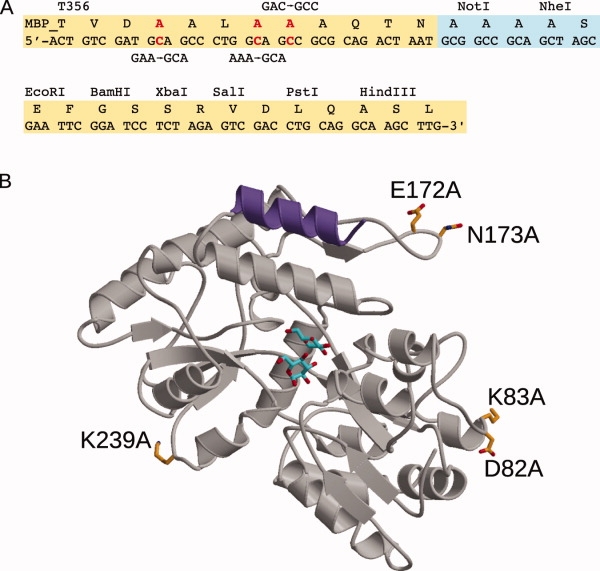
The tandem fixed-arm MBP/SER System. A: Schematic of the pMALX polylinker region. Sequences in yellow originated from the commercially available pMAL-c2X vector (New England Biolabs). Alanine substitutions in the C-terminal α-helix are highlighted in red with the nucleotide changes shown above or below in black. The sequence boxed in light blue shows the point of truncation of the MBP sequence after Asn367, deletion of the Factor Xa cleavage site, and insertion of NotI and NheI restriction sites. B: Ribbon diagram of the structure of MBP (PDB ID code 1HSJ38) showing the residues included in the crystallization cassettes. The C-terminal α-helix bearing three alanine substitutions is shown in purple. The bound maltose is drawn in stick (cyan). The positions of the residues that were substituted with alanine for the SER mutations are drawn in stick in orange. This figure was generated using MolScript.75
Creation of SER mutations in MBP
The pMALX vector was used as a template to create the SER mutations, using the QuikChange Mutagenesis protocol (Stratagene). By examining the published structure of MBP in the presence of maltose (PDB ID code 1HSJ38), we generated several mutations on solvent-exposed loop regions in different areas of the protein [Fig. 1(B) and Table II]. These mutations were generated either singly or in pairs on a particular loop, and were used in a combinatorial fashion to create five interchangeable cassettes: pMALX(A) with mutations D82A/K83A, pMALX(B) with mutations E172A/N173A, pMALX(C) with mutations D82A/K83A/K239A, pMALX(D) with mutations E172A/N173A/K239A, and pMALX(E) with mutations D82A/K83A/E172A/N173A/K239A [Fig. 1(B) and Table II].
Rationale of construct design
To maximize the chances of successful crystallization using this technique, initial construct design is crucial. The length of the linker region between the MBP and the protein of interest has been shortened to the extent that it should function more like a “fixed arm,” merging the two proteins into an inflexible crystallization unit (henceforth referred to as the “fusion protein”). Because the exact structure of the N-terminal end of the target protein is unknown, several N-terminal truncation constructs should be screened. If the linker is too long, the fusion protein may have undesired conformational flexibility. Yet, if the linker is not long enough, the connection to the MBP C-terminal α-helix may disrupt the structure of the downstream target protein, leading to misfolding or insolubility.
There are many tools to aid in construct design. We combine available biochemical data for the target protein and structural information from related or orthologous proteins with secondary structure prediction. Given known structural information on plausible domain organization, we design several constructs, utilizing different N-terminal truncations. This method maximizes the likelihood of successfully melding the N-terminal secondary structure of the target protein with the C-terminal α-helix of MBP. If structures of homologous proteins exist, manual docking of the N-terminus of the target protein to the MBP C-terminal α-helix can provide a visual aid to construct design.
Cloning and expression
Multiple constructs of the target protein are fused to the MBP carrier protein using traditional cloning methods. Restriction sites are added to the coding sequence of the target protein by PCR. The PCR products are digested with the appropriate restriction enzyme and ligated into the pMALX(wt) vector. All constructs are sequenced to ensure the absence of undesirable mutations, then transformed into an appropriate bacterial expression cell line. Small-scale test expressions (1L cultures) are performed to determine which constructs express soluble protein. In short, the cells are grown to density (OD600 = 0.5–1.0) at 37°C, at which time the temperature is decreased to 18°C for 30 min. Protein expression is induced by addition of isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 0.2–0.5 mM and incubation proceeds for 14–16 h. The cells are pelleted by centrifugation and lysed by sonication in an appropriate buffer. The soluble fraction is obtained by centrifugation of the lysate, then bound in-batch to amylose resin (New England Biolabs). Unbound protein is washed from the resin and the bound protein eluted by addition of 40 mM D-(+)-maltose dissolved in sonication buffer. The eluted protein is concentrated and loaded onto a Superdex200 16/60 size exclusion column (GE Healthcare) equilibrated in elution buffer containing 40 mM D-(+)-maltose. The ligand is added to the chromatography buffer for optimal protein separation because the MBP fusion protein can exhibit nonspecific interaction with the column matrix in the absence of maltose (data not shown). Size exclusion chromatography is likely crucial for success using the MBP/SER technique. Because the presence of MBP dramatically increases the expression and solubility of the resulting fusion protein, a substantial population of the protein can be incorrectly folded or aggregated. Such protein appears in the void volume peak from a size exclusion column and is easily separated from the more well-behaved and crystallizable target population.
As a general rule, the smallest target protein constructs with the shortest linker, and exhibiting the highest level of desired activity or function are used for crystallization. Active, soluble, and well-behaved constructs are transferred to the pMALX vectors harboring MBP/SER mutations by simple restriction and ligation. All constructs, including the wildtype, are then prepared in a large-scale purification (12 L cultures), using the same protocol as described above.
In many cases, the fusion protein is sufficiently pure for crystallization after the two-step purification protocol of affinity and size exclusion chromatography. However, further chromatography steps can be carried out if a higher extent of purification is required. After purification, the fusion protein is dialyzed into a buffer appropriate for crystallization—a low salt, non-phosphate buffer, with the addition of 5 mM D-(+)-maltose. Maltose is included in the crystallization buffer because the MBP exhibits conformational heterogeneity dependent on the presence or absence of ligand.76,77 Crystallization trials are then carried out using commercially available screens. Crystals obtained in the initial screens are subsequently optimized for size and quality and then used for data collection.
Solving the phasing problem with molecular replacement
A key advantage of the MBP fusion system is that the MBP protein structure can be used to solve the crystallographic phase problem by molecular replacement. Once molecular replacement solutions for the MBP have been found, these molecules can be fixed, and a second molecular replacement search for the target protein can be performed if quality search models exist. However, if no such search model exists, the phase information gained from the MBP may be sufficient for the target molecule to be built manually, utilizing an iterative process of density modification and solvent flattening to improve the maps, followed by model building and refinement. Alternatively, automated model building programs such as AutoBuild78 in Phenix79 have been tested and shown in some cases to provide helpful starting models for chain tracing the target protein if no acceptable molecular replacement models exist.
Case study #1: 2OST from Gallus gallus
Heparan sulfate (HS) is a highly sulfated linear polysaccharide found ubiquitously on the surface of cells, and in the surrounding extracellular matrix. Specifically sulfated saccharide sequences act as binding sites for proteins involved in a myriad of biological processes, ranging from embryonic development, inflammatory response, and blood coagulation (reviewed in80). 2-O-sulfotransferase (2OST) is involved in HS biosynthesis, transferring a sulfo group to the 2-OH position of iduronic or glucuronic acid within an HS chain.81 This sulfation is critical for substrate recognition in blood coagulation and for signal transduction through fibroblast growth factor-mediated pathways.82
We recently determined the crystal structure of chicken 2OST using the MBP/SER system.42 Initial attempts to structurally characterize 2OST involved crystallization of a His6x-tagged construct of the catalytic domain (amino acids Arg63-Asn356) of hamster 2OST (94% similarity to chicken 2OST). This construct formed somewhat disordered crystalline rods which diffracted poorly to 3.3 Å. The structure could not be solved from these data. To generate a maximal change in the crystallization surface, while preserving physiological relevance, the catalytic domain (Asp69-Asn356) of chicken 2OST (92% similarity to human 2OST) was fused downstream of the MBP carrier protein. Asp69 was selected as the starting residue based on conserved secondary structural elements found in both cytosolic and membrane-bound sulfotransferases. The sequence encoding these amino acids was inserted between the NotI and BamHI restriction sites in the pMALX(wt) vector.42
Expression of the MBP-2OST fusion protein gave a greatly increased yield of soluble protein, compared to the His6x-tagged protein (unpublished data). The fusion protein also exhibited markedly improved solubility, both in solution and during crystallization trials, and proved active in sulfotransferase assays.42 The MBP-2OST protein crystals were obtained at 4°C using the sitting-drop vapor diffusion method.42,83
The published structure of MBP (PDB ID code 1HSJ) was used to solve the phase problem via molecular replacement with the program MOLREP from the CCP4 Suite.84 A single molecule of MBP was found within the asymmetric unit in this crystal form. A generic search model comprised of the conserved secondary structural elements distinctive to known sulfotransferases was generated using amino acids Asp39-Tyr62, Arg78-Glu83, Ile104-Ile146, Leu189-Phe210, and Pro269-Lys285 from estrogen sulfotransferase (EST, PDB ID code 1AQU)85 and was used to determine the relative position of 2OST to MBP with a second round of molecular replacement in MOLREP. Density modification in CNS86 was utilized to improve the phases so that model building could be carried out. The final model includes all residues in MBP and residues Asp69-Lys354 of chicken 2OST [Fig. 2(A)].
Figure 2.
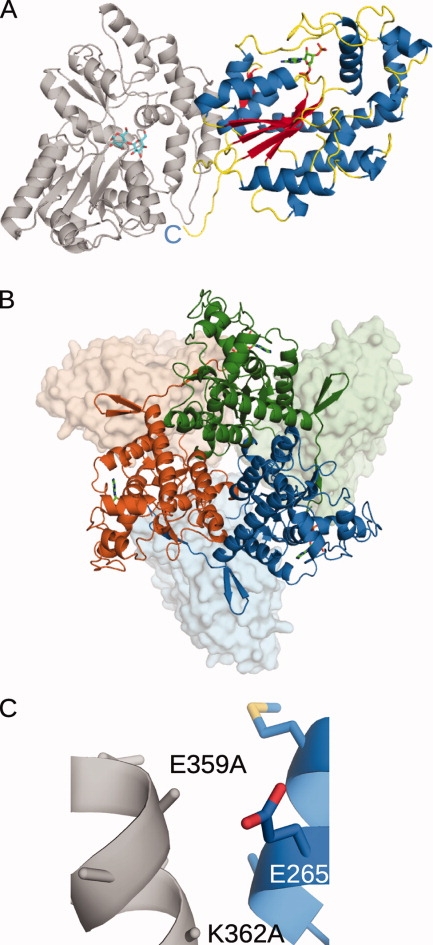
Crystal-structure of MBP(wt)-2OST (PDB ID code 3F5F). The maltose bound to the MBP is drawn in stick in cyan. The PAP co-factor bound to the 2OST is drawn in stick in green. Ribbon diagrams were generated using PyMOL.87 A: Ribbon diagram of the crystal structure of the MBP(wt)-2OST fusion protein. The structure of the MBP is shown in gray. α-helices from the 2OST structure are shown in blue, β-strands are shown in red, and the random coil regions are shown in yellow. B: Ribbon diagram of the trimeric MBP(wt)-2OST. Molecule 1 is shown in green, Molecule 2 in blue, and Molecule 3 is shown in orange. 2OST molecules are drawn in ribbon and the MBP molecules are shown as surface renderings. C: Ribbon diagram of the MBP(wt)-2OST interaction surface. Secondary structural elements of the 2OST are shown in blue. The C-terminal α-helix of MBP is shown in gray, with the K359A and K362A mutations drawn in stick.
Though only a single molecule of MBP-2OST is present in the asymmetric unit in this space group, examination of the crystal packing reveals a three-fold crystallographic axis running through the center of a trimer [Fig. 2(B)]. Size exclusion chromatography of MBP-2OST suggests that the protein behaves as a trimer in solution. The trimeric interface is mediated by C-terminal residues Asn345-Tyr352, which form an additional β-strand antiparallel to strand β5 of another monomer. 2OST truncation mutants lacking the C-terminal residues exhibited greatly decreased trimer formation, and concomitantly reduced sulfotransferase activity. These observations indicate that the trimeric form may be the biologically active unit, and most noteably, that addition of the MBP carrier protein did not disrupt this interaction42 [Fig. 2(B)]. The position of all three MBP molecules, grouped proximally at the N-termini of the 2OST molecules, could be viewed as a possible arrangement of the N-terminal transmembrane domains present in the native proteins. Such an arrangement is consistent with current knowledge of the physiological behavior of 2OST. In vivo, this sulfotransferase is associated with the luminal side of the Golgi membrane, where it modifies oligosaccharide groups on glycoproteins later bound for the cell membrane and the extracellular matrix.88
In this case study, SER mutations were not required to obtain diffraction quality crystals. The E359A mutation within the C-terminal α-helix of MBP was fortuitous in this instance, as deletion of the wildtype glutamate residue allows a tighter interaction between the MBP and 2OST molecules [Fig. 2(C)]. A glutamate at position 359 would have likely created an electrostatic clash with the adjacent Glu265 residue on the 2OST molecule, preventing domain arrangement and crystallization.
Case study #2: RACK1A from Arabidopsis thaliana
The receptor for activated C-kinase 1 (RACK1) belongs to the WD40 repeat family of β-propeller proteins and is highly conserved in eukaryotes. RACK1 is believed to function as a scaffold protein, mediating multiple simultaneous protein-protein interactions involved in diverse cellular processes including regulation of transcription, signal transduction pathways, and ribosome assembly (reviewed in Refs. 89 and 90). RACK1 is highly conserved in eukaryotes—mouse and chicken orthologs are identical to the human protein. RACK1A, one of three RACK1 isoforms in A. thaliana, exhibits 66% sequence conservation to human RACK1. The high degree of sequence conservation across these diverse species suggests a conserved function in eukaryotes.
Initial crystallization attempts with untagged RACK1A failed to produce crystals with commercially available crystallization matrix screens. Therefore, the sequence encoding RACK1A amino acids Gly4-Tyr327 was cloned into the pMALX expression system, using the NotI and BamHI restriction sites, leaving the shortest possible linker between the two proteins. Gly4 was chosen as the starting residue based on secondary structure prediction, and by manually docking the N-terminus of related β-propeller proteins to the C-terminus of the MBP.43
The structure of the MBP(C)-RACK1A is shown in Figure 3(A). A detailed analysis of the structure within the crystal lattice identifies several key features. Shortening the linker between the MBP and the RACK1A was effective in this fusion protein—only one residue at the junction (Gly4) was not involved in a secondary structural element. Given the brevity of the linker connecting the two proteins, there are surprisingly few inter-domain interactions between MBP and RACK1A [Fig. 3(A)]. Residues along the interface are a minimum of 5 Å apart. In addition, all interactions forming the crystal lattice are either MBP-MBP or MBP-RACK1A. There are no RACK1A-RACK1A interactions, which may explain the failure of crystallization attempts with RACK1A alone.
Figure 3.
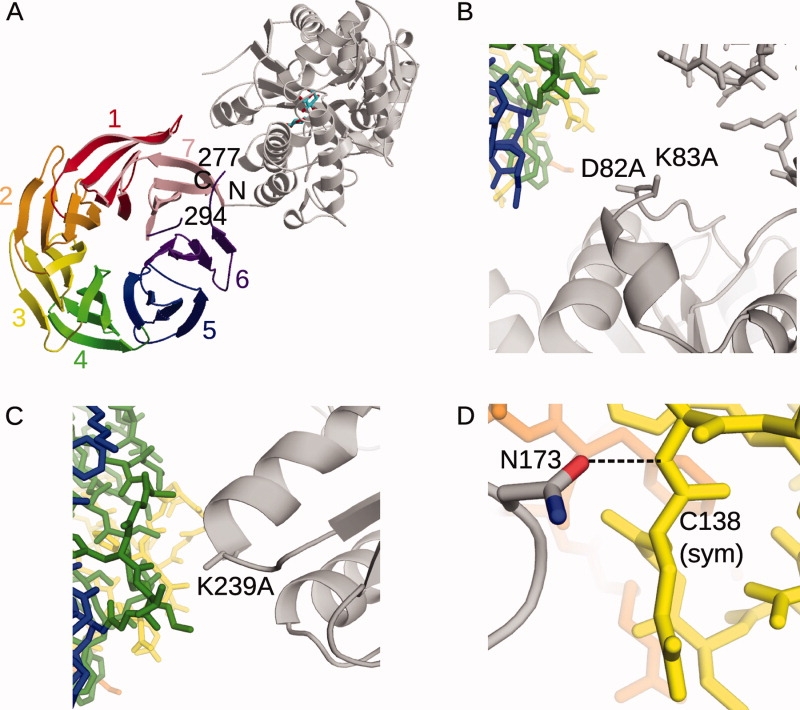
Crystal structure of the MBP(C)-RACK1A fusion protein (PDB ID code 3DM0). A: Ribbon diagram of the MBP(C)-RACK1A fusion protein. The bound maltose is drawn in stick (cyan). The structure of the MBP is shown in gray, relative to that of the RACK1A (multicolored). The β-strands of the RACK1A β-propeller domain are shown as ribbons, with each WD motif colored and labeled. The residues of the disordered loop (Lys277-Lys294) in WD6 (purple) are labeled. B,C, and D: Secondary structural elements of the MBP(C) protein are drawn as gray ribbons, with the SER mutations drawn in stick. The positions of symmetry-related MBP molecules (gray) and RACK1A (green, blue, yellow) are drawn in stick. B: Position of the D82A/K83A SER mutations in crystal lattice formation. C: Position of the K239A mutation in crystal lattice formation. D: Role of Asn173 in crystal lattice formation.
The position of the D82A/K83A/K239A mutations within the lattice suggests a preference for crystal formation with these specific mutations over other surface mutations [Fig. 3(B,C)]. D82A and K83A are located on the surface of the MBP, near two different symmetry-related molecules [Fig. 3(B)]. The presence of large aspartate and lysine residues could have negatively influenced crystal packing. Similarly, the loop containing K239A forms a tight crystal lattice contact, and a lysine side chain could have disrupted crystal formation [Fig. 3(C)]. Conversely, Asn173 participates in a hydrogen bonding lattice contact with the backbone of Cys138 of a symmetry-related RACK1A molecule [Fig. 3(D)]. An alanine substitution at this position could have been detrimental to formation of this lattice contact. Because of the impossibility of determining beforehand which residues may be either advantageous or detrimental to lattice formation, the availability of multiple cassettes with different combinations of SER mutations improves the odds of crystallization.
Case study #3: Der p 7 from Dermatophagoides pteronyssinus
Conditions stemming from allergic response—that is, rhinitis, asthma, anaphylaxis—are currently a major health concern. One of the most common sources of indoor allergens is the house dust mite. To date, multiple groups of dust mite allergens have been identified, many of which have been found to be enzymes.91 Allergens belonging to Groups 5 and 7 are proteins of unknown function.92 Der p 7-reactive IgE serum antibodies are found in only 53% of allergic patients,93 however, the scale of the response can be as great as for the more major allergens.94 To gain some insight into the abnormal patient response, and the natural protein function in dust mites, we solved the structure of the Group 7 allergen from Dermatophagoides pteronyssinus, Der p 7 (Mueller et al, doi:10.1016).
Based on sequence alignment, Der p 7 is not related to any known protein. However, analyzing the protein sequence with the GenTHREADER alignment server95 suggested that Der p 7 may have a fold similar to two lipid-binding proteins—human Bactericidal/permeability-increasing protein (BPI, PDB ID code 1EWF96) and the Takeout 1 protein from Epiphyas postvittana (PDB ID code 3E8T97)—though with expectedly low sequence identities (14% and 12%, respectively).
His6x-tagged Der p 7 was readily expressed in soluble form in E. coli, and quickly generated a variety of crystals. However, these crystals were poorly ordered, and did not yield quality diffraction. Der p 7 residues Asp18-Gln215 were cloned into the pMALX cassettes using the NotI and BamHI restriction sites. The N-terminal region of Der p 7 carries a signal peptide, which was removed in this construct. Residue 18 was used as the starting residue for this construct based on secondary structure prediction. The fusion protein behaved similarly to the natural allergen in IgE binding assays (Mueller et al, doi:10.1016). Crystals of MBP-Der p 7 were obtained using the MBP cassette bearing the D82A/K83A/E172A/N173A/K239A SER mutations. These crystals diffracted at 2.35 Å (Mueller et al, doi:10.1016).
In this crystal form, there are three MBP(E)-Der p 7 molecules within the asymmetric unit. Following positioning of the MBP molecules using molecular replacement with an MBP model (PDB ID code 3DM043), discontinuous electron density was visible for the Der p 7 molecule. Although molecular replacement models using the structurally related proteins BPI (PDB ID code 1EWF96) and Takeout1 (PDB ID code 3E8T97) were unsuccessful, these structures provided a guide for chain tracing and manual model building. Molecule A from the final MBP(E)-Der p 7 model is shown in Figure 4(A). All three of the Der p 7 molecules superimpose well (RMSD of less than 0.51Å over 197 Cα atoms). Though Der p 7 currently has no known enzymatic activity, the structural similarities between Der p 7 and BPI or Takeout1 suggested a possible role in binding of hydrophobic ligands. Subsequent HSQC and STD-NMR experiments have demonstrated interaction of the bacterial lipopeptide polymixin B with Der p 7 (Mueller et al, doi:10.1016). Such interactions with hydrophobic ligands may provide insight into the allergenicity of Der p 7. The binding of hydrophoboic ligands is thought to inappropriately stimulate an allergic immune response.98
Figure 4.
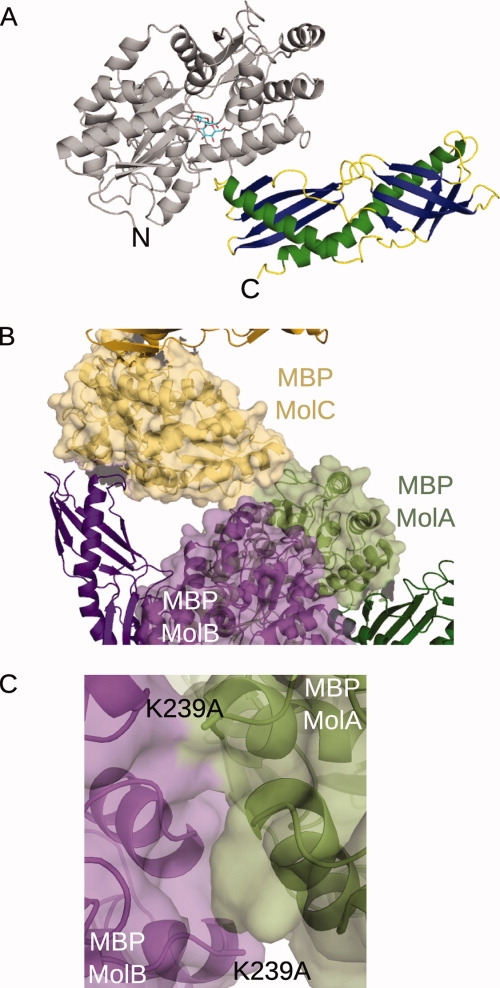
Crystal structure of the MBP(E)-Der p 7 fusion protein (PDB ID code 3H4Z). A: The ribbon diagram of Molecule A of MBP(E)-Der p 7. The structure of the MBP is shown in gray, with the bound maltose drawn in stick (cyan). α-helices of the Der p 7 structure are shown in green, β-strands in dark blue, and random coil regions in yellow. B: Intermolecular interactions comprising the asymmetric unit of the MBP(E)-Der p 7 crystals. Surface renderings of the MBP molecules (Molecule A in light green, Molecule B in light purple, Molecule C in light gold) illustrating the extensive MBP-MBP interactions within the asymmetric unit. Minimal interactions between the Der p 7 of Molecule B (dark purple) with the MBP of Molecule C (yellow). C: Position of the K239A mutation in the crystal lattice. The K239A mutations of Molecules A (green) and B (purple) lie along an intermolecular interaction surface.
A detailed analysis of the MBP(E)-Der p 7 crystal packing yielded some interesting observations. The most extensive interactions appear to involve MBP-MBP interactions [Fig. 4(B)], which, as for 2OST and for RACK1A, appear to be the driving force for crystal lattice formation. All permutations of MBP-MBP, MBP-Der p 7, and Der p 7-Der p 7 interactions exist between symmetry-related molecules.
Determining the role of the SER mutations is more complex for the Der p 7 crystal lattice than for either 2OST or RACK1A. For both the Molecules A and B, K239A lies along the interaction surface between the two MBP molecules. The presence of a lysine side chain at this junction could have disrupted lattice packing if the original lysine residue were still present [Fig. 3(C)]. For the D82A/K83A and E172A/N173A mutations, there would have been ample space within the crystal to accommodate the large side chains. The general reduction of side chain entropy could have been a contributing factor. In the case of K239A, however, there is clear evidence that this particular SER mutation may have played a key role in forming this crystal lattice.
Acknowledgments
The authors thank Traci Hall and Michael Murray for critical reading and thoughtful comments on the manuscript. The expression vectors are freely available upon request.
References
- 1.Means GE, Feeney RE. Reductive alkylation of proteins. Anal Biochem. 1995;224:1–16. doi: 10.1006/abio.1995.1001. [DOI] [PubMed] [Google Scholar]
- 2.Rayment I. Reductive alkylation of lysine residues to alter crystallization properties of proteins. Methods Enzymol. 1997;276:171–179. [PubMed] [Google Scholar]
- 3.Walter TS, Meier C, Assenberg R, Au KF, Ren J, Verma A, Nettleship JE, Owens RJ, Stuart DI, Grimes JM. Lysine methylation as a routine rescue strategy for protein crystallization. Structure. 2006;14:1617–1622. doi: 10.1016/j.str.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hegy GB, Shackleton CH, Carlquist M, Bonn T, Engstrom O, Sjoholm P, Witkowska HE. Carboxymethylation of the human estrogen receptor ligand-binding domain-estradiol complex: HPLC/ESMS peptide mapping shows that cysteine 447 does not react with iodoacetic acid. Steroids. 1996;61:367–373. doi: 10.1016/0039-128x(96)00042-6. [DOI] [PubMed] [Google Scholar]
- 5.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–937. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 6.Dong A, Xu X, Edwards AM. In situ proteolysis for protein crystallization and structure determination. Nat Methods. 2007;4:1019–1021. doi: 10.1038/nmeth1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wernimont A, Edwards A. In situ proteolysis to generate crystals for structure determination: an update. PLoS ONE. 2009;4(4):e5094. doi: 10.1371/journal.pone.0005094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobo-Molina A, Ding J, Nanni RG, Clark AD, Jr, Lu X, Tantillo C, Williams RL, Kamer G, Ferris AL, Clark P, Hizi A, Hughes SH, Arnold E. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 A resolution shows bent DNA. Proc Natl Acad Sci USA. 1993;90:6320–6324. doi: 10.1073/pnas.90.13.6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nermut MV, Hockley DJ, Jowett JB, Jones IM, Garreau M, Thomas D. Fullerene-like organization of HIV gag-protein shell in virus-like particles produced by recombinant baculovirus. Virology. 1994;198:288–296. doi: 10.1006/viro.1994.1032. [DOI] [PubMed] [Google Scholar]
- 10.Iwata S, Ostermeier C, Ludwig B, Michel H. Structure at 2.8 A resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 11.Ostermeier C, Iwata S, Ludwig B, Michel H. Fv fragment-mediated crystallization of the membrane protein bacterial cytochrome c oxidase. Nat Struct Biol. 1995;2:842–846. doi: 10.1038/nsb1095-842. [DOI] [PubMed] [Google Scholar]
- 12.Ostermeier C, Harrenga A, Ermler U, Michel H. Structure at 2.7 A resolution of the Paracoccus denitrificans two-subunit cytochrome c oxidase complexed with an antibody FV fragment. Proc Natl Acad Sci USA. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]
- 14.Hunte C, Michel H. Crystallisation of membrane proteins mediated by antibody fragments. Curr Opin Struct Biol. 2002;12:503–508. doi: 10.1016/s0959-440x(02)00354-8. [DOI] [PubMed] [Google Scholar]
- 15.Shore DA, Teyton L, Dwek RA, Rudd PM, Wilson IA. Crystal structure of the TCR co-receptor CD8alphaalpha in complex with monoclonal antibody YTS 105.18 Fab fragment at 2.88 A resolution. J Mol Biol. 2006;358:347–354. doi: 10.1016/j.jmb.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 16.Shore DA, Issafras H, Landais E, Teyton L, Wilson IA. The crystal structure of CD8 in complex with YTS156.7.7 Fab and interaction with other CD8 antibodies define the binding mode of CD8 αβ to MHC class I. J Mol Biol. 2008;384:1190–1202. doi: 10.1016/j.jmb.2008.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Solmaz SR, Hunte C. Structure of complex III with bound cytochrome c in reduced state and definition of a minimal core interface for electron transfer. J Biol Chem. 2008;283:17542–17549. doi: 10.1074/jbc.M710126200. [DOI] [PubMed] [Google Scholar]
- 18.Kovari LC, Momany C, Rossmann MG. The use of antibody fragments for crystallization and structure determinations. Structure. 1995;3:1291–1293. doi: 10.1016/s0969-2126(01)00266-0. [DOI] [PubMed] [Google Scholar]
- 19.Shea C, Bloedorn L, Sullivan MA. Rapid isolation of single-chain antibodies for structural genomics. J Struct Funct Genomics. 2005;6:171–175. doi: 10.1007/s10969-005-5246-6. [DOI] [PubMed] [Google Scholar]
- 20.Lam AY, Pardon E, Korotkov KV, Hol WG, Steyaert J. Nanobody-aided structure determination of the EpsI: EpsJ pseudopilin heterodimer from Vibrio vulnificus. J Struct Biol. 2009;166:8–15. doi: 10.1016/j.jsb.2008.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Venturi M, Hunte C. Monoclonal antibodies for the structural analysis of the Na+/H+ antiporter NhaA from Escherichia coli. Biochim Biophys Acta. 2003;1610:46–50. doi: 10.1016/s0005-2736(02)00713-7. [DOI] [PubMed] [Google Scholar]
- 22.Rothlisberger D, Pos KM, Pluckthun A. An antibody library for stabilizing and crystallizing membrane proteins—selecting binders to the citrate carrier CitS. FEBS Lett. 2004;564:340–348. doi: 10.1016/S0014-5793(04)00359-X. [DOI] [PubMed] [Google Scholar]
- 23.Smyth DR, Mrozkiewicz MK, McGrath WJ, Listwan P, Kobe B. Crystal structures of fusion proteins with large-affinity tags. Protein Sci. 2003;12:1313–1322. doi: 10.1110/ps.0243403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Saul FA, Vulliez-le Normand B, Lema F, Bentley GA. Crystal structure of a recombinant form of the maltodextrin-binding protein carrying an inserted sequence of a B-cell epitope from the preS2 region of hepatitis B virus. Proteins. 1997;27:1–8. doi: 10.1002/(sici)1097-0134(199701)27:1<1::aid-prot2>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 25.Wiltzius JJ, Sievers SA, Sawaya MR, Eisenberg D. Atomic structures of IAPP (amylin) fusions suggest a mechanism for fibrillation and the role of insulin in the process. Protein Sci. 2009;18:1521–1530. doi: 10.1002/pro.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mendillo ML, Putnam CD, Kolodner RD. Escherichia coli MutS tetramerization domain structure reveals that stable dimers but not tetramers are essential for DNA mismatch repair in vivo. J Biol Chem. 2007;282:16345–16354. doi: 10.1074/jbc.M700858200. [DOI] [PubMed] [Google Scholar]
- 27.Ke A, Wolberger C. Insights into binding cooperativity of MATa1/MATalpha2 from the crystal structure of a MATa1 homeodomain-maltose binding protein chimera. Protein Sci. 2003;12:306–312. doi: 10.1110/ps.0219103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim MW, Chelliah Y, Kim SW, Otwinowski Z, Bezprozvanny I. Secondary structure of Huntingtin amino-terminal region. Structure. 2009;17:1205–1212. doi: 10.1016/j.str.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kawano S, Yamano K, Naoe M, Momose T, Terao K, Nishikawa S, Watanabe N, Endo T. Structural basis of yeast Tim40/Mia40 as an oxidative translocator in the mitochondrial intermembrane space. Proc Natl Acad Sci USA. 2009;106:14403–14407. doi: 10.1073/pnas.0901793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kobe B, Center RJ, Kemp BE, Poumbourios P. Crystal structure of human T cell leukemia virus type 1 gp21 ectodomain crystallized as a maltose-binding protein chimera reveals structural evolution of retroviral transmembrane proteins. Proc Natl Acad Sci USA. 1999;96:4319–4324. doi: 10.1073/pnas.96.8.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilbreth RN, Esaki K, Koide A, Sidhu SS, Koide S. A dominant conformational role for amino acid diversity in minimalist protein-protein interfaces. J Mol Biol. 2008;381:407–418. doi: 10.1016/j.jmb.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Potter JA, Randall RE, Taylor GL. Crystal structure of human IPS-1/MAVS/VISA/Cardif caspase activation recruitment domain. BMC Struct Biol. 2008;8:11. doi: 10.1186/1472-6807-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pioszak AA, Parker NR, Suino-Powell K, Xu HE. Molecular recognition of corticotropin-releasing factor by its G-protein-coupled receptor CRFR1. J Biol Chem. 2008;283:32900–32912. doi: 10.1074/jbc.M805749200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monne M, Han L, Schwend T, Burendahl S, Jovine L. Crystal structure of the ZP-N domain of ZP3 reveals the core fold of animal egg coats. Nature. 2008;456:653–657. doi: 10.1038/nature07599. [DOI] [PubMed] [Google Scholar]
- 35.Chao JA, Prasad GS, White SA, Stout CD, Williamson JR. Inherent protein structural flexibility at the RNA-binding interface of L30e. J Mol Biol. 2003;326:999–1004. doi: 10.1016/s0022-2836(02)01476-6. [DOI] [PubMed] [Google Scholar]
- 36.Center RJ, Kobe B, Wilson KA, Teh T, Howlett GJ, Kemp BE, Poumbourios P. Crystallization of a trimeric human T cell leukemia virus type 1 gp21 ectodomain fragment as a chimera with maltose-binding protein. Protein Sci. 1998;7:1612–1619. doi: 10.1002/pro.5560070715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laporte SL, Forsyth CM, Cunningham BC, Miercke LJ, Akhavan D, Stroud RM. De novo design of an IL-4 antagonist and its structure at 1.9 A. Proc Natl Acad Sci USA. 2005;102:1889–1894. doi: 10.1073/pnas.0408890102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y, Manna A, Li R, Martin WE, Murphy RC, Cheung AL, Zhang G. Crystal structure of the SarR protein from Staphylococcus aureus. Proc Natl Acad Sci USA. 2001;98:6877–6882. doi: 10.1073/pnas.121013398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song JJ, Liu J, Tolia NH, Schneiderman J, Smith SK, Martienssen RA, Hannon GJ, Joshua-Tor L. The crystal structure of the Argonaute2 PAZ domain reveals an RNA binding motif in RNAi effector complexes. Nat Struct Biol. 2003;10:1026–1032. doi: 10.1038/nsb1016. [DOI] [PubMed] [Google Scholar]
- 40.Pioszak AA, Xu HE. Molecular recognition of parathyroid hormone by its G protein-coupled receptor. Proc Natl Acad Sci USA. 2008;105:5034–5039. doi: 10.1073/pnas.0801027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kukimoto M, Nureki O, Shirouzu M, Katada T, Hirabayashi Y, Sugiya H, Furuyama S, Yokoyama S, Hara-Yokoyama M. Crystallization and preliminary X-ray diffraction analysis of the extracellular domain of the cell surface antigen CD38 complexed with ganglioside. J Biochem. 2000;127:181–184. doi: 10.1093/oxfordjournals.jbchem.a022592. [DOI] [PubMed] [Google Scholar]
- 42.Bethea HN, Xu D, Liu J, Pedersen LC. Redirecting the substrate specificity of heparan sulfate 2-O-sulfotransferase by structurally guided mutagenesis. Proc Natl Acad Sci USA. 2008;105:18724–18729. doi: 10.1073/pnas.0806975105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ullah H, Scappini EL, Moon AF, Williams LV, Armstrong DL, Pedersen LC. Structure of a signal transduction regulator, RACK1, from Arabidopsis thaliana. Protein Sci. 2008;17:1771–1780. doi: 10.1110/ps.035121.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang DT, Hunt HW, Zhuang M, Ohi MD, Holton JM, Schulman BA. Basis for a ubiquitin-like protein thioester switch toggling E1-E2 affinity. Nature. 2007;445:394–398. doi: 10.1038/nature05490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim K, Ho JX, Keeling K, Gilliland GL, Ji X, Ruker F, Carter DC. Three-dimensional structure of Schistosoma japonicum glutathione S-transferase fused with a six-amino acid conserved neutralizing epitope of gp41 from HIV. Protein Sci. 1994;3:2233–2244. doi: 10.1002/pro.5560031209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ware S, Donahue JP, Hawiger J, Anderson WF. Structure of the fibrinogen gamma-chain integrin binding and factor XIIIa cross-linking sites obtained through carrier protein driven crystallization. Protein Sci. 1999;8:2663–2671. doi: 10.1110/ps.8.12.2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Z, Devarajan P, Dorfman AL, Morrow JS. Structure of the ankyrin-binding domain of α-Na, K-ATPase. J Biol Chem. 1998;273:18681–18684. doi: 10.1074/jbc.273.30.18681. [DOI] [PubMed] [Google Scholar]
- 48.Tang L, Guo B, Javed A, Choi JY, Hiebert S, Lian JB, van Wijnen AJ, Stein JL, Stein GS, Zhou GW. Crystal structure of the nuclear matrix targeting signal of the transcription factor acute myelogenous leukemia-1/polyoma enhancer-binding protein 2alphaB/core binding factor α2. J Biol Chem. 1999;274:33580–33586. doi: 10.1074/jbc.274.47.33580. [DOI] [PubMed] [Google Scholar]
- 49.Kuge M, Fujii Y, Shimizu T, Hirose F, Matsukage A, Hakoshima T. Use of a fusion protein to obtain crystals suitable for X-ray analysis: crystallization of a GST-fused protein containing the DNA-binding domain of DNA replication-related element-binding factor, DREF. Protein Sci. 1997;6:1783–1786. doi: 10.1002/pro.5560060822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nauli S, Farr S, Lee YJ, Kim HY, Faham S, Bowie JU. Polymer-driven crystallization. Protein Sci. 2007;16:2542–2551. doi: 10.1110/ps.073074207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hanson MA, Cherezov V, Griffith MT, Roth CB, Jaakola VP, Chien EY, Velasquez J, Kuhn P, Stevens RC. A specific cholesterol binding site is established by the 2.8 A structure of the human β2-adrenergic receptor. Structure. 2008;16:897–905. doi: 10.1016/j.str.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, Stevens RC. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, Ijzerman AP, Stevens RC. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Corsini L, Hothorn M, Scheffzek K, Sattler M, Stier G. Thioredoxin as a fusion tag for carrier-driven crystallization. Protein Sci. 2008;17:2070–2079. doi: 10.1110/ps.037564.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stoll VS, Manohar AV, Gillon W, MacFarlane EL, Hynes RC, Pai EF. A thioredoxin fusion protein of VanH, a d-lactate dehydrogenase from Enterococcus faecium: cloning, expression, purification, kinetic analysis, and crystallization. Protein Sci. 1998;7:1147–1155. doi: 10.1002/pro.5560070508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Yao XJ, Weis WI, Stevens RC, Kobilka BK. GPCR engineering yields high-resolution structural insights into β-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 57.Braun P, Hu Y, Shen B, Halleck A, Koundinya M, Harlow E, LaBaer J. Proteome-scale purification of human proteins from bacteria. Proc Natl Acad Sci USA. 2002;99:2654–2659. doi: 10.1073/pnas.042684199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hammarstrom M, Hellgren N, van Den Berg S, Berglund H, Hard T. Rapid screening for improved solubility of small human proteins produced as fusion proteins in Escherichia coli. Protein Sci. 2002;11:313–321. doi: 10.1110/ps.22102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shih YP, Kung WM, Chen JC, Yeh CH, Wang AH, Wang TF. High-throughput screening of soluble recombinant proteins. Protein Sci. 2002;11:1714–1719. doi: 10.1110/ps.0205202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kapust RB, Waugh DS. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sachdev D, Chirgwin JM. Fusions to maltose-binding protecontrol of folding and solubility in protein purification. Methods Enzymol. 2000;326:312–321. doi: 10.1016/s0076-6879(00)26062-x. [DOI] [PubMed] [Google Scholar]
- 62.Edwards AM, Arrowsmith CH, Christendat D, Dharamsi A, Friesen JD, Greenblatt JF, Vedadi M. Protein production: feeding the crystallographers and NMR spectroscopists. Nat Struct Biol Suppl. 2000;7:970–972. doi: 10.1038/80751. [DOI] [PubMed] [Google Scholar]
- 63.Stevens RC. Design of high-throughput methods of protein production for structural biology. Structure. 2000;8:R177–R185. doi: 10.1016/s0969-2126(00)00193-3. [DOI] [PubMed] [Google Scholar]
- 64.Grisshammer R, Little J, Aharony D. Expression of rat NK-2 (neurokinin A) receptor in E. coli. Receptors Channels. 1994;2:295–302. [PubMed] [Google Scholar]
- 65.Chen GQ, Gouaux JE. Overexpression of bacterio-opsin in Escherichia coli as a water-soluble fusion to maltose binding proteefficient regeneration of the fusion protein and selective cleavage with trypsin. Protein Sci. 1996;5:456–467. doi: 10.1002/pro.5560050307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kanamori M, Kamata H, Yagisawa H, Hirata H. Overexpression of the alanine carrier protein gene from thermophilic bacterium PS3 in Escherichia coli. J Biochem. 1999;125:454–459. doi: 10.1093/oxfordjournals.jbchem.a022308. [DOI] [PubMed] [Google Scholar]
- 67.White JF, Trinh LB, Shiloach J, Grisshammer R. Automated large-scale purification of a G protein-coupled receptor for neurotensin. FEBS Lett. 2004;564:289–293. doi: 10.1016/S0014-5793(04)00195-4. [DOI] [PubMed] [Google Scholar]
- 68.Derewenda ZS. Rational protein crystallization by mutational surface engineering. Structure. 2004;12:529–535. doi: 10.1016/j.str.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 69.Derewenda ZS, Vekilov PG. Entropy and surface engineering in protein crystallization. Acta Crystallogr D Biol Crystallogr. 2006;62:116–124. doi: 10.1107/S0907444905035237. [DOI] [PubMed] [Google Scholar]
- 70.Cooper DR, Boczek T, Grelewska K, Pinkowska M, Sikorska M, Zawadzki M, Derewenda Z. Protein crystallization by surface entropy reduction: optimization of the SER strategy. Acta Crystallogr D Biol Crystallogr. 2007;63:636–645. doi: 10.1107/S0907444907010931. [DOI] [PubMed] [Google Scholar]
- 71.Vekilov PG, Feeling-Taylor AR, Yau ST, Petsev D. Solvent entropy contribution to the free energy of protein crystallization. Acta Crystallogr D Biol Crystallogr. 2002;58:1611–1616. doi: 10.1107/s0907444902014312. [DOI] [PubMed] [Google Scholar]
- 72.Vekilov PG. Solvent entropy effects in the formation of protein solid phases. Methods Enzymol. 2003;368:84–105. doi: 10.1016/S0076-6879(03)68006-7. [DOI] [PubMed] [Google Scholar]
- 73.Avbelj F, Fele L. Role of main-chain electrostatics, hydrophobic effect and side-chain conformational entropy in determining the secondary structure of proteins. J Mol Biol. 1998;279:665–684. doi: 10.1006/jmbi.1998.1792. [DOI] [PubMed] [Google Scholar]
- 74.Mateja A, Devedjiev Y, Krowarsch D, Longenecker K, Dauter Z, Otlewski J, Derewenda ZS. The impact of Glu-->Ala and Glu-->Asp mutations on the crystallization properties of RhoGDI: the structure of RhoGDI at 1.3 A resolution. Acta Crystallogr D Biol Crystallogr. 2002;58:1983–1991. doi: 10.1107/s090744490201394x. [DOI] [PubMed] [Google Scholar]
- 75.Kraulis P. MOLSCRIPT: a program to produce both detailed and schematic plots of proteins. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 76.Spurlino JC, Lu GY, Quiocho FA. The 2.3-A resolution structure of the maltose- or maltodextrin-binding protein, a primary receptor of bacterial active transport and chemotaxis. J Biol Chem. 1991;266:5202–5219. doi: 10.2210/pdb1mbp/pdb. [DOI] [PubMed] [Google Scholar]
- 77.Sharff AJ, Rodseth LE, Spurlino JC, Quiocho FA. Crystallographic evidence of a large ligand-induced hinge-twist motion between the two domains of the maltodextrin binding protein involved in active transport and chemotaxis. Biochemistry. 1992;31:10657–10663. doi: 10.1021/bi00159a003. [DOI] [PubMed] [Google Scholar]
- 78.Terwilliger TC, Grosse-Kunstleve RW, Afonine PV, Moriarty NW, Zwart PH, Hung LW, Read RJ, Adams PD. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild Wizard. Acta Crystallogr D Biol Crystallogr. 2008;64:61–69. doi: 10.1107/S090744490705024X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Adams PD, Grosse-Kunstleve RW, Hung LW, Loerger TR, McCoy AJ, Moriarty NW, Read RJ, Saccjettomo JC, Sauter NK, Terwilliger TC. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 80.Bishop JR, Schuksz M, Esko JD. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature. 2007;446:1030–1037. doi: 10.1038/nature05817. [DOI] [PubMed] [Google Scholar]
- 81.Rong J, Habuchi H, Kimata K, Lindahl U, Kusche-Gullberg M. Substrate specificity of the heparan sulfate hexuronic acid 2-O-sulfotransferase. Biochemistry. 2001;40:5548–5555. doi: 10.1021/bi002926p. [DOI] [PubMed] [Google Scholar]
- 82.Kreuger J, Salmivirta M, Sturiale L, Gimenez-Gallego G, Lindahl U. Sequence analysis of heparan sulfate epitopes with graded affinities for fibroblast growth factors 1 and 2. J Biol Chem. 2001;276:30744–30752. doi: 10.1074/jbc.M102628200. [DOI] [PubMed] [Google Scholar]
- 83.Chayen NE. Comparative studies of protein crystallization by vapour-diffusion and microbatch techniques. Acta Crystallogr D Biol Crystallogr. 1998;54:8–15. doi: 10.1107/s0907444997005374. [DOI] [PubMed] [Google Scholar]
- 84.Vagin ATA. MOLREP: An automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 85.Kakuta Y, Pedersen LG, Carter CW, Negishi M, Pedersen LC. Crystal structure of estrogen sulphotransferase. Nat Struct Biol. 1997;4:904–908. doi: 10.1038/nsb1197-904. [DOI] [PubMed] [Google Scholar]
- 86.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography and NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 87.DeLano WL. The PyMOL molecular graphics system. San Carlos, CA: DeLano Scientific; 2002. [Google Scholar]
- 88.Pinhal MA, Smith B, Olson S, Aikawa J, Kimata K, Esko JD. Enzyme interactions in heparan sulfate biosynthesis: uronosyl 5-epimerase and 2-O-sulfotransferase interact in vivo. Proc Natl Acad Sci USA. 2001;98:12984–12989. doi: 10.1073/pnas.241175798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McCahill A, Warwicker J, Bolger GB, Houslay MD, Yarwood SJ. The RACK1 scaffold protea dynamic cog in cell response mechanisms. Mol Pharmacol. 2002;62:1261–1273. doi: 10.1124/mol.62.6.1261. [DOI] [PubMed] [Google Scholar]
- 90.Nilsson J, Sengupta J, Frank J, Nissen P. Regulation of eukaryotic translation by the RACK1 protea platform for signalling molecules on the ribosome. EMBO Rep. 2004;5:1137–1141. doi: 10.1038/sj.embor.7400291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chapman MD, Pomes A, Breiteneder H, Ferreira F. Nomenclature and structural biology of allergens. J Allergy Clin Immunol. 2007;119:414–420. doi: 10.1016/j.jaci.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 92.Thomas WR. Mite allergens groups I-VII. A catalogue of enzymes. Clin Exp Allergy. 1993;23:350–353. doi: 10.1111/j.1365-2222.1993.tb00338.x. [DOI] [PubMed] [Google Scholar]
- 93.Shen HD, Chua KY, Lin KL, Hsieh KH, Thomas WR. Molecular cloning of a house dust mite allergen with common antibody binding specificities with multiple components in mite extracts. Clin Exp Allergy. 1993;23:934–940. doi: 10.1111/j.1365-2222.1993.tb00278.x. [DOI] [PubMed] [Google Scholar]
- 94.Shen HD, Chua KY, Lin WL, Chen HL, Hsieh KH, Thomas WR. IgE and monoclonal antibody binding by the mite allergen Der p 7. Clin Exp Allergy. 1996;26:308–315. [PubMed] [Google Scholar]
- 95.Jones DT. GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol. 1999;287:797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- 96.Kleiger G, Beamer LJ, Grothe R, Mallick P, Eisenberg D. The 1.7 A crystal structure of BPI: a study of how two dissimilar amino acid sequences can adopt the same fold. J Mol Biol. 2000;299:1019–1034. doi: 10.1006/jmbi.2000.3805. [DOI] [PubMed] [Google Scholar]
- 97.Hamiaux C, Stanley D, Greenwood DR, Baker EN, Newcomb RD. Crystal structure of Epiphyas postvittana takeout 1 with bound ubiquinone supports a role as ligand carriers for takeout proteins in insects. J Biol Chem. 2009;284:3496–3503. doi: 10.1074/jbc.M807467200. [DOI] [PubMed] [Google Scholar]
- 98.Trompette A, Divanovic S, Visintin A, Blanchard C, Hegde RS, Madan R, Thorne PS, Wills-Karp M, Gioannini TL, Weiss JP, Karp CL. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature. 2009;457:585–588. doi: 10.1038/nature07548. [DOI] [PMC free article] [PubMed] [Google Scholar]