

Abstract
Modeling of protein binding site flexibility in molecular docking is still a challenging problem due to the large conformational space that needs sampling. Here, we propose a flexible receptor docking scheme: A dihedral restrained replica exchange molecular dynamics (REMD), where we incorporate the normal modes obtained by the Elastic Network Model (ENM) as dihedral restraints to speed up the search towards correct binding site conformations. To our knowledge, this is the first approach that uses ENM modes to bias REMD simulations towards binding induced fluctuations in docking studies. In our docking scheme, we first obtain the deformed structures of the unbound protein as initial conformations by moving along the binding fluctuation mode, and perform REMD using the ENM modes as dihedral restraints. Then, we generate an ensemble of multiple receptor conformations (MRCs) by clustering the lowest replica trajectory. Using RosettaLigand, we dock ligands to the clustered conformations to predict the binding pose and affinity. We apply this method to postsynaptic density-95/Dlg/ZO-1 (PDZ) domains; whose dynamics govern their binding specificity. Our approach produces the lowest energy bound complexes with an average ligand root mean square deviation of 0.36 Å. We further test our method on (i) homologs and (ii) mutant structures of PDZ where mutations alter the binding selectivity. In both cases, our approach succeeds to predict the correct pose and the affinity of binding peptides. Overall, with this approach, we generate an ensemble of MRCs that leads to predict the binding poses and specificities of a protein complex accurately.
Keywords: protein flexibility, PDZ domain, docking, selectivity, RosettaLigand
Introduction
Molecular docking is an important tool in studying protein-ligand or protein–protein interactions and designing new drugs. Computer methods have long been useful for “docking” due to the discovery of ligands that can bind to proteins as lead candidates for drugs.1–6 Majority of the current binding/docking methods attempt to predict the bound ligand by keeping the protein (receptor) fixed and moving the target ligand around the binding site while performing an energy minimization. The major problems of this approach are: (i) proteins are dynamic, flexible, and deformable, so keeping them fixed often misses the correct binding modes, (ii) relying on pure energy minimization is insufficient to predict correct binding affinities. Docking algorithms predict the incorrect binding pose for about 50–70% of all ligands when the receptor is kept in single position.7 Overall, induced flexibility is the key ingredient to understand the physical principles of molecular recognition between ligand and receptor. It has been shown that even small changes in receptor upon binding can be important in computing the binding affinities. On the other hand, modeling of receptor flexibility is still a challenging problem due to the need of large conformational space that must be sampled. In order to overcome this challenge, multiple receptor conformations (MRCs) are generated in several ways: (i) using multiple conformations from molecular dynamics (MD) snapshots,8–13 (ii) applying principal component analysis to MD trajectories,14 (iii) using the snapshots based on geometry-based simulation techniques,15 (iv) detecting rigid and hinge regions with the Gaussian Network Model and the Elastic Network Model (ENM), respectively,16,17 (v) perturbing receptor conformations along different normal modes directions,18–21 (vi) using normal modes as additional flexible variables during docking simulations,22–25 and (vii) using multiple structures of the protein receptor obtained from experimental studies, for example X-ray crystallography or NMR analysis.26–28 The majority of these types of approaches are very computationally intensive. Furthermore, the success of these types of ensemble docking approaches depends on two features of the set of receptor conformations: (i) they should include a wide range of binding site conformations realized in nature and (ii) they should exclude artifact conformations that can lead to the prediction of incorrect poses or false positives in virtual screening (i.e., prediction of a ligand with a high binding score when it does not bind in nature). Thus, the crucial ingredient in generating MRCs is to mimic nature and sample binding induced conformations using smart sampling strategies. Recently, Abagyan and collaborators29 were able to incorporate the multiple receptor conformational ensemble in a single docking simulation and reduce the sampling time with their approach.
In this present study, we come up with a novel sampling strategy by combining the ENM with replica exchange molecular dynamics (REMD)30 to generate “binding induced” MRCs. Clustering molecular dynamics snapshots8–13 and generating conformations by perturbing the conformation along normal modes18,19,24,31–33 have been separately applied to incorporate the receptor flexibility in many docking studies. Here, by merging these two approaches, our goal is to bias MD search towards more binding-related conformations through normal mode predicted fluctuation profiles, which in return, to increase the efficiency and accuracy in generating the multiple receptor ensemble.
Although molecular dynamics is the most reliable method to sample conformations within the flexibility of protein structure, it is quite challenging to sample the rare events of large amplitude fluctuations (i.e., binding induced conformational changes) especially for the large protein molecules. On the other hand, the ENM34–41 which is based on a purely mechanical model, and view a protein structure as an elastic network, have been applied to many proteins to obtain slowest (i.e., functionally related) fluctuations. The nodes of the elastic network are α-carbons where identical springs connect the “interacting” α-carbons in their native fold. The model has the advantages that (i) it determines the functionally related motions (even large ones) without using any energy functions and (ii) it is computationally much more efficient than molecular dynamics. Most importantly, it has been shown that a few lowest frequency modes (or their linear combinations) of unbound conformations obtained by ENMs can capture the conformational change upon binding.42–44 Recently, Dobbins et al.19 applied ENM to a set of proteins from docking benchmark and found that the modes with certain characteristic frequencies can provide guidelines to predict the conformational change on protein–protein docking.
With the insights from these studies, we incorporate the modes that are related to the binding induced conformational changes into the REMD sampling search. Thus, in our approach, we use a restrained REMD search where we bias the search toward modal directions by using dihedral restraints. Our approach has the following advantages over using straightforward molecular dynamics runs: (i) REMD can help to overcome potential barriers with high temperature replicas, (ii) with biasing towards normal modes, binding induced conformational changes (i.e., similar to bound conformations) can be sampled much more efficiently than the straightforward sampling strategies, and (iii) it is computationally faster, as it is well suited for parallel computing. Moreover, our approach has another benefit over generating multiple flexible receptor conformations using only normal modes (i.e., generating conformations by perturbation of normal modes): coupling REMD with normal mode analysis (NMA) enables us to mimic a wide range of binding site conformations realized in nature. In addition normal mode by itself cannot really sample native-like bound conformations, it just gives us a crude estimate. Thus coupling REMD with normal modes can help us to predict binding affinities much more accurately.
We test our approach in analyzing the binding selectivity of postsynaptic density-95/Dlg/ZO-1 (PDZ) domain proteins (PDZs). PDZs, which are distributed diversely in the genome, play critical roles in (i) targeting proteins to specific membrane compartments, (ii) assembling proteins into supramolecular complexes, and (iii) regulating the function of their ligands in cellular signaling pathways. This ties them directly to the most puzzling diseases such as Alzheimer's, Parkinson's, cancer, and diabetes. PDZs perform their job by binding the C-terminal peptide of specific protein partners. PDZ domains have been categorized into two main classes according to the specificity of the interaction depending on its C-terminal four amino acids of their binding ligands.45 Class I type PDZs bind to a C-terminal motif with the sequence [Ser/Thr-X-φ-COOH] and Class II type PDZs prefer the sequence [φ-X-φ-COOH] where X is any amino acid and φ is any hydrophobic amino acid. All PDZs share about 25% identity in the sequence, similar secondary and tertiary structures with an average backbone root mean square deviation (RMSD) of around 1.4 Å and highly conserved C-terminal peptide interactions.46 Although PDZ binding site is well defined and PDZ motifs are classified based on their sequence type, there is still little information available on the binding affinity and stoichiometry of PDZ binding motifs and blocking peptides. This is partly because their dynamics rather than the structure or sequence determines the binding affinity as indicated with very recent study of Petit et al.47 In their study, they investigated how binding affinity alters, when they remove distal the third helix (α3) of third PDZ domain of PSD-95 which is not observed in any other PDZs and is not necessary to maintain the structure. Strikingly, removal of α3 reduces the binding affinity by 21-fold, even though it lies outside of the binding site and does not make direct contact with the binding C-terminal peptide. This result along with many others supports the notion that different PDZs evolve to have different dynamics properties tailored to mediate different functions in the cell, despite the fact that they all have same conserved structure and sequence.48
By analyzing the conformational dynamics of the unbound PDZs, it may be possible to determine their binding specificities. In our previous study, we showed that the most collective fluctuation profile obtained by modified version of ENM can capture on the average 60% of the binding induced conformational changes in PDZ domains.49 We also investigated the role of binding induced conformational dynamics of PDZs in their peptide selectivity and classification. By clustering the binding induced fluctuation profiles of a diverse set of PDZ domains, we showed that ENM predicted normal modes not only can identify the structural regions and types of correlated fluctuations critical for binding of Class I and Class II peptides but also predicts binding selectivity of PDZs.49
The challenge of PDZ domain (i.e., the reasons behind their selectivity and promiscuity) and their link to many different diseases has led to a number of important experimental50,51 and computational49,52–58 studies. Niv and Weinstein also developed a flexible docking scheme (called PDZDocScheme),56 which is based on simulated annealing molecular dynamics with the soft core potential or flexible binding site side chains, followed by rotamer optimization. When they apply their protocol to the original bound structures (self-docking), their scheme reproduces the structures of PDZ complexes with peptides 4–8 amino acids long within 2 Å, except for Syntenin (the RMSD of the best score is 3.7 Å when they redock Class II peptide to native bound structure). However, when unbound or homology models are used for docking, their flexible docking scheme can only predict the docked peptides within an average RMSD of 3 Å. Here, we would like to apply our flexible approach based on ENM-guided REMD to improve upon the PDZ-binding affinities, especially for the cases where only unbound and homology structures are available.
In our flexible docking procedure, we first generate an initial set of conformations by perturbing the unbound structure and homology models using the weighted average profile of significant lowest frequency ENM modes responsible for Class I and Class II types binding in PDZs.49 Then, we further sample these two sets of structures by running a dihedral restrained REMD where the restraints are set with respect to the binding induced fluctuation profile of ENM. After restrained REMD, the snapshots of the lowest replica are clustered and each individual structure from this ensemble of conformations is docked against different peptides using RosettaLigand.59,60 The overall average ligand RMSD values of redocking native peptides to native bound structures and unbound structures are 0.31 Å and 0.78 Å, respectively. However, when the native peptides are docked to unbound structures with our flexible approach, we obtain lowest energy models with an average ligand RMSD of 0.36 Å from the experimental structure, which is as good as redocking peptides to bound structures. Although docking methods provide low RMSD values for the docked poses of the ligands, they are generally unreliable for prediction of binding affinities. There are different approaches to overcome this deficiency using empirical scoring functions (DrugScore61 and XScore62) and more physically realistic methods such as molecular mechanics-generalized Born (GB) surface area (SA) technique.63 While RosettaLigand is successful in obtaining good RMSD values for best poses, we prefer using a knowledge-based scoring function (DrugScore)61 to estimate the binding affinities of ligands to a particular receptor with reasonable accuracy. Thus, at the end of Rosetta run, we re-evaluate the binding energy of the ligand bound complex structure with the lowest Rosetta binding score. Our results indicate that the binding preference of different peptides in PDZs can be determined using the docking approach based on ENM-guided REMD. This would enable us to validate its applicability to include protein flexibility in docking studies. In addition, we further apply our method on (i) homolog structures of PDZs whose binding selectivities are verified experimentally and (ii) mutant structures of PDZ where mutations alter the binding selectivity. We find that the inclusion of backbone flexibility through MRCs, obtained by the normal mode incorporated REMD runs, help us to discriminate the binding preference for homology models and mutated structures, which are much more challenging cases for docking.
Materials and Methods
Benchmark
The four PDZs (PSD-95, GRIP, Syntenin, and Erbin) with available unbound (apo) and bound (holo) structures analyzed in this study are listed in Table I. The crystal structures of unbound and bound proteins are retrieved from the PDB (http://www.rcsb.org).64 The PDB codes, the names of the corresponding proteins, the class of PDZ domain according to their binding specificity and the sequences of the binding peptides are displayed in the columns of the table.
Table I.
The PDB Codes, the Peptide Sequences, and the Binding Preferences of Proteins
| PDB code |
Peptide |
||||
|---|---|---|---|---|---|
| Protein | Unbound (apo) | Bound (holo) | Name | Class | Sequence |
| PSD-95 | 1BFE | 1BE9 | CRIPT | I | KQTSV |
| GRIP | 1N7E | 1N7F | Liprin | II | ATVRTYSC |
| Syntenin | 1NTE | 1OBX | Interleukin 5 receptor alpha peptide | I | DSVF |
| 1OBY | Syndecan-4 | II | NEFYA | ||
| Erbin | 2H3L | 1N7T | A phage-derived peptide | I | TGWETWV |
| 1MFG | ErbB2 receptor | II | EYLGLDVPV | ||
The ENM-guided REMD method for docking
With our docking scheme, called the ENM-guided REMD method (ENM-REMD), our goal is to generate an ensemble of a given receptor conformation which can mimic the nature and includes the conformations that are sampled through the binding process. Thus, we generate a set of conformations from molecular dynamics trajectories where we bias the search toward binding induced dynamics. We achieve this in three steps: first using ENM, we obtain the binding induced fluctuation profile of a protein by analyzing the slowest modes of unbound structure. Then, we obtain the deformed structures of the unbound structure by perturbing the unbound structure along the binding fluctuation modes of ENM (see details in subsection “Elastic Network Model and generating new conformations using ENM”). The perturbed/deformed structures are then used as initial structures in our restrained REMD simulation where we define dihedral restraints based on the ENM predicted binding induced fluctuation profile (i.e., the regions predicted as rigid/less flexible by ENM mode are assigned with stronger restraints whereas the regions with high flexibility are assigned with weaker restraints). After defining the restraints and creating the perturbed structures as initial structures based on ENM predict binding induced fluctuation profile, as a second step, we perform a dihedral restrained REMD simulation (see the details of the simulation parameter in the subsection “Dihedral restrained replica exchange molecular dynamics”). Having our simulation restrained with respect to binding induced fluctuation profiles helps us to sample towards correct bound-like conformations. Once the simulation is done, an ensemble of MRCs is generated by clustering the conformations sampled at the lowest replica. As a final step, each structure in the ensemble is used for docking and the best energy score docked pose is chosen as the best result. Figure 1(A) displays each step in our flexible docking protocol and the docking performance of the conformations obtained after the each step is also presented in Figure 1(B) for the case of PSD-95. PSD-95 only binds to Class I type of peptides. Shown in Figure 1(B), as we further proceed the steps, the Class I type binding affinity becomes more significant while the RMSD value of the Class I peptide decreases.
Figure 1.

(A) The procedure of the docking protocol. The deformed structures of the unbound structure are obtained moving along the mode with ENM. Then, a large set of conformations starting from these structures is generated using the dihedral restrained REMD. RosettaLigand is used for self- and cross-docking runs (see “Materials and Methods” for details). (B) As case study of PSD95, we tested the docking performance of the conformations obtained at each step shown in (A) by simply docking Class I and Class I peptides. The RMSD values of the peptides and binding energy values are given. Overall, the docking performance gets better as we further follow the steps. [Color figure can be viewed in the online issue, which is available at http://www.interscience.wiley.com.]
We carry out four different tests: First, each ligand is redocked into a known PDZ domain of the bound conformation (self-docking). Second, different ligands are docked into the original bound conformation (cross-docking). Third, the docking procedure applied to bound structures is also repeated for the unbound structures. Fourth, ligands are docked against the multiple conformations obtained with our flexible docking protocol (ENM-REMD). For further testing, we apply the ENM-REMD method to two protein structures (Lrcc7 and Cipp-PDZ9) generated from homology modeling and mutants of another PDZ domain, PICK1 that have dual (Class I and Class II types) binding specificity. All docking analysis in this study is performed using RosettaLigand where the ligand flexibility is established by changing torsional angles, and the backbone of the ligand and the whole protein are held fixed throughout the docking simulation.
Elastic Network Model and generating new conformations using ENM
The normal modes that correspond to Class I- and Class II-type peptide binding fluctuations are obtained using the Anisotropic Network Model (also referred to as the ENM).34 ENM is equivalent to a NMA with an ENM at the Cα level and the Hessian is based on a harmonic potential form.41 ENM uses the coordinates of a structure from PDB database;64 with the adjacent residues connected based on a cut-off distance with a spring to build a Hessian connectivity matrix. A cutoff value of 16 Å is used in this study.
The diagonalized of the inverse of Hessian yields it into 3N–6 eigenvalues (λn) and their corresponding eigenvectors un, and we take the lowest frequency (most global) eigenmodes. To find the binding induced fluctuations, we focus on the contribution of each mode, weighted by the inverse of corresponding eigenvalue. The distribution of mode frequencies (eigenvalues) are evaluated to address how many modes contribute to the binding dynamics. A subset of modes whose eigenvalues are dispersed from those of the other modes is identified.49 In this approach, first, the histogram of the eigenvalues is generated and then the bin size is computed based on the highest dispersion in eigenvalue spectrum (i.e., the first gap in the eigenvalue spectrum). The eigenvalues correspond to the first bin are used along with their eigenmodes to compute the weighted sum of the square fluctuations obtained from
| (1) |
The weighted average of each mode corresponds to a fluctuation between two oppositely directed motions. We generate two sets of deformations for each mode k as
| (2) |
where t is a scaling parameter of the deformation43 and we used t = 25 for the proteins tested in this study. Each obtained structure that contains distorted bond lengths and angles is further subjected to an energy minimization of 50 steepest descent iterations followed by 1000 conjugate gradient iterations using the AMBER96 forcefield65 along with a GBSA solvation model.66
It is also possible to use a set of experimental structures in different liganded complexes (if they are available) to compute the binding induced dynamics.67–70 The recent study of Bakan and Bahar has shown that ENM predicted modes are very well correlated with the principal components of the conformational change for a large set of different liganded experimental structures of a given protein.71 We have repeated the same analysis for our test protein: PDZ domain. Thus, we collect the different complex structure of PSD-95 available in the Protein Data Bank. After structurally aligning these experimental structures, we compute the mean position of each Cα atom (〈ΔRi〉) and construct the covariance matrix based on the Cα deviations of protein structures. Then, we obtain the principal components of the structural change by decomposing this covariance matrix. The comparison of principal modes with ENM modes of our calculations is also highly correlated (see Supporting Information Fig. S1). This indicates that ENM predicted normal modes of unliganded (unbound) structure intrinsically captures the dynamics that lead to the liganded (bounded) conformation, therefore they can be used in docking to guide the unbound structure towards binding induced dynamics.
Homology modeling and mutated proteins
The protein sequences are obtained from the SMART (a Simple Modular Architecture Research Tool) database72 and submitted to “The Structure Prediction Meta Server”73,74 with the option of an automated homology model program ESyPref3D. ESyPref3D is based on a strategy using neural networks to evaluate sequence alignments75 and uses the MODELLER program76,77 to build the final structural model. Homology models are constructed with MODELLER with a minimal sequence similarity of 50% to the target. To obtain mutated structure of the PICK1 (PDZ protein interacting with C kinase 1), a computational point mutation is introduced into the X-ray structure via Swiss PDB Viewer.78 Before applying ENM, homolog and mutated structures are subjected to an energy minimization of 50 steepest descent iterations followed by 1000 conjugate gradient iterations using AMBER 96 force field65 along with a GB solvation model.66
Dihedral restrained replica exchange molecular dynamics
We apply a dihedral restrained REMD30 to generate a large set of conformations starting from deformed structures obtained with ENM. First, dihedral restraints are applied to move the conformations along its binding induced fluctuations. The strength of each dihedral restraint is adjusted with respect to the fluctuation profile of normal modes obtained by ENM, where the regions shown as the most flexible in ENM analysis have weaker restraints compared to rigid parts. This type of restraint biases our sampling towards the directionality of the binding induced fluctuations. Second, rather than using unbound conformation as initial conformation, we generate a set of conformations by perturbing the unbound conformations along the normal mode vectors as explained in “Elastic Network Model and generating new conformations using ENM” section of “Materials and Methods”. The advantage of using different initial conformations in REMD is that it speeds up our search towards correct binding site conformations.
The AMBER96 forcefield with a GB implicit solvent model66 and a SA penalty term of 5 cal/mol Å2 are used in REMD. Backbone torsional restraints that hold specific phi/psi angles in the same conformation are applied to all replicas. We simulate each protein with 26 replicas for 5 ns/replica ranging from temperatures 270–450 K. Each structure is subjected to an energy minimization of 50 steepest descent iterations followed by 1000 conjugate gradient iterations prior to dynamics. The number of replicas is arranged according to the acceptance ratio of 48% conformational swap change between replicas. At the end of restrained REMD runs, trajectory of the lowest replica are clustered to ˜1 Å RMSD by using a modified k-means algorithm. Then, docking into available ligands is carried out on each member of the clustered ensembles.
Docking with RosettaLigand
We minimize each clustered conformation with a short equilibration MD so that they can relieve some of the strain of the system. Then, the individual structures from the ensemble of PDZ domain conformations obtained with REMD is docked into several C-terminal peptides using RosettaLigand59,60 protocol in the ROSETTA package. RosettaLigand is a method specifically developed for docking ligands into protein binding sites. The method uses a Monte Carlo minimization protocol to optimize the rigid body position and orientation of the ligand and the protein side chain conformations. The energy function includes van der Waals interactions, an implicit solvation model, an electrostatics model, an explicit orientation hydrogen bonding potential, and an empirically derived torsional potentials. The RosettaLigand protocol we apply is substantially the same as described in the study of Meiler and Baker.60 The coordinates of the peptides are taken from the crystallographic complexes and they are treated as a single residue. The peptide flexibility is introduced by changing torsional angles and the backbone is held fixed throughout the docking simulation. In this study, we perturb the ligand position and orientation randomly with the translation of mean 0.1 Å and the rotations of mean 3o, respectively. We compute 10,000 trajectories to generate a comprehensive ensemble of conformations of the receptor-ligand complex for each peptide. The formation of a distinct binding funnel in binding energy/RMSD plots is an indication of successful docking and the final docked conformations are selected based on the lowest free energy pose in the protein-binding site.
Assessing the scoring accuracy with DrugScore
After selecting the pose corresponding to the lowest free energy of binding at the end of Rosetta docking, we reassess the binding energy score of this complex using DrugScore.61 Thus, we submit the receptor part of the docked pose in PDB format and the ligand coordinates in mol2 format into DrugScore online (http://www.agklebe.de). DrugScore, a knowledge-based scoring function for protein-ligand interactions, employs statistically derived pair potentials using the distance-dependent occurrence frequencies by which a particular ligand atom type is found in contact with a protein atom type. Wang and Wang79 studied the accuracy of different knowledge-based scoring functions for docking of 100 complexes. They showed that only four, X-Score, DrugScore, PLP (piece-wise linear potential), and G-Score among 11 various postdocking ligand-protein scoring functions give moderate correlations with the experimentally determined protein-ligand binding affinities. Gohlke et al.61 and Velec et al.80 have also reported that DrugScore shows better ranking than the scoring functions within the docking programs. Furthermore, a recent study Dokholyan and colleagues81 showed that the success rate of their docking results was improved to 85% by consensus scoring with DrugScore for a docking decoy set consisting of 100 complexes. Likewise, our binding affinities got improved by rescoring the lowest energy pose by DrugScore. Higher negative values indicate a higher binding affinity prediction. With DrugScore, the binding selectivity preferences for Class I and Class II peptides become more significant.
Results and Discussion
The proposed methodology is applied to the PDZ structures in Table I. First, we check the performance of RosettaLigand by self-docking and cross-docking tests for experimentally known bound structures. Docking becomes more challenging when the experimental structures of bound complexes are not available, especially when homology models need to be used. For those cases, the success of docking depends whether the right fluctuation profiles toward bound-like conformations can be incorporated accurately in docking computations. To test this, we generate MRCs of the unbound structure with the ENM-guided dihedral restraint REMD (ENM-REMD) method described in “Materials and Methods” and shown schematically in Figure 1(A). In all tables, we present RMSD values with respect to the heavy atoms between the ligand position of the lowest energy docking pose (i.e., the first rank based on energy score) and that of the crystal structure. This provides a measurement for the accuracy of any given docking attempt, as most of the docking approaches; the ligand is being kept flexible, while keeping the receptor rigid. Strikingly, in most of the docking tests, we observe that the lowest energy score obtained with our ENM-REMD method also has the lowest peptide RMSD value indicating the success of our approach in sampling the correct peptide poses.
Self-docking and cross-docking of bound complex structures
As the ultimate purpose of protein-ligand docking studies is to reproduce the bound complex structures from the unbound conformations, it is important to check the performance of docking protocol on the experimentally known bound structures as a first test. Thus, we perform docking using bound conformations while the peptide and the backbone of bound structure are kept rigid. The peptide RMSD values of the poses with the best score for each PDZ are summarized in Table II. The average RMSD of the self-docking tests is 0.62 Å. Note that overall RMSDs of up to 2 Å are generally accepted as near native in docking.82 We also check the docking scores of each bound structure with different peptides. We find that the average RMSD values of the cross-docking models (0.98 Å) are slightly higher than that of the self-docking tests (0.62 Å). Overall, we obtain quite low RMSD values with RosettaLigand in case of docking “bound” structures. Furthermore, the low energy scores of the lowest RMSD structures obtained with self-docking prove the binding preference of each PDZs. It has already been shown that RosettaLigand, which includes side chain conformations and ligand flexibility, is a successful approach providing an accurate prediction compared to other existing docking methods in self-docking experiments60 and the results of our self-docking and cross-docking of bound structures also agree with these findings.
Table II.
Docking Rigid Ligand Conformations to their Native (Bound) Structures (Self-Docking and Cross-Docking)
| Peptide |
RosettaLigand |
|||||
|---|---|---|---|---|---|---|
| Protein | PDB code | Class | Sequence | RMSD (Å)a | Ebind (kcal/mol) | DRUGSCORECSDEbind (kcal/mol)b |
| PSD-95 | 1BE9 | I | KQTSV | 0.70 | −13.21 | −195.32 |
| II | EYLGLDVPV | 2.07 | −14.26 | −213.24 | ||
| GRIP | 1N7F | I | KQTSV | 0.90 | −15.33 | −153.86 |
| II | ATVRTYSC | 0.90 | −21.96 | −256.95 | ||
| Syntenin | 1OBX | I | DSVF | 0.37 | −15.83 | −183.48 |
| II | NEFYA | 0.60 | −17.73 | −187.15 | ||
| 1OBY | I | DSVF | 0.80 | −14.32 | −184.83 | |
| II | NEFYA | 0.56 | −22.44 | −269.06 | ||
| Erbin | 1N7T | I | TGWETWV | 0.83 | −16.50 | −232.20 |
| II | EYLGLDVPV | 2.0 | −11.56 | −177.19 | ||
| 1MFG | I | TGWETWV | 0.67 | −15.07 | −196.94 | |
| II | EYLGLDVPV | 0.35 | −21.71 | −308.70 | ||
RMSD values between the top scoring pose in Ångstroms measured over all heavy atoms of the peptide and the peptide's position in the crystal structure.
Calculated with DrugScore online (http://pc1664.pharmazie.uni-marburg.de/drugscore/). Higher negative values indicate a higher-affinity prediction.
Evaluation of results of docking jobs based on ENM-guided dihedral restrained REMD snapshots
For all PDZs, we dock natural binding peptides along with different peptides using unbound structures in order to test if we can predict the binding selectivities of PDZs with our approach. First, we generate deformed conformations by perturbing the unbound conformation along binding-related fluctuation profiles using a few slowest modes of ENM. Different sets of slowest modes (i.e., the slowest modes that are highly coupled with binding related fluctuations) are used for each PDZ by carefully analyzing the eigenvalue spectrum as explained in method section. Thus, the weighted averages of five (1–5), three (1–3), five (1–5), and two (1–2) ENM modes are used for PSD-95, GRIP, Syntenin, and Erbin, respectively to generate the deformed conformations (see “Methods” for details). The overall average backbone RMSD between deformed ones and unbound conformations of PDZs based on the α-carbons is 0.84 Å, while average RMSD between deformed ones and bound conformations is only 1.06 Å. These structures are used as initial structures in the dihedral restrained REMD simulations. Finally, the lowest replica trajectories of the dihedral restrained REMD are clustered to generate a MRC set for the peptide docking. The lowest binding energy scores and corresponding peptide RMSDs are summarized in Table III for two different docking cases (i) using only unbound experimental structure for docking and (ii) ensemble of structures obtained by ENM-guided REMD for docking. The selected best-scoring poses (i.e., the poses correspond to the lowest binding energy) are highlighted with gray color. The successful docking runs are frequently accompanied with the formation of a distinct binding funnel in the binding energy/RMSD plots.60 Figure 2(A) shows the binding energy score versus RMSD of the docked complex for PSD-95. PSD-95 only binds to Class I peptides. Simply, using only unbound experimental structure of PSD95 for docking to Class I and Class II peptides does not yield the selectivity preference [brown and red dots in Fig. 2(A)]. However, when MRCs obtained by ENM-guided REMD are used for docking, we clearly see that our flexible scheme does a better job in indicating the binding selectivity of PSD-95 (blue and cyan dots in the plots). The best-scoring poses for unbound conformation and ENM-guided REMD snapshot obtained from our docking scheme are superimposed on the original Class I peptide positions. Illustrated in Figure 2(A), green represents the original peptide (i.e., from the complex crystal structure) while blue and brown represent the pose with the best score of the ENM-guided REMD snapshot and unbound conformation, respectively. We obtain an excellent agreement with the ENM-guided REMD snapshot.
Table III.
Docking of Peptides to Unbound Structures and ENM-REMD Snapshots
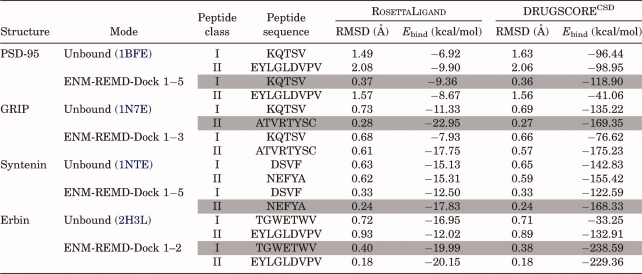 |
Figure 2.
The binding energy score versus RMSD of the docked complex for (A) PSD-95 and (B) GRIP. Docking Class I and Class II peptides to the unbound conformation of PSD-95 does not discriminate the selectivity preference [brown (Class I) and red (Class II) dots in the plots]. However, when ENM-REMD snapshots are used (i.e., when the backbone flexibility is also considered), our flexible scheme does a better job than simply docking to the unbound structure of PSD-95 [blue (Class I) and cyan (Class II) dots in the plots]. (C) The comparison of docking Class I peptide to the unbound structure (upper figure) and the ENM-REMD snapshot (lower figure) is shown as ribbon diagrams. Green represents the actual binding mode in both docking, whereas blue and brown ones are for the docked peptide conformations corresponding to the lowest binding energies of ENM-REMD snapshot and the unbound structure, respectively. (D) Ribbon diagrams for docking Class II type of peptide to the unbound conformation and ENM-REMD conformation by RosettaLigand. [Color figure can be viewed in the online issue, which is available at http://www.interscience.wiley.com.]
For the unbound GRIP docked to its original ligand (Class II), we obtain 0.29 Å as the RMSD of best score compared to that of 1.8 Å in PDZDocScheme of Niv and Weinstein.56 Surprisingly, the unbound structure of GRIP provides a good initial structure for docking. It might be expected that the prediction accuracy of the docking calculations decreases with the quality of the receptor from the bound (holo) protein to the unbound (apo) protein to the modeled structures.83 In the case of the unbound structure of GRIP, its conformation may be adequate to accommodate a ligand. Figure 2(B) displays the comparison of the binding energy score versus RMSD of the docked complex. With our docking scheme, we also find that its binding specificity is towards Class II type peptide. The difference of the binding energy score obtained from DrugScore between Class I and Class II peptides is 34.13 kcal/mol when the unbound structures are used for docking. However, the difference in docking to ENM-guided REMD snapshots is 98.61 kcal/mol, which is much more significant (see Table III).
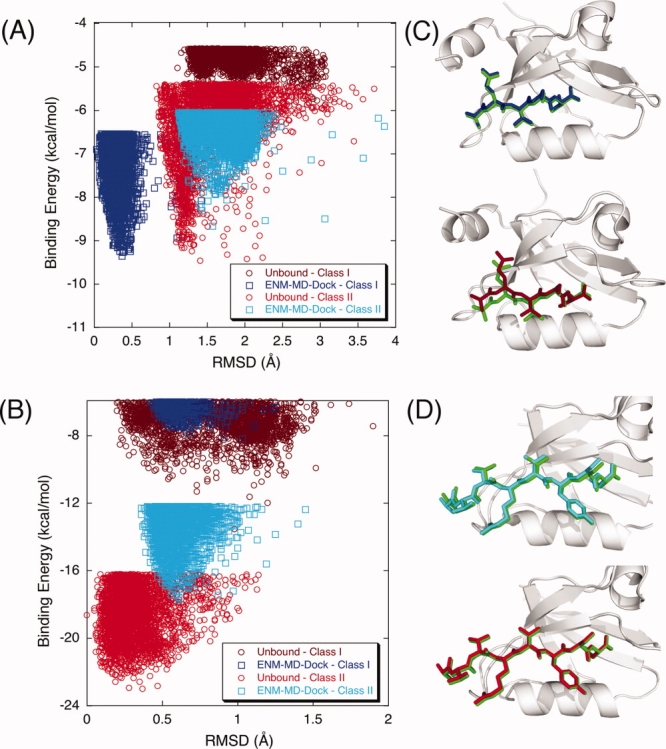
In Figures 3(A,B), we present the binding energy score versus RMSD of two PDZs (Syntenin and Erbin) which exhibit Class I and II dual specificities. First, flexible docking scheme can indicate the dual specificities of these two PDZs (i.e., the DrugScore values of lowest energy complexes obtained by RosettaLigand are very close as seen in Table III) whereas using only unbound structure in RosettaLigand fails to discriminate the dual specificity of Erbin. Moreover, using ENM-guided REMD snapshots as multiple receptor ensembles also provide the discrimination of binding affinities for Class I and Class II types of PDZs. Figure 3(A) shows that Syntenin binds Class II peptide with higher affinity than Class I peptide, which is consistent with experimental observation indicating that the PDZ2 domain of Syntenin binds slightly better to the Class II than to the Class I peptide84 (the binding energies of the poses are –168.33 kcal/mol and –122.59 kcal/mol for docking into Class II and Class I peptides, respectively). When we compare the binding energy values of each peptide for Erbin PDZ, Erbin binds Class I peptide with higher affinity than Class II peptide [Fig. 3(B)]. This agrees with the intrinsic binding affinities between the Erbin PDZ domain and peptides measured using ELISA. The affinity of the phage-selected peptide (TGWETWV) binding at a submicromolar level is higher than the affinities measured for the ErbB2 peptide (EYLGLDVPV).85,86 Overall, incorporating flexibility with ENM-guided REMD runs predicts binding selectivities of PDZ domains very accurately.
Figure 3.
The binding energy score versus RMSD of the docked complex for (A) Syntenin and (B) Erbin that have dual specificities. Docking Class I and Class II peptides to the unbound conformations of Syntenin and Erbin does not discriminate the selectivity preference [brown (Class I) and red (Class II) dots in the plots]. When ENM-REMD conformations are used, Syntenin binds slightly better to the Class II peptide (cyan dots) than to the Class I peptide (blue dots) whereas Erbin binds Class I peptide with higher affinity than the Class II peptide (blue and cyan dots in the plots). (C) The corresponding lowest energy structures of docking Class II peptide to the unbound structure (upper figure) and the ENM-REMD snapshot (lower figure) of Syntenin are displayed as ribbon diagrams along with actual peptide (green). (D) Docking Class I peptide to the best pose obtained for the unbound structure (upper figure) and the ENM-REMD snapshot (lower figure) of Erbin are presented. [Color figure can be viewed in the online issue, which is available at http://www.interscience.wiley.com.]
The advantage of docking procedure
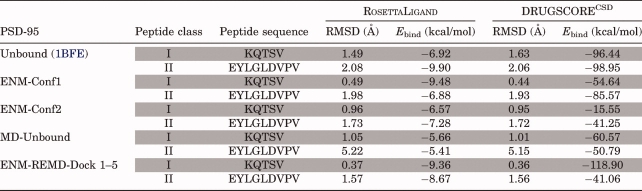
We believe the success of our method relies on sampling the bound-like conformations through performing an REMD simulation guided by the binding-related normal modes. To test the efficiency of our docking scheme, we compare our schemes with docking the peptides into (i) the unbound structure, (ii) conformations generated by perturbing the unbound conformation using the binding-related normal modes of ENM, and (iii) an ensemble of conformations obtained by the snapshot of the REMD simulation started from unbound experimental structure. Table IV presents the comparison of the docking results of PSD-95 with these different approaches. PSD-95 prefers Class I type of binding, therefore docking scores of Class I peptide should be lower than those of Class II peptide. As expected, docking Class I peptide to the unbound structure of PSD-95 provides the worst RMSD value (1.49 Å). Moreover, docking of Class II peptide to the unbound structure gives a lower energy score than that of Class I peptide. Docking studies using the conformations generated by the perturbation of ENM mode is computationally less expensive and provides good RMSD values (0.44 Å and 0.65 Å for two different deformed structures). However, the corresponding binding energy values obtained with DrugScore do not clearly indicate the binding affinity of PSD-95 (i.e., DrugScore binding energies are –54.64 kcal/mol for docking of Class I peptide and –85.57 kcal/mol for docking of Class II peptide). Thus, incorporating only ENM modes to docking does not differentiate the binding preference of PSD-95. Likewise, the clustered conformations of straightforward REMD simulation do not discriminate the binding preference and this approach also fails to capture the high-resolution low RMSD pose of PSD-95 complex. We get 1.01 Å and –60.57 kcal/mol by docking the snapshots of the REMD simulation starting from an unbound experimental structure. However, our docking approach successfully not only distinguishes the binding preference but also predicts the binding pose of receptor accurately. In our docking protocol, first, we generate new conformations by perturbing the unbound structure along the weighted averages of first five modes (1–5) of ENM, second, we use these conformations as initial structures for the restrained REMD simulations where the dihedral restraints are imposed with respect to average normal mode of ENM. Finally, clustered structures of the REMD simulation are docked into peptides. The pose obtained by our new protocol does not only provide the lowest binding energy (–118.90 kcal/mol) but also gives the best RMSD value of the receptor and significant binding energy differences between Class I and Class II peptides. These results indicate that REMD conformational search along the modes of ENM based on a dihedral restrained REMD improves the docking prediction accuracy by sampling native-like bound conformations. This observation is consistent with the other PDZs we tested as explained above.
Table IV.
Docking Ligands into Unbound and Structures Obtained with ENM, MD Only, and ENM-Guided REMD for PSD-95
 |
Flexible docking for homolog and mutant structures
In a more stringent test, we applied our flexible approach to homologs of PDZs whose binding selectivities are verified experimentally and also to PDZs whose binding specificities are altered upon mutation. Both Class I and Class II peptides were docked to homolog structures obtained using MODELLER. Then, we also obtained MRCs for homolog structures by our ENM-guided REMD approach using MODELLER prediction as a starting conformation. The lowest binding energy scores of the docked poses for both homolog structures and their corresponding RMSD values are presented in Table V. The docked pose of Lrcc7 which has Class I binding specificity has very low RMSD values (0.29 Å when homology model is used for docking, and 0.23 Å when MRCs of ENM-REMD approach are used for docking). Likewise, docking Class II peptide to Cipp-PDZ9, exhibiting Class II type binding affinity, yields a 1.12 Å RMSD docked pose when homology model is simply used for docking. On the other hand, docking Class II peptide to of Cipp-PDZ9 using our flexible approach gives consistently lower RMSD of 0.23 Å. As observed in earlier PDZ docking cases, the inclusion of backbone flexibility through ENM-REMD snapshots does a better job in discriminating that that Lrcc7 has affinity towards Class I peptide, and likewise Cipp-PDZ9 prefers Class II peptide (see Table V for DrugScore energy scores).
Table V.
Prediction for Homolog and Mutated PICK1 Structures
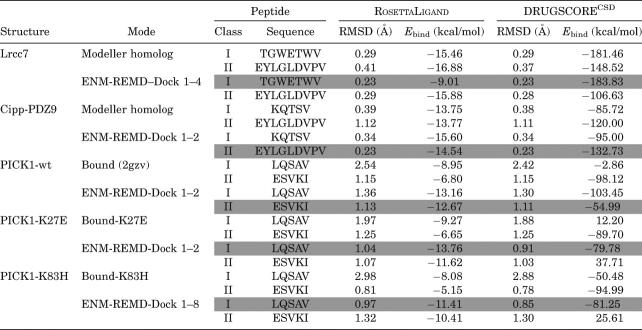 |
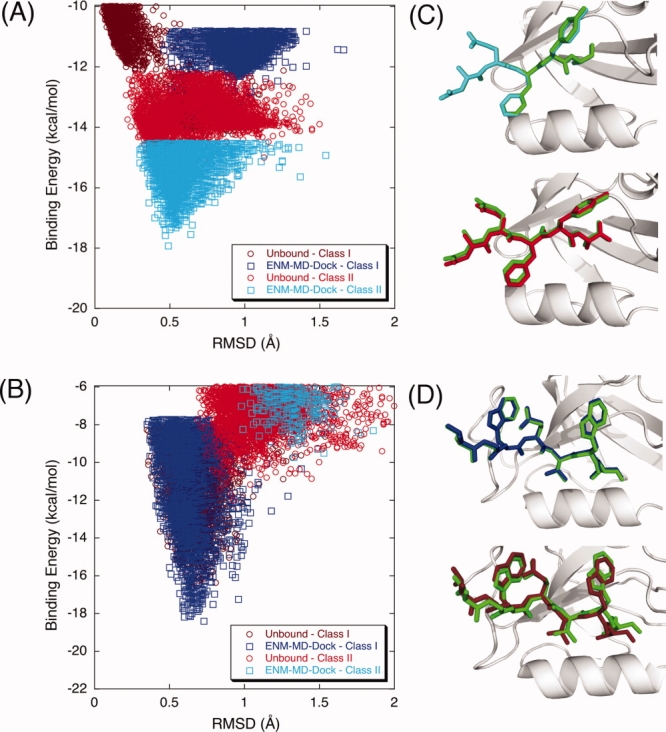
We further test our flexible docking protocol by examining the binding characteristics of wild-type PICK1 and PICK1 carrying various mutations. The wild type of PICK1 can bind both Class I (PKCα) and Class II (GluR2) peptides.87–89 Yet, there are several mutations that have been reported previously, such as the mutation of lysine 27 (K27E) to glutamic acid alone or together with aspartic acid 28 (D28A) to alanine, which completely disrupt the interaction with both GluR2 and PKCα.89 Moreover, the experimental study of Dev et al.87 has shown that mutating lysine 27 to glutamic acid, a point mutation on the βA–βB loop, changes the binding selectivity of PICK1 to exhibit only Class I behavior. Another mutation study that replaces the residue in αB helix (lysine 83 to histidine) by Madsen et al.88 showed that the preference of PICK1 reverts to that of a Class I motif. Docking Class I and Class II peptides to the unbound conformation of wild and mutant PICK1 does not show the change in selectivity upon mutation [brown and red dots in the Figure 4(A–C)]. However, when the ENM-guided REMD snapshot are used, the wild type has a higher affinity for Class II peptide [cyan on (A)] whereas both mutants prefer Class I peptide [blue on Fig. 4(B,C)]. From the binding energy score versus RMSD of the docked complex for PICK1, we can clearly see that wild-type PICK1 [Fig. 4(A)] prefers Class II type of peptide sequence while K25E mutation [Fig. 4(B)] and K83H [Fig. 4(C)] on PICK1 alters its binding specificity to Class I type of peptide sequences as observed experimentally.
Figure 4.

The binding energy score versus RMSD of the docked complex for PICK1. Wild-type PICK1 (A) prefers Class II type of peptide sequence while K25E mutation (B) and K83H mutation (C) on PICK1 alter its binding specificity to Class I type of peptide sequences. Docking Class I and Class II peptides to the unbound conformation of wild and mutant PICK1 does not show the change in selectivity upon mutation [brown (Class I) and red (Class II) dots in the plots]. However, when REMD-ENM conformations are used, wild type has a higher affinity for Class II type peptide binding [cyan on (A)] whereas both mutants prefer Class I type of peptide [blue on (B) and (C)]. The corresponding lowest energy structures of PDZ-peptide complexes are represented as ribbon diagrams along with experimental peptide position. [Color figure can be viewed in the online issue, which is available at http://www.interscience.wiley.com.]
Conclusions
We present a new flexible docking scheme which biases REMD search towards binding induced conformational changes by incorporating the fluctuation profiles obtained by slowest eigenvectors of ENM called ENM-guided REMD-DOCK method. We applied our method to a set of PDZs, which shows different affinities to different peptide types. Rigorous analysis has shown that generating MRCs using ENM-guided REMD can be a useful tool to predict binding poses and selectivities accurately. In our method, the backbone flexibility is introduced by perturbing the receptor structure along its relevant normal modes and the conformational space is explored with the help of a dihedral restrained REMD simulation, which enables us to sample the conformational space towards bound-like conformations. The individual snapshots of the receptor are docked against various ligands to generate a collection of docked complexes of different stabilities by using a docking program (RosettaLigand). The final docked complex of the lowest binding energy score is also coupled with an independent scoring method (DrugScore) to increase accuracy in docking scores. Our results show that (i) the receptor flexibility plays a key role in determining the binding selectivities of the PDZs, (ii) incorporating flexibility in PDZ-peptide docking predicts the docked poses accurately with an RMSD of >1 Å, (iii) the experimental binding affinities are captured qualitatively (including the higher affinity of the Erbin for the Class I peptide, and that of the Syntenin for Class II peptide) when the MRCs are generated by REMD sampling conformational space along binding induced fluctuation profiles of normal modes, and (iv) the final screening process using DrugScore enables us to discriminate the binding specificity of ligands in PDZs accurately.
Acknowledgments
We gratefully acknowledge Dr. Michael Feig, Dr. Ozlem Keskin, Tyler Glembo, and Ashini Bolia for their valuable comments. We also thank the Fulton High Performance Computing Initiative at Arizona State University and NCSA teragrid for computer time.
Glossary
Abbreviations:
- ENM
Elastic Network Model
- MRCs
multiple receptor conformations
- NMA
normal mode analysis
- PDZ
postsynaptic density-95/Dlg/ZO-1
- PDZs
PDZ domains
- REMD
replica exchange molecular dynamics
- RMSD
root mean square deviation.
References
- 1.Cozzini P, Kellogg GE, Spyrakis F, Abraham DJ, Costantino G, Emerson A, Fanelli F, Gohlke H, Kuhn LA, Morris GM, Orozco M, Pertinhez TA, Rizzi M, Sotriffer CA. Target flexibility: an emerging consideration in drug discovery and design. J Med Chem. 2008;51:6237–6255. doi: 10.1021/jm800562d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Halperin I, Ma B, Wolfson H, Nussinov R. Principles of docking: an overview of search algorithms and a guide to scoring functions. Proteins: Struct Funct Bioinf. 2002;47:409–443. doi: 10.1002/prot.10115. [DOI] [PubMed] [Google Scholar]
- 3.Leach AR. Molecular Modelling: Principles and Applications. New York: Prentice Hall; 2001. [Google Scholar]
- 4.Mohan V, Gibbs AC, Cummings MD, Jaeger EP, DesJarlais RL. Docking: successes and challenges. Curr Pharm Des. 2005;11:323–333. doi: 10.2174/1381612053382106. [DOI] [PubMed] [Google Scholar]
- 5.Sousa SF, Fernandes PA, Ramos MJ. Protein-ligand docking: current status and future challenges. Proteins: Struct Funct Bioinf. 2006;65:15–26. doi: 10.1002/prot.21082. [DOI] [PubMed] [Google Scholar]
- 6.Wong CF, McCammon JA. Protein flexibility and computer-aided drug design. Annu Rev Pharmacol Toxicol. 2003;43:31–45. doi: 10.1146/annurev.pharmtox.43.100901.140216. [DOI] [PubMed] [Google Scholar]
- 7.Totrov M, Abagyan R. Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr Opin Struct Biol. 2008;18:178–184. doi: 10.1016/j.sbi.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlson HA, McCammon JA. Accommodating protein flexibility in computational drug design. Mol Pharmacol. 2000;57:213–218. [PubMed] [Google Scholar]
- 9.Gorfe AA, Caflisch A. Functional plasticity in the substrate binding site of beta-secretase. Structure. 2005;13:1487–1498. doi: 10.1016/j.str.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 10.Lin JH, Perryman AL, Schames JR, McCammon JA. Computational drug design accommodating receptor flexibility: the relaxed complex scheme. J Am Chem Soc. 2002;124:5632–5633. doi: 10.1021/ja0260162. [DOI] [PubMed] [Google Scholar]
- 11.Mangoni M, Roccatano D, Di Nola A. Docking of flexible ligands to flexible receptors in solution by molecular dynamics simulation. Proteins: Struct Funct Bioinf. 1999;35:153–162. doi: 10.1002/(sici)1097-0134(19990501)35:2<153::aid-prot2>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 12.Nakajima N, Higo J, Kidera A, Nakamura H. Flexible docking of a ligand peptide to a receptor protein by multicanonical molecular dynamics simulation. Chem Phys Lett. 1997;278:297–301. [Google Scholar]
- 13.Wong CF, Kua J, Zhang Y, Straatsma TP, McCammon JA. Molecular docking of balanol to dynamics snapshots of protein kinase A. Proteins: Struct Funct Bioinf. 2005;61:850–858. doi: 10.1002/prot.20688. [DOI] [PubMed] [Google Scholar]
- 14.Zacharias M. Rapid protein-ligand docking using soft modes from molecular dynamics simulations to account for protein deformability: binding of FK506 to FKBP. Proteins: Struct Funct Bioinf. 2004;54:759–767. doi: 10.1002/prot.10637. [DOI] [PubMed] [Google Scholar]
- 15.Jolley CC, Wells SA, Hespenheide BM, Thorpe MF, Fromme P. Docking of photosystem I subunit C using a constrained geometric simulation. J Am Chem Soc. 2006;128:8803–8812. doi: 10.1021/ja0587749. [DOI] [PubMed] [Google Scholar]
- 16.Andrusier N, Nussinov R, Wolfson HJ. FireDock: fast interaction refinement in molecular docking. Proteins: Struct Funct Bioinf. 2007;69:139–159. doi: 10.1002/prot.21495. [DOI] [PubMed] [Google Scholar]
- 17.Schneidman-Duhovny D, Nussinov R, Wolfson HJ. Automatic prediction of protein interactions with large scale motion. Proteins: Struct Funct Bioinf. 2007;69:764–773. doi: 10.1002/prot.21759. [DOI] [PubMed] [Google Scholar]
- 18.Cavasotto CN, Kovacs JA, Abagyan RA. Representing receptor flexibility in ligand docking through relevant normal modes. J Am Chem Soc. 2005;127:9632–9640. doi: 10.1021/ja042260c. [DOI] [PubMed] [Google Scholar]
- 19.Dobbins SE, Lesk VI, Sternberg MJ. Insights into protein flexibility: the relationship between normal modes and conformational change upon protein-protein docking. Proc Natl Acad Sci USA. 2008;105:10390–10395. doi: 10.1073/pnas.0802496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Floquet N, Marechal JD, Badet-Denisot MA, Robert CH, Dauchez M, Perahia D. Normal mode analysis as a prerequisite for drug design: application to matrix metalloproteinases inhibitors. Febs Lett. 2006;580:5130–5136. doi: 10.1016/j.febslet.2006.08.037. [DOI] [PubMed] [Google Scholar]
- 21.Mustard D, Ritchie DW. Docking essential dynamics eigenstructures. Proteins: Struct Funct Bioinf. 2005;60:269–274. doi: 10.1002/prot.20569. [DOI] [PubMed] [Google Scholar]
- 22.Lindahl E, Delarue M. Refinement of docked protein-ligand and protein-DNA structures using low frequency normal mode amplitude optimization. Nucleic Acids Res. 2005;33:4496–4506. doi: 10.1093/nar/gki730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.May A, Zacharias M. Accounting for global protein deformability during protein–protein and protein-ligand docking. Biochim Biophys Acta. 2005;1754:225–231. doi: 10.1016/j.bbapap.2005.07.045. [DOI] [PubMed] [Google Scholar]
- 24.May A, Zacharias M. Energy minimization in low-frequency normal modes to efficiently allow for global flexibility during systematic protein–protein docking. Proteins: Struct Funct Bioinf. 2008;70:794–809. doi: 10.1002/prot.21579. [DOI] [PubMed] [Google Scholar]
- 25.May A, Zacharias M. Protein-ligand docking accounting for receptor side chain and global flexibility in normal modes: evaluation on kinase inhibitor cross docking. J Med Chem. 2008;51:3499–3506. doi: 10.1021/jm800071v. [DOI] [PubMed] [Google Scholar]
- 26.Barril X, Morley SD. Unveiling the full potential of flexible receptor docking using multiple crystallographic structures. J Med Chem. 2005;48:4432–4443. doi: 10.1021/jm048972v. [DOI] [PubMed] [Google Scholar]
- 27.Damm KL, Carlson HA. Exploring experimental sources of multiple protein conformations in structure-based drug design. J Am Chem Soc. 2007;129:8225–8235. doi: 10.1021/ja0709728. [DOI] [PubMed] [Google Scholar]
- 28.Philippopoulos M, Lim C. Exploring the dynamic information content of a protein NMR structure: comparison of a molecular dynamics simulation with the NMR and X-ray structures of Escherichia coli ribonuclease HI. Proteins: Struct Funct Bioinf. 1999;36:87–110. doi: 10.1002/(sici)1097-0134(19990701)36:1<87::aid-prot8>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 29.Bottegoni G, Kufareva I, Totrov M, Abagyan R. Four-dimensional docking: a fast and accurate account of discrete receptor flexibility in ligand docking. J Med Chem. 2009;52:397–406. doi: 10.1021/jm8009958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugita Y, Okamoto Y. Replica-exchange molecular dynamics method for protein folding. Chem Phys Lett. 1999;314:141–151. [Google Scholar]
- 31.Kovacs JA, Cavasotto CN, Abagyan R. Conformational sampling of protein flexibility in generalized coordinates: application to ligand docking. J Comput Theor Nanosci. 2005;2:354–361. [Google Scholar]
- 32.Kovacs JA, Chacon P, Abagyan R. Predictions of protein flexibility: first-order measures. Proteins: Struct Funct Bioinf. 2004;56:661–668. doi: 10.1002/prot.20151. [DOI] [PubMed] [Google Scholar]
- 33.Rueda M, Bottegoni G, Abagyan R. Consistent improvement of cross-docking results using binding site ensembles generated with elastic network normal modes. J Chem Inf Model. 2009;49:716–725. doi: 10.1021/ci8003732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Atilgan AR, Durell SR, Jernigan RL, Demirel MC, Keskin O, Bahar I. Anisotropy of fluctuation dynamics of proteins with an Elastic Network Model. Biophys J. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bahar I, Atilgan AR, Demirel MC, Erman B. Vibrational dynamics of folded proteins: significance of slow and fast motions in relation to function and stability. Phys Rev Lett. 1998;80:2733–2736. [Google Scholar]
- 36.Bahar I, Atilgan AR, Erman B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold Des. 1997;2:173–181. doi: 10.1016/S1359-0278(97)00024-2. [DOI] [PubMed] [Google Scholar]
- 37.Hinsen K. Structural flexibility in proteins: impact of the crystal environment. Bioinformatics. 2008;24:521–528. doi: 10.1093/bioinformatics/btm625. [DOI] [PubMed] [Google Scholar]
- 38.Rader AJ, Chennubhotla C, Yang LW, Bahar I. The Gaussian Network Model: theory and applications. In: Cui Q, Bahar I, editors. Normal mode analysis. Theory and applications to biological and chemical systems. Boca Raton, FL: CRC Press, Taylor & Francis Group; 2006. pp. 41–64. [Google Scholar]
- 39.Song G, Jernigan RL. vGNM: a better model for understanding the dynamics of proteins in crystals. J Mol Biol. 2007;369:880–893. doi: 10.1016/j.jmb.2007.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tama F, Sanejouand YH. Conformational change of proteins arising from normal mode calculations. Protein Eng. 2001;14:1–6. doi: 10.1093/protein/14.1.1. [DOI] [PubMed] [Google Scholar]
- 41.Tirion MM. Large amplitude elastic motions in proteins from a single-parameter, atomic analysis. Phys Rev Lett. 1996;77:1905–1908. doi: 10.1103/PhysRevLett.77.1905. [DOI] [PubMed] [Google Scholar]
- 42.Keskin O. Binding induced conformational changes of proteins correlate with their intrinsic fluctuations. BMC Struct Biol. 2007;7:31–41. doi: 10.1186/1472-6807-7-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tobi D, Bahar I. Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc Natl Acad Sci USA. 2005;102:18908–18913. doi: 10.1073/pnas.0507603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng W, Brooks BR. Normal-modes-based prediction of protein conformational changes guided by distance constraints. Biophys J. 2005;88:3109–3117. doi: 10.1529/biophysj.104.058453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 46.Ranganathan R, Ross EM. PDZ domain proteins: scaffolds for signaling complexes. Curr Biol. 1997;7:R770–R773. doi: 10.1016/s0960-9822(06)00401-5. [DOI] [PubMed] [Google Scholar]
- 47.Petit CM, Zhang J, Sapienza PJ, Fuentes EJ, Lee AL. Hidden dynamic allostery in a PDZ domain. Proc Natl Acad Sci USA. 2009;106:18249–18254. doi: 10.1073/pnas.0904492106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smock RG, Gierasch LM. Sending signals dynamically. Science. 2009;324:198–203. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gerek ZN, Keskin O, Ozkan SB. Identification of specificity and promiscuity of PDZ domain interactions through their dynamic behavior. Proteins: Struct Funct Bioinf. 2009;77:796–811. doi: 10.1002/prot.22492. [DOI] [PubMed] [Google Scholar]
- 50.Fuentes EJ, Der CJ, Lee AL. Ligand-dependent dynamics and intramolecular signaling in a PDZ domain. J Mol Biol. 2004;335:1105–1115. doi: 10.1016/j.jmb.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 51.Stiffler MA, Chen JR, Grantcharova VP, Lei Y, Fuchs D, Allen JE, Zaslavskaia LA, MacBeath G. PDZ domain binding selectivity is optimized across the mouse proteome. Science. 2007;317:364–369. doi: 10.1126/science.1144592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Basdevant N, Weinstein H, Ceruso M. Thermodynamic basis for promiscuity and selectivity in protein–protein interactions: PDZ domains, a case study. J Am Chem Soc. 2006;128:12766–12777. doi: 10.1021/ja060830y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Los Rios P, Cecconi F, Pretre A, Dietler G, Michielin O, Piazza F, Juanico B. Functional dynamics of PDZ binding domains: a normal-mode analysis. Biophys J. 2005;89:14–21. doi: 10.1529/biophysj.104.055004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dhulesia A, Gsponer J, Vendruscolo M. Mapping of two networks of residues that exhibit structural and dynamical changes upon binding in a PDZ domain protein. J Am Chem Soc. 2008;130:8931–8939. doi: 10.1021/ja0752080. [DOI] [PubMed] [Google Scholar]
- 55.Kurakin A, Swistowski A, Wu SC, Bredesen DE. The PDZ domain as a complex adaptive system. PloS one. 2007;2:e953, 1–11. doi: 10.1371/journal.pone.0000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Niv MY, Weinstein H. A flexible docking procedure for the exploration of peptide binding selectivity to known structures and homology models of PDZ domains. J Am Chem Soc. 2005;127:14072–14079. doi: 10.1021/ja054195s. [DOI] [PubMed] [Google Scholar]
- 57.Shan JF, Zheng J. Optimizing Dvl PDZ domain inhibitor by exploring chemical space. J Comput Aided Mol Des. 2009;23:37–47. doi: 10.1007/s10822-008-9236-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharp K, Skinner JJ. Pump-probe molecular dynamics as a tool for studying protein motion and long range coupling. Proteins: Struct Funct Bioinf. 2006;65:347–361. doi: 10.1002/prot.21146. [DOI] [PubMed] [Google Scholar]
- 59.Davis IW, Baker D. RosettaLigand docking with full ligand and receptor flexibility. J Mol Biol. 2009;385:381–392. doi: 10.1016/j.jmb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 60.Meiler J, Baker D. RosettaLigand: protein-small molecule docking with full side-chain flexibility. Protein Sci. 2006;65:538–548. doi: 10.1002/prot.21086. [DOI] [PubMed] [Google Scholar]
- 61.Gohlke H, Hendlich M, Klebe G. Knowledge-based scoring function to predict protein-ligand interactions. J Mol Biol. 2000;295:337–356. doi: 10.1006/jmbi.1999.3371. [DOI] [PubMed] [Google Scholar]
- 62.Wang R, Lai L, Wang S. Further development and validation of empirical scoring functions for structure-based binding affinity prediction. J Comput Aided Mol Des. 2002;16:11–26. doi: 10.1023/a:1016357811882. [DOI] [PubMed] [Google Scholar]
- 63.Graves AP, Shivakumar DM, Boyce SE, Jacobson MP, Case DA, Shoichet BK. Rescoring docking hit lists for model cavity sites: predictions and experimental testing. J Mol Biol. 2008;377:914–934. doi: 10.1016/j.jmb.2008.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kollman PA, Dixon R, Cornell W, Vox T, Chipot C, Pohorille A. The development/application of a "minimalist" organic/biochemical molecular mechanical force field using a combination of ab Initio calculations and experimental data. In: van Gunsteren WF, Weiner PK, Wilkinson AJ, editors. Computer simulation of biomolecular systems theoretical and experimental applications. Boston, MA: Kluwer; 1997. pp. 83–96. [Google Scholar]
- 66.Tsui V, Case DA. Molecular dynamics simulations of nucleic acids with a generalized Born solvation model. J Am Chem Soc. 2000;122:2489–2498. [Google Scholar]
- 67.Liu L, Koharudin LMI, Gronenborn AM, Bahar I. A comparative analysis of the equilibrium dynamics of a designed protein inferred from NMR, X-ray, and computations. Proteins: Struct Funct Bioinf. 2009;77:927–939. doi: 10.1002/prot.22518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang L, Song G, Carriquiry A, Jernigan RL. Close correspondence between the motions from principal component analysis of multiple HIV-1 protease structures and elastic network modes. Structure. 2008;16:321–330. doi: 10.1016/j.str.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang LW, Eyal E, Bahar I, Kitao A. Principal component analysis of native ensembles of biomolecular structures (PCA_NEST): insights into functional dynamics. Bioinformatics. 2009;25:606–614. doi: 10.1093/bioinformatics/btp023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang LW, Eyal E, Chennubhotla C, Jee J, Gronenborn AM, Bahar I. Insights into equilibrium dynamics of proteins from comparison of NMR and X-ray data with computational predictions. Structure. 2007;15:741–749. doi: 10.1016/j.str.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bakan A, Bahar I. The intrinsic dynamics of enzymes plays a dominant role in determining the structural changes induced upon inhibitor binding. Proc Natl Acad Sci USA. 2009;106:14349–14354. doi: 10.1073/pnas.0904214106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: Identification of signaling domains. Proc Natl Acad Sci USA. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bujnicki JM, Elofsson A, Fischer D, Ryclewski L. Structure prediction meta server. Bioinformatics. 2001;17:750–751. doi: 10.1093/bioinformatics/17.8.750. [DOI] [PubMed] [Google Scholar]
- 74.Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003;19:1015–1018. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- 75.Lambert C, Leonard N, De Bolle X, Depiereux E. ESyPred3D: prediction of proteins 3D structures. Bioinformatics. 2002;18:1250–1256. doi: 10.1093/bioinformatics/18.9.1250. [DOI] [PubMed] [Google Scholar]
- 76.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 77.Shen MY, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–2524. doi: 10.1110/ps.062416606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 79.Wang R, Lu Y, Wang S. Comparative evaluation of 11 scoring functions for molecular docking. J Med Chem. 2003;46:2287–2303. doi: 10.1021/jm0203783. [DOI] [PubMed] [Google Scholar]
- 80.Velec HF, Gohlke H, Klebe G. DrugScore(CSD)-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction. J Med Chem. 2005;48:6296–6303. doi: 10.1021/jm050436v. [DOI] [PubMed] [Google Scholar]
- 81.Yin S, Biedermannova L, Vondrasek J, Dokholyan NV. MedusaScore: an accurate force field-based scoring function for virtual drug screening. J Chem Inf Model. 2008;48:1656–1662. doi: 10.1021/ci8001167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ferrara P, Gohlke H, Price DJ, Klebe G, Brooks CL. Assessing scoring functions for protein-ligand interactions. J Med Chem. 2004;47:3032–3047. doi: 10.1021/jm030489h. [DOI] [PubMed] [Google Scholar]
- 83.McGovern SL, Shoichet BK. Information decay in molecular docking screens against holo, apo, and modeled conformations of enzymes. J Med Chem. 2003;46:2895–2907. doi: 10.1021/jm0300330. [DOI] [PubMed] [Google Scholar]
- 84.Kang BS, Cooper DR, Devedjiev Y, Derewenda U, Derewenda ZS. Molecular roots of degenerate specificity in syntenin's PDZ2 domareassessment of the PDZ recognition paradigm. Structure. 2003;11:845–853. doi: 10.1016/s0969-2126(03)00125-4. [DOI] [PubMed] [Google Scholar]
- 85.Birrane G, Chung J, Ladias JA. Novel mode of ligand recognition by the Erbin PDZ domain. J Biol Chem. 2003;278:1399–1402. doi: 10.1074/jbc.C200571200. [DOI] [PubMed] [Google Scholar]
- 86.Laura RP, Witt AS, Held HA, Gerstner R, Deshayes K, Koehler MF, Kosik KS, Sidhu SS, Lasky LA. The Erbin PDZ domain binds with high affinity and specificity to the carboxyl termini of delta-catenin and ARVCF. J Biol Chem. 2002;277:12906–12914. doi: 10.1074/jbc.M200818200. [DOI] [PubMed] [Google Scholar]
- 87.Dev KK, Nakanishi S, Henley JM. The PDZ domain of PICK1 differentially accepts protein kinase C-alpha and GluR2 as interacting ligands. J Biol Chem. 2004;279:41393–41397. doi: 10.1074/jbc.M404499200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Madsen KL, Beuming T, Niv MY, Chang CW, Dev KK, Weinstein H, Gether U. Molecular determinants for the complex binding specificity of the PDZ domain in PICK1. J Biol Chem. 2005;280:20539–20548. doi: 10.1074/jbc.M500577200. [DOI] [PubMed] [Google Scholar]
- 89.Staudinger J, Lu J, Olson EN. Specific interaction of the PDZ domain protein PICK1 with the COOH terminus of protein kinase C-alpha. J Biol Chem. 1997;272:32019–32024. doi: 10.1074/jbc.272.51.32019. [DOI] [PubMed] [Google Scholar]