

Abstract
Monoclonal antibodies (Mabs) are a favorite drug platform of the biopharmaceutical industry. Currently, over 20 Mabs have been approved and several hundred others are in clinical trials. The anti-LINGO-1 Mab Li33 was selected from a large panel of antibodies by Fab phage display technology based on its extraordinary biological activity in promoting oligodendrocyte differentiation and myelination in vitro and in animal models of remyelination. However, the Li33 Fab had poor solubility when converted into a full antibody in an immunoglobulin G1 framework. A detailed analysis of the biochemical and structural features of the antibody revealed several possible reasons for its propensity to aggregate. Here, we successfully applied three molecular approaches (isotype switching, targeted mutagenesis of complementarity determining region residues, and glycosylation site insertion mutagenesis) to address the solubility problem. Through these efforts we were able to improve the solubility of the Li33 Mab from 0.3 mg/mL to >50 mg/mL and reduce aggregation to an acceptable level. These strategies can be readily applied to other proteins with solubility issues.
Keywords: antibody, aggregation, improved solubility, isotype switching, CDR mutagenesis, glycoengineering, antibody engineering, drug design
Introduction
Immunoglobulin (Ig)Gs are the most abundant class of antibodies in serum and have become a favorite drug platform for the biopharmaceutical industry. Humans have four IgG isotypes: IgG1, IgG2, IgG3, and IgG4.1,2 All are heterotetrameric with two identical heavy (HC) and light chains (LC) linked by intrachain disulfides. IgGs contain two identical Fabs and an Fc connected by a flexible hinge region. The Fab contains six variable regions [complementarity determining region (CDR) sequences] that lead to the extraordinary target diversity of antibodies. The Fc contains binding sites for Fc(gamma) receptors, FcRN, and C1q complement that impart effector function. The hinges differ in size and disulfide patterns, which impact the flexibility and spatial arrangement of the Fabs relative to each other and to the Fc.3–5 Enormous strides have been made in the development and optimization of therapeutic antibodies with the IgG1, IgG2, and IgG4 frameworks being successfully exploited for drug development.1,2 Targeted molecular designs that either eliminate the Fc glycosylation sites to reduce effector function or that stabilize the IgG4 hinge to prevent conversion of IgG4 antibodies into functionally monovalent forms in vivo are two examples of the many engineering achievements.1,3,6–9 Perhaps the most common and challenging issue encountered in developing protein biopharmaceuticals is aggregation, which can be exacerbated by the need to develop high protein concentration formulations to support systemic administration.10 The size and complexity of proteins makes aggregation a difficult issue to address. Aggregates can be classified by whether they are soluble or insoluble, reversible or irreversible, or whether they lead to denaturation or the formation of covalent linkages. Hydrophobic interactions appear to be a major contributor,10,11 however; the literature is full of examples where other factors were identified that led to aggregation. Biochemical and biophysical techniques that assess size, aggregation state, structure, and intermolecular contacts provide a useful battery of methods to evaluate aggregation and the types of aggregates that form.10,12,13 Formulation development is routinely used to minimize aggregation and other degradation pathways to avoid loss of activity, instability, and immunogenicity.10,11 Nevertheless, improved engineering strategies along with better understanding of the pathways leading to aggregation are needed to address this important issue.
LINGO-1 (GI:15029689) is a surface glycoprotein selectively expressed in neurons and oligodendrocytes in the CNS where it acts as an endogenous inhibitor of myelination.14 Loss of LINGO-1 function leads to robust myelination, suggesting that antagonists to the pathway may provide new therapeutic opportunities for the treatment of CNS demyelination disorders.14–16 To test this possibility anti-LINGO-1 antibodies were generated that were screened for affinity to human LINGO-1, selectivity, and their ability to promote myelination in vitro and promote CNS myelin repair in vivo in animal models of demylination. Biological studies utilizing two of these antibodies (1A7, Li33) were previously described.15,16 In vivo studies with the mouse anti-human LINGO-1 1A7 antibody were hampered by its poor affinity for rodent LINGO-1.15,16 The Li33 antibody identified by Fab phage display technology was more potent than the 1A7 monoclonal antibody (Mab) at promoting myelination and had high affinity for both human and rodent LINGO-1. However, conversion of the Li33 Fab to an IgG1 Mab resulted in an antibody with surprisingly poor solubility. Here, we used a combination of biochemical, structural, and molecular approaches to investigate the pathways that led to the formation of Li33 Mab aggregates and to develop solutions to the solubility problem. The types of aggregates that formed with the Li33 Mab were impacted by construct design and formulation. Particularly striking was the impact of the IgG framework as simple replacement of the IgG1 with an IgG2 framework resulted in an Li33Mab with excellent solubility. Other versions of the Li33 Mab with improved solubility were generated using rational design and provided further insights into the aggregation phenomenon. The Li33 IgG2 Mab was highly efficacious in animal models of demyelination15 and has proven to be an important reagent for probing LINGO-1 biology.
Results
Impact of IgG framework on Li33 solubility
The anti-LINGO-1 Li33 Fab was converted into a full human antibody and expressed in mammalian cells. Three different IgG frameworks (IgG1, IgG2, and IgG4) were evaluated both in wild-type and aglycosyl forms. For the IgG2 framework, the V234A/G237A mutation was also evaluated as an alternative to the glycosylation site mutation to eliminate FcRIIa binding.17 SDS–PAGE gel analysis of the seven purified constructs is shown in Figure 1. All constructs displayed typical antibody features under reducing and non-reducing conditions. Two bands corresponding to the HC (50 kDa) and LC (25 kDa) were observed under reducing conditions. The slight increase in mobility of the HC for the three aglycosyl constructs is consistent with loss of the glycan due to the glycosylation site mutation. A single prominent band with molecular mass of 150 kDa was observed under non-reducing conditions resulting from the formation of the tetrameric 2HC–2LC complex. All three aglycosyl forms show a minor band at 75 kDa under nonreducing conditions, which corresponds to HC–LC dimer. Higher levels of the HC–LC dimer occur in the wild-type IgG4 preparation consistent with published observations.18
Figure 1.

Characterization of Mabs by SDS–PAGE. Li33 Mabs (4 μg/lane) were subjected to SDS–PAGE under reducing and nonreducing conditions and stained with Coomassie brilliant blue. Molecular weight markers and their apparent molecular masses are show in the first lane.
The ability of the antibodies to bind LINGO-1 was assessed in an ELISA format. The seven Mabs showed similar EC50 values for binding to LINGO-1 (Table I) with apparent affinities of 0.12 nM for the IgG1wt and agly, ∼0.24 nM for IgG2wt and agly, and ∼0.36 nM for IgG4wt and agly. The activity differences, while reproducible, were not large enough to influence framework selection.
Table I.
Impact of Li33 IgG Frameworks on Solubility
| Stability (°C) |
|||||
|---|---|---|---|---|---|
| Li33 isotype | Solubilitya (mg/mL) | LINGO-1 binding, EC50 (nM) | TM1 | TM2 | SEC, % monomer |
| IgG1wt pH 7.0 | 0.9 | 0.12 | 69 | 76 | 99 |
| IgG1agly pH 7.0 | 0.3 | 0.12 | 60 | 77 | >99 |
| IgG4wt pH 7.0 | >30 | 0.35 | 64 | 72 | 98 |
| IgG4agly pH 7.0 | 0.3 | 0.37 | 56 | 73 | 95 |
| IgG2wt pH 7.0 | >50 | 0.23 | 69 | 76 | 96 |
| IgG2agly pH 7.0 | 0.2 | 0.26 | 59 | 76 | 98 |
| IgG2-V234A/G237A pH 7.0 | 5.6 | 0.19 | 69 | 76 | 95 |
| IgG1 Fab2 pH 7.0 | 0.3 | 0.10 | – | 77 | 98 |
| IgG2 Fab2 pH 7.0 | >50 | 0.39 | – | 77 | 98 |
| IgG1 Fab pH 7.0 | >50 | 0.68 | – | 76 | 95 |
| IgG1agly reduced pH 7.0 | >40 | 0.12 | 55 | 75 | 98 |
| IgG1wt reduced pH 7.0 | >50 | 0.15 | 63 | 75 | 98 |
| IgG1wt pH 6.5 | 1.7 | 0.10 | 69 | 77 | |
| IgG1wt pH 6.0 | 2.4 | 0.10 | 69 | 78 | |
| IgG1wt pH 5.5 | 30 | 0.16 | 66 | 81 | |
| IgG1wt pH 5.0 | >50 | 0.45 | 66 | 81 | |
| IgG1wt pH 4.5 | >50 | 2.1 | 62 | 82 | |
| IgG1wt pH 4.0 | >50 | 16 | 54 | 78 | |
| IgG1wt pH 3.5 | >50 | 34 | 46/66 | 74 | |
| IgG1 wt pH 3.0 | >50 | >100 | 34/52 | 72 | |
Li33 Mabs were characterized for the limit in their solubility at 4°C, apparent binding affinity for LINGO-1, thermal stability, and aggregation state by SEC (samples loaded at 300 μg/mL). Samples at pH 7.0 were prepared in 20 mM sodium phosphate, 150 mM NaCl. Samples at other pHs were prepared in 10 mM sodium citrate, 150 mM NaCl.
The protocol for assessing solubility is described in the Materials and Methods section.
The most striking feature of the seven Li33 Mab preparations was the differences in their solubility in 20 mM NaH2PO4, 150 mM NaCl at pH 7.0 (PBS), which was the maximum concentration each protein could be stored in PBS at 4°C without precipitating (see Table I). All three aglycosyl antibodies had poor solubility with extensive precipitation at concentrations >0.3 mg/mL Mab. The solubility of the IgG1wt Mab was slightly improved, 0.9 mg/mL, while the IgG2wt and IgG4wt Mabs were soluble at the highest concentration tested. The >50 mg/mL solubility of the IgG2wt Mab represents more than a 250-fold increase over the aglycosyl version of the same construct. The IgG2 V234A/G237A variant was intermediate in terms of its solubility. Below the solubility limits the antibodies were stable to prolonged storage at 4°C and to freeze thaw. The insoluble aggregates that formed with the IgG1wt and all the agly Mabs at pH 7.0 were irreversible and could not be resolubilized by sample dilution.
The purity and aggregation state of the antibodies were studied by size exclusion chromatography (SEC) and analytical ultracentrifugation (AUC). All constructs eluted from the SEC column as a single prominent peak with an apparent molecular mass of 150 kDa and >95% purity (Table I). Example SEC profiles for the IgG1wt and IgG2wt Mabs are shown in Figure 2. The IgG1wt Mab contained 99% monomer with no evidence of soluble aggregates, whereas the IgG2wt Mab contained 96% monomer, 2% dimer and 2% higher molecular mass aggregates. AUC studies were performed at low and much higher protein concentration to better evaluate aggregation that might occur under routine storage conditions. AUC results for the IgG2wt Mab revealed a concentration-dependent formation of soluble aggregates that were reversible upon sample dilution [Fig. 3(A–C), Table II]. The smallest and most prominent aggregation product appears to correspond to a dimer (see Supporting Information Table I). A coarse estimate of its apparent KD based on the concentration at which 50% of Mab was in this aggregate form is 50 μM. Other much larger and more heterogeneous aggregates were also observed. An interesting behavior of these latter aggregates is the significant decrease both in sedimentation coefficients and polydispersity with increasing protein concentration (Fig. 3 and Supporting Information Table I). It is unclear if this behavior is due to a unique mechanism of aggregation or to nonideality effects that are not accounted for by the data processing software.19 AUC data for the IgG1wt Mab were similar to SEC results and showed no significant evidence of reversible aggregation at a concentration that was at its limit of solubility (Table II). Thus, while the IgG2wt framework prevented the transition to an insoluble aggregate seen with the IgG1wt framework, the protein self-associated to form soluble aggregates. The lack of consistency between SEC and AUC results although not that common demonstrates a potential issue in trying to extrapolate dilution solution behavior of SEC to the high concentrations found in formulated biopharmaceutical samples.10,12,20 The further reduction in AUC values for % aggregate for the 0.5 mg/mL IgG2 Mab sample at pH 5.0, 5% versus 14% for the 0.3 mg/mL sample at pH 7.0 (discussed below), suggests that even after dilution to 0.3 mg/mL, dissociation of the reversible aggregates was incomplete at pH 7.0.
Figure 2.

Characterization by SEC. Li33 IgG1wt and IgG2wt Mabs were subjected to SEC on a Superdex 200 column (samples loaded at 300 μg/mL). The column effluent was monitored at 280 nm. The elution profile of gel filtration standards and their molecular masses are shown at the top of the panel.
Figure 3.

Evaluating the aggregation state of Li33 IgG2wt by AUC. Data from velocity sedimentation centrifugations studies on IgG2wt Mab at pH 7.0 at concentrations of 0.4 (A), 7 (B), and 24 (C) mg/mL and at pH 5.0 at 19 mg/mL (D) are presented. Left panels show either the raw UV absorbance data or noise corrected refractometric data scans as a function of time. Right panels show sample distribution of sedimentation coefficients [c(s) vs. S] that were derived from the corresponding UV absorbance or refractometric data scans with SEDFIT. Peak 1, monomer. Peak 2, smallest aggregation product. S values for AUC peaks are tabulated in Supporting Information Table I.
Table II.
Aggregation Data Obtained by AUC for Different Versions of the Li33 Antibody as a Function of Protein Concentration and Buffer Conditions
| Sample | Concentration (mg/mL) | % Apparent dimera | % Total aggregate |
|---|---|---|---|
| IgG2wt pH 7.0 | 0.3 | 2 | 14 |
| IgG2wt pH 7.0 | 7.0 | 37 | 65 |
| IgG2wt pH 7.0 | 24 | 53 | 83 |
| IgG2wt pH 5.0 | 0.5 | 3 | 5 |
| IgG2wt pH 5.0 | 19 | 2 | 8 |
| IgG1wt pH 7.0 | 0.9b | 4 | 6 |
| IgG1wt pH 5.5 | 2.4b | 2 | 3 |
| IgG1wt pH 5.0 | 0.5 | 2 | 2 |
| IgG1wt pH 5.0 | 25 | 1 | 3 |
| IgG1wt pH 7.0 + TCEP | 12 | 20 | 31 |
| IgG1 Fab pH 7.0 | 0.5 | 10 | 10 |
| IgG1 Fab pH 7.0 | 29 | 60 | 74 |
| IgG1 Fab pH 5.0 | 0.5 | 13 | 13 |
| IgG1 Fab pH 5.0 | 26 | 6 | 6 |
| IgG2–234A/237A TripleA | 6.0 | 2 | 9 |
| IgG2–234A/237A TripleS | 6.0 | 2 | 5 |
| IgG1wt A120N pH 7.0 | 7.0 | 5 | 5 |
Percent apparent dimer and total aggregate were calculated from integrated areas of the distribution of sedimentation coefficient plot generated for each sample.
Defined as the first peak that appears after the monomer peak with a larger sedimentation coefficient.
Samples tested at the limit of their solubility.
Stability measurements can be used to identify features that impact the solubility of a protein. The thermal stability of all the Li33 constructs was measured by differential scanning fluorometry (Table I, Fig. 4). Melting temperature (Tm) values for the Fab region (TM2) were 76–77°C for all of the IgG1 and IgG2 constructs and 72–73°C for the IgG4wt and agly Mabs. Tm values for the CH2 domain (TM1) were variable and 8–10°C lower for each of the agly constructs versus their wild-type counterparts. The IgG4wt and agly constructs were the least stable as indicated by their lower Tm values. No obvious trends were observed in the stability data that could explain the dramatic solubility differences between the IgG1 and IgG2 Mabs.
Figure 4.
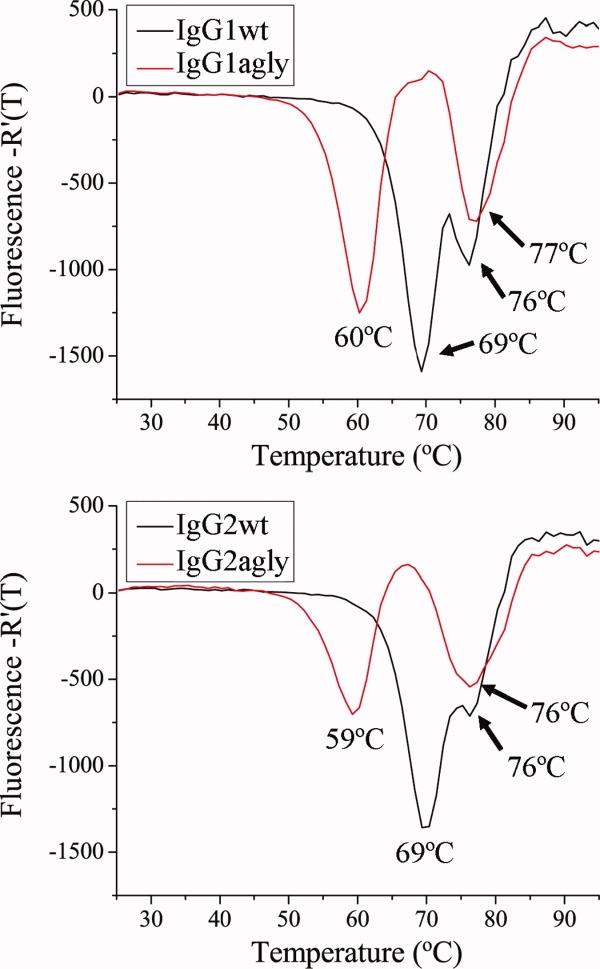
Thermal denaturation studies. IgG1wt and IgG2wt Mabs were analyzed by DSF. Data for fluorescence as a function of temperature are plotted as first derivative curves.
To further assess features of the antibodies that were affecting solubility a variety of conditions and fragmentation products were tested (Table I). Fab2 and Fab fragments were generated enzymatically with pepsin and papain, respectively. The solubility of the IgG2 Fab2 was >50 mg/mL whereas the solubility of the IgG1 Fab2 was only 0.3 mg/mL. Thus the solubility differences observed with the IgG1wt and IgG2wt Mabs were maintained in the corresponding Fab2 fragments. The IgG1 Fab was soluble to >50 mg/mL. Fragmentation had little impact on stability. No TM1 signal was observed for the Fab2 and Fab moieties as expected, as the TM1 transition is produced from the CH2 domain. The higher values for EC50 for the binding of the Fab to LINGO-1 are consistent with monovalent binding for the Fab versus bivalent binding for the intact Mab and Fab2 fragments. SEC results for the IgG1 Fab2 and Fab, and IgG2 Fab2 showed purities of >95%. AUC analysis of the IgG1 Fab [Table II, Supporting Information Fig. 1(A)] revealed that it displayed concentration-dependent aggregation in PBS similar to that observed for the IgG2wt Mab. A crude estimate of the apparent KD for dimer formation of the Fab is approximately 300 μM. Thus, the aggregation seen with the Mabs could be anticipated from the AUC behavior of the Fab itself at high concentrations.
Reduction of the interchain disulfides also improved the solubility of the IgG1 Mabs, switching the aggregation pathway from the formation of insoluble to soluble aggregates (Tables I and II). Reduction had no impact on LINGO-1 binding or aggregation state as determined by SEC and had only a slight effect on thermal stability (Table I). AUC analysis showed that the reduced IgG1wt Mab at 12 mg/mL formed soluble aggregates with an apparent dimer being the predominant species [Table II, Supporting Information Fig. 1(C)].
Impact of formulation on Li33 solubility
Formulation development is routinely used to optimize storage conditions that maintain activity, provide stability, and minimize product degradation at protein concentrations useful for both storage and delivery. pH had a very dramatic effect on solubility of the IgG1wt Mab where below pH 5.5 the antibody was very soluble and above pH 5.5 it had poor solubility (Table I). Other excipients that were evaluated; salt, sucrose, and Tween 80, had no significant effect on solubility. Thermal stability measurements at the various pH treatments revealed that the Fab region (TM2) was stable under all test conditions. The CH2 domain (TM1) was stable from pH 7.0 to 5.0 but then exhibited a sharp decrease in stability as the pH was further reduced. For these reasons, pH 5.0 was chosen for additional studies. AUC data for the IgG1wt Mab at pH 5.0 revealed a low level of stable aggregates (<3%) with no significant signs of concentration dependent aggregation [Table II, Supporting Information Fig. 1(D)]. A similar AUC result was also seen with the IgG1 Fab [Table II, Supporting Information Fig. 1(B)]. As the solubility of a protein can be significantly reduced at pHs near its isoelectric point, pI values for the Li33 Mabs were determined by isoelectric focusing. All of the antibodies had pI values of >pH 8.2 and consequently this could not account for the instances where poor solubility was observed.
The apparent concentration-dependent dimerization seen with the IgG2wt Mab by AUC was also greatly reduced by pH 5.0 treatment [Table II, Fig. 3(D)] in a manner similar to that seen for the IgG1wt Mab and IgG1 Fab. Whereas soluble aggregate levels at pH 7 increased with Mab concentration from 14% at 0.3 mg/mL to 65% at 7 mg/mL and to 83% at 24 mg/mL Mab, soluble aggregate levels at pH 5.0 were 5% at 0.5 mg/mL Mab and 8% at 19 mg/mL. An unexpected feature of IgG2wt Mab at pH 5.0 was that temperature now affected its solubility where the solubility was >50 mg/mL at room temperature, but 14 mg/mL at 4°C. The drop in solubility at 4°C was accompanied by formation of reversible insoluble aggregates that went back into solution at room temperature. In fact the sample that was characterized by AUC at room temperature had undergone this temperature dependent precipitation and resolubilization step.
Crystallization of the IgG2 Fab2
Crystallization efforts were used to gain insights into features of the antibody that might impact solubility. The Fab2 structure was solved by molecular replacement (data collection and refinement statistics are summarized in Table III). IgG2 Fab2 crystals diffracted to 2.6 Å and were in a P1 space group with eight Fabs per unit cell. An unusual feature of the Li33 structure was the presence of a region of hydrophobic CDR residues that form intermolecular contacts. Figure 5(A) shows representative electron density data for this region of the structure. Five residues, W50, W104, I57, and P54 from the HC and W94 from the LC are highlighted in the figure. Fab packing arrangements indicated two different types of interactions: CDR–CDR [Fig. 5(B)] and CDR framework [Fig. 5(C)]. The chains that use a CDR–CDR arrangement are A/B with E/F, and C/D and G/H with crystal contacts to Fabs outside of the unit cell. The chains that use a CDR-framework packing interaction include chain J with the F chain framework, P with the D chain framework, N with the H chain framework, and L with a crystal contact framework outside of the unit cell.
Table III.
Data Collection and Refinement Statistics
| Data collection | IgG1 Fab | IgG2 Fab2 |
|---|---|---|
| Space group | P6522 | P1 |
| Cell dimensions: a, b, c (Å), α, β, γ (°) | 90.6, 90.6, 215.0, 90, 90, 90 | 91.7,109.5, 118.4, 61.5, 79.3, 87.6 |
| Wavelength (Å) | 0.9797 | 0.9797 |
| Resolution (Å) | 50–3.2 | 47.6–2.62 |
| Rsym (%)a | 22.1 (57.1) | 13.2 (62.4) |
| I/σ(I)a | 13.4 (4.9) | 3.5 (2.0) |
| Multiplicitya | 17.8 (17.2) | 1.9 (1.7) |
| Total no. of reflections/no. of unique reflections | 889,690/9331 | 221,192/115,601 |
| Completeness (%)a | 100 (100) | 96.8 (96.8) |
| Refinement | ||
| Resolution (Å) | 44.3–3.2 | 20–2.6 |
| No. of reflections | 8815 | 109,594 |
| Rwork (Rfree)b (%) | 19.4 (29.0) | 26.3 (34.1) |
| No. of Fabs per asymmetric unit | 1 | 8 |
| No. of atoms: protein, sulfates, water | 3290, 5, 0 | 26395, 0, 0 |
| Ave. B-factor(Å2): protein, sulfates, water | 18.3, 41.8, 0 | 48.3, 0, 0 |
| R.m.s deviations: bond (Å), bond angles (°) | 0.023, 2.38 | 0.013, 1.16 |
| Ramachandron plot: allowed, generously allowed, not allowed (%) | 98.1, 1.1, 0.8 | 94.9, 2.7, 2.4 |
The value in parentheses is for the highest resolution bin (approximate interval, 0.1 Å), Rsym = Σ|Ihkl − 〈Ihkl〉|/ΣIhkl.
Rwork = ΣIhkl||Fo − |Fc||/ ΣIhkl|Fo| for all data except 5% which is used for the Rfree calculation.
Figure 5.
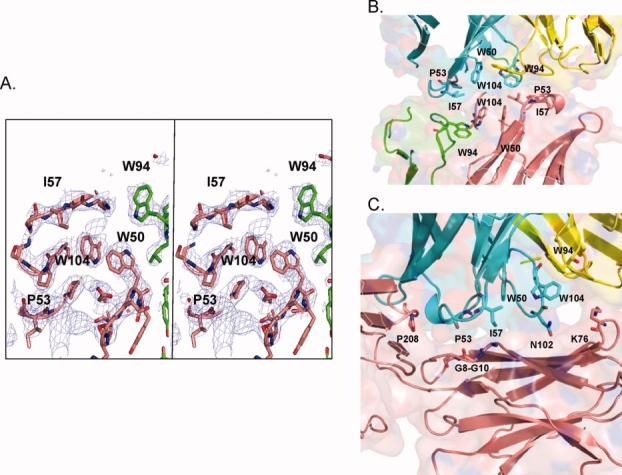
Identification of intermolecular CDR contacts in the IgG2 Fab2 crystal structure. The eight Fabs in the asymmetric unit of the Fab2 structure, designated A/B, C/D, E/F, G/H, I/J, K/L, M/N, and O/P (LC/HC), displayed two types of packing interactions. (A) Stereo electron density at 1 sigma of the cluster of hydrophobic CDR residues that formed intermolecular contacts. (B) Crystal packing of CDR–CDR contacts of Fab A/B with Fab E/F, highlighting residues considered for targeted mutagenesis. HC residues I57, W104, P53, and W94 are in pink and blue, and LC W94 in green and yellow. (C) Crystal packing of the CDR-framework interactions of the J chain CDRs (blue) of Fab I/J with F chain (pink) framework residues G8-G10 and K76 of Fab E/F.
The crystal structure of the IgG1 Fab was solved to 3.2 Å by molecular replacement (Table III, Fig. 6). Overall the IgG1 Fab and IgG2 Fab2 structures were very similar. The IgG1 structure differs from both of the IgG2 structures as its elbow angle is much smaller than either of the IgG2 subtypes, although it more closely resembles the A/B subtype (data not shown). As a result, the packing of side chains at the interface such as W94 of the LC and W104 of the HC shift significantly between the two subtypes. Crystal contacts in the IgG1 structure were different than those detected in the IgG2 structure.
Figure 6.

Crystal structure of the Li33IgG1 Fab. (A) Stereo electron density at 1 sigma of the cluster of hydrophobic CDR residues that formed intermolecular contacts. (B) Mapping of the four asparagine attachment sites for N-glycosylation (spheres) that were engineered in the CH1 domain by targeted mutagenesis on Fab HC (pink). (C) Crystal packing showing the location of the engineered glycosylation sites on the CH1 domain (pink) and their proximity to other Fabs in the structure (gray). Distances from closest crystal contacts (indicated by dashed lines) are denoted.
Improving solubility of Li33 by targeted mutagenesis
The structural data provided an opportunity to use rational design to solve the solubility issue. Two different strategies were used. First, site-directed mutagenesis of the hydrophobic CDR residues was used to directly test the involvement of these residues on aggregation [Fig. 5(B,C)]. For the CDR mutagenesis work, W94, W104, I57, and P54 were replaced with less hydrophobic amino acids. W50 was not targeted because it formed intra-Fab HC/LC contacts that presumably stabilized the Fab. All P54 substitutions significantly impacted LINGO-1 binding and were not further evaluated. For the W94, I57, and W104 substitutions, functional data from expressed constructs with single point mutations were used to design double and triple mutants. Selected results from the targeted CDR mutagenesis strategy are shown in Table IV. The IgG1agly W94V/I57-V,-S,-P, and -T double mutants all had improved solubility with no impact on LINGO-1 binding, stability, or on the level of aggregation detected by SEC. The W104Q/W94V/I57-S and -A triple mutants also had improved solubility with no impact on stability or on the level of aggregation detected by SEC, but the EC50 for LINGO-1 binding was reduced to 2–3 nM (Table IV). Characterization of the triple mutants by AUC (Table II) revealed very low dimer levels at concentrations that promoted reversible dimer formation with the IgG2wt Mab. Limiting amounts of the antibodies prevented us from testing them at higher concentrations. These studies confirm that the hydrophobic CDR residues identified as crystallization contact points directly contribute to the aggregation seen with the Li33 Mab.
Table IV.
Impact of Targeted Mutagenesis on Li33 Solubility
| Stability (°C) |
|||||
|---|---|---|---|---|---|
| Li33 Isotype | Solubilitya (mg/mL) | LINGO-1 binding, EC50 (nM) | TM1 | TM2 | SEC, % monomer |
| CDR targeted mutagenesis | |||||
| IgG1agly | 0.3 | 0.12 | 60 | 77 | >99 |
| IgG1aglyW94V/I57V | >10 | 0.15 | 59 | 74/82 | 99 |
| IgG1aglyW94V/I57S | >7 | 0.38 | 58 | 75/82 | 99 |
| IgG1aglyW94V/I57P | >8 | 0.07 | 58 | 75/83 | 99 |
| IgG1aglyW94V/I57T | >7 | 0.13 | 59 | 75/83 | 99 |
| IgG2wt | >50 | 0.23 | 69 | 76 | 96 |
| IgG2agly | 0.3 | 0.26 | 59 | 76 | 98 |
| IgG2-V234A/G237A | 5.6 | 0.19 | 69 | 76 | 95 |
| +W94V/W104Q/I57S | >50 | 2.9 | 67 | 74 | 95 |
| +W94V/W104Q/I57A | >50 | 2.4 | 69 | 73 | 95 |
| Glycosylation site insertion mutants | |||||
| IgG1wt | 0.9 | 0.12 | 69 | 76 | 99 |
| IgG1wt A120N | >50 | 0.10 | 69 | 75 | 95 |
| IgG1wt Q177N | 4.9 | 0.15 | 69 | 75 | 96 |
| IgG1wt T197N | >50 | 0.12 | 69 | 75 | 95 |
| IgG1wt K212S | 3.7 | 0.12 | 69 | 75 | 85 |
Samples were characterized for the limit in their solubility at 4°C, apparent binding affinity for LINGO-1, thermal stability, and aggregation state by SEC. All samples were analyzed at pH 7.0 in PBS.
Protocol for assessing solubility is described in the Materials And Methods section.
Second, N-glycosylation sites were inserted into the Li33 IgG1 CH1 framework at positions where the added glycan could potentially disrupt aggregation through steric effects (Fig. 6). The use of engineered glycans to improve solubility as well as other protein attributes is well documented.21–23N-glycosylation sites were incorporated into surface residues where NXS/T motifs could be introduced by single amino acid substitution and the added glycans would project away from the CDRs in orthogonal directions where they each could disrupt different contact surfaces. The four residues targeted, A120N, Q177N, T197N, and K212S, are mapped on the Li33 IgG1 Fab structure in Figure 6. The added glycans all produced a decrease in mobility of the HC by SDS–PAGE and an acidic shift in the Mab pI values by IEF relative to the mobility of the parent IgG1wt Mab. Characterization data for the mutants are summarized in Table IV. Binding affinity and protein stability were not affected by the mutations. The A120N and T197N mutations resulted in proteins with excellent solubility. By AUC, the preparations were largely monomer with low levels of soluble aggregates that were irreversible (Table II). The other two mutations produced only slight improvements in solubility. The K212S mutant protein was least soluble and showed substantial levels of aggregation by SEC. The sites with the greatest improvement in solubility correlated with their proximity to crystal contacts in the IgG1 structure [Fig. 6(C)]. Because the glycoengineering approach targets framework residues, it can be applied to any antibody though the specific site(s) that enable improved solubility could differ in different antibodies.
Discussion
We have applied a variety of biochemical, biophysical, and molecular engineering approaches to understand factors that contributed to the complex solubility properties of the Li33 Mabs. Two different aggregation pathways were identified. One pathway led to formation of insoluble aggregates where the protein irreversibly precipitated. Samples that underwent irreversible aggregation typically had a solubility limit of <1 mg/mL. For the purpose of our analyses we focused on the IgG1wt Mab as a prototypic example of this type of behavior. The other pathway led to formation of soluble aggregates that were reversible following sample dilution. The prominent aggregate intermediate observed in samples that formed soluble aggregate appeared to be a dimer. The IgG2wt Mab was utilized to understand factors that led to this behavior. For samples that formed soluble aggregates a solubility limit was not achieved and most samples were soluble at >50 mg/mL. Many unrelated factors contributed to aggregation of the Li33 Mabs. For example, removal of the glycan from the IgG2wt Mab caused a >250-fold drop in solubility and the formation of insoluble aggregates, while reduction of the IgG1wt Mab increased solubility by >50-fold and the formation of soluble aggregates. Thus, the types of aggregates that formed could be switched by simple sample manipulations.
The presence of soluble aggregates in the IgG1 Fab indicated that a key determinant leading to aggregation lies within the Fab. The most unusual feature in the Li33 Fab2 structure was a patch of hydrophobic CDR residues that formed CDR–CDR and CDR-framework contacts (Fig. 5). The large number of hydrophobic residues found in the Li33 variable sequences is abnormal for an antibody and could provide an early nucleation step leading to aggregation. To test this possibility, we produced and characterized a large panel of mutants targeting these sites. Replacement of the hydrophobic CDR residues with less hydrophobic amino acids eliminated the aggregation seen under physiological conditions with the parent constructs, verifying their involvement in the aggregation of the Li33 Mab.
The pH 5.0 formulation provided a simple solution to resolve the solubility and aggregation issues seen with the Li33 Mabs. These effects seem to arise from contacts within the Fab, as the IgG1 Fab was also less aggregated at pH 5.0. The sharp transition at pH 5.5 is indicative of the titration of the imidazole proton on a histidine (pKa = 6.0). H101 in CDR3 of the HC was of particular interest because it formed a H101–H101 contact of CDRs and with framework in the IgG2 Fab2 structure. H101 is in close proximity to the hydrophobic patch targeted by CDR mutagenesis and in its unprotonated state could provide additional hydrophobic contacts. As the pH is dropped and H101 becomes protonated, the added charges could destabilize the hydrophobic lattice. Thus, the impact of pH on aggregation may be analogous to the effect of the CDR mutagenesis approach in destabilizing intermolecular hydrophobic interactions. Attempts to study H101 by targeted mutagenesis failed because constructs carrying H101 mutations expressed poorly and could not be evaluated.
The difference in the solubility of the IgG1wt, IgG2wt, and IgG4wt Mabs implies that factors outside of the Fab also contribute to aggregation. Since the solubility of IgG1 and IgG2 Fab2 fragments differed by >150-fold, another key determinant in solubility presumably lies within the hinge/interchain disulfide networks that make up the Fab2 portion of the constructs. One structural difference between IgG1 and the other frameworks is the positioning of the interchain disulfides that connect the HC and LC. The IgG1 antibodies utilize cysteine-5 in the HC whereas IgG2 and IgG4 frameworks utilize cysteine-3. When the IgG1wt and agly samples were treated with reductant to break interchain disulfides, they were now soluble, supporting that structural constraints within the IgG1 framework lead to its poor solubility. Alternatively, the heterogeneity of the wt IgG2 might contribute to its increased solubility. Consistent with published data for IgG2 Mabs,5 the Li33 IgG2wt Mab contains four interchain disulfide variants (data not shown). Like most Mabs, the IgG2wt Mab also contains over a dozen glycoforms that are predominated by core biantennary structures. The disulfide and glycan heterogeneity could prevent the formation of ordered structures that lead to precipitation. Interestingly, when the eight Fabs in the IgG2 Fab2 crystal structure were superimposed, the relative orientation of the light and HCs fell into two classes that correlated with the types of CDR contacts made by the two different subtypes. In the A/B subtype, the HC rotates relative to the LC to provide a more acute elbow angle than in the O/P subtype. More extensive evaluation of the crystal data is needed to assess if these differences are caused by the disulfide heterogeneity of the IgG2 hinge or if they are caused by the crystallization procedure.
The reduced solubility of aglycosyl constructs implies that other interactions can also contribute to the aggregation seen with the Li33 Mabs. Since successful application of aglycosyl versions of antibodies is well documented,3,6 the poor solubility characteristics of the Li33 aglycosyl versions are likely augmented by issues associated with the Li33 Fab. The success of the glycosylation site engineering strategy at preventing aggregation of the Li33 Mab, provides a clear demonstration of how added glycans can sterically block protein contacts from forming.
In summary, we have investigated a number of approaches to alter the physicochemical properties of the Li33 Mab to eliminate its tendency to self-associate. Many variables can impact the multiple aggregation pathways a protein can take. Some of these approaches can be additive, have no effect, or can lead to counterproductive outcomes. Such complex behavior highlights the difficult nature in addressing this general problem. Furthermore, we show care needs to be taken in using simple absorbance and SEC measurements to assess what is happening in a protein sample in terms of its solution properties, especially as it relates to solubility and aggregation. For the Li33 Mabs, CDR–CDR and CDR-framework interactions revealed by the Li33 Fab crystal structure appear to be the major pathway that leads to the poor solubility of the Li33 IgG1 Mab. Mutations at these sites improve solubility. Alternatively, these interactions could be masked by insertion of glycosylation sites in the CH1 domain near the CDR-framework junction. Most surprising was the impact of isotype switching as different frameworks displayed a >250-fold range in solubility. In terms of the latter we believe spatial/steric factors may play an important role facilitating or limiting protein interactions that lead to self-association. The strategies we developed and discussed in this article to improve the solubility and reduce aggregation of the Li33 Mab should be readily applicable to other proteins with unacceptable biochemical properties.
Methods
Molecular Cloning of the Li33 Mabs
The HC and LC variable regions of the Li33 antibody were isolated utilizing the semi-synthetic human antibody Fab phage display library technology from Dyax (Cambridge, MA). Native human kappa LC and HC signal peptides were used to direct secretion of Li33 light and HCs, respectively in mammalian cell hosts. The variable domain fragment of the LC was subcloned into a shuttle vector containing the intact signal peptide and LC kappa chain constant regions. The variable domain fragment of the HC was subcloned into shuttle vectors containing the intact signal peptide and IgG1wt, IgG1agly, IgG4wt, IgG4agly, IgG2wt, IgG2agly, and IgG2 V234A/G237A HC constant region. Point mutations were generated by site directed mutagenesis. Aglycosyl constructs all utilized a threonine to alanine mutation in the conserved CH2 domain NST glycosylation motif (T297A in the IgG2agly Mab). The mature protein sequence for the Li33IgG2 construct is shown below. CDR sequences are underlined.
Light chain
DIQMTQSPGTLSLSPGERATLSCRASQSVSSYLAWYQQKPGQAPRLLIYDASNRATGIPARFSGSGSGTEFTLTISSLQSEDFAVYYCQQYDKWPLTFGGGTKVEIKRTVAAPSVFIFPPSDEQLKSGTASVVCLLNNFYPREAKVQWKVDNALQSGNSQESVTEQDSKDSTYSLSSTLTLSKADYEKHKVYACEVTHQGLSSPVTKSFNRGEC
Heavy chain
EVQLLESGGGLVQPGGSLRLSCAASGFTFSIYPMFWVRQAPGKGLEWVSWIGPSGGITKYADSVKGRFTISRDNSKNTLYLQMNSLRAEDTATYYCAREGHNDWYFDLWGRGTLVTVSSASTKGPSVFPLAP CSRSTSESTAALGCLVKDYFPEPVTVSWNSGALTSGVHTFPAVLQSSGLYSLSSVVTVPSSNFGTQTYTCNVDHKPSNTKVDKTVERKCCVECPPCPAPPVAGPS VFLFPPKPKDTLMISRTPEVTCVVVDVSHEDPEVQFNWYVDGVEVHNAKTKPREEQFNSTFRVVSVLTVVHQDWLNGKEYKCKVSNKGLPAPIEKTISKTKGQPREPQVYTLPPSREEMTKNQVSLTCLVKGFYPSDIAVEWESNGQPENNYKTTPPMLDSDGSFLYSKLTVDKSRWQQGNVFSCSVMHEALHNHYTQKSLSLSPGK
Purification
Li33 Mabs were expressed in Chinese hamster ovary cells and purified from clarified and filtered culture supernatants on recombinant Protein A Sepharose Fast Flow (GE Heathcare) at 10 mg Mab/mL resin. The protein content of the eluted samples was estimated from absorbance spectra. Due to the limited solubility of Li33IgG1wt and agly samples, they were stored in the 10 mM citrate pH 4.7, 25 mM NaCl buffer. All other samples were dialyzed into 20 mM sodium phosphate pH 7.0, 150 mM NaCl (PBS). Samples were filtered through 0.22 Mμ units, aliquoted and stored at −70°C. Reduced Li33IgG1wt and agly preparations were generated by incubating samples with 1 mM Tris(2-carboxyethyl)phosphine (TCEP) for 60 min at 37°C in PBS.
IgG1 and IgG2 Fab2 fragments were generated with pepsin at an enzyme:protein ratio of 1:100. Samples in 10 mM sodium acetate pH 3.6 were incubated at 37°C for 6 h for IgG2 or 4 h IgG1 for complete conversion of the Mab to Fab2. Fab2 fragments were purified on Protein A Sepharose followed by chromatography on a Fractogel EMD SO4 column.
IgG1 Fab was generated with papain at an enzyme:Mab ratio of 1:100. Samples in 10 mM Na2HPO4 pH 7.5, 5 mM EDTA pH 7.5, 20 mM cysteine–HCl, 20 mM NaOH were incubated at 37°C for 2.5 h and quenched with 35 μg/mL E64. Fab fragments were purified on Protein A Sepharose followed by chromatography on a Fractogel EMD SO4 column.
SDS–polyacrylamide gel electrophoresis
Samples were subjected to SDS–PAGE on 4–20% tris-glycine gradient gels from Invitrogen. Nonreduced samples were treated with 5 mMN-ethyl maleimide for 5 min at room temperature, diluted with Laemmli nonreducing sample buffer and heated at 95°C for 2 min prior to analysis. Reduced samples were treated with sample buffer containing 2% 2-mercaptoethanol and heated.
Solubility studies
Samples were concentrated in Amicon YM30 centrifugal filter devices until visible precipitates formed or a predefined maximum concentration was achieved. Protein concentrations were determined from absorbance spectra taken immediately after concentration and then after 5 days at 4°C. Prior to each absorbance measurement, samples were filtered through 0.45 μm membranes. If the absorbance decreased during storage, the samples were further incubated at 4°C until a solubility limit had been achieved. When possible samples were concentrated to 50 mg/mL; however, due to the small sample size of some of the purified constructs they were only concentrated to the amount indicated.
Characterization by SEC
Samples (100 μg in 300 μL of column buffer) were subjected to SEC at room temperature on a Superdex 200 HR10/30 FPLC column (GE Heathcare) using PBS as the mobile phase. The column was run at 0.3 mL/min. The column effluent was monitored by UV detection at 280 nm and purity assessed by peak height.
Analytical ultracentrifugation
Sedimentation velocity experiments were run at 20°C, 40,000 or 50,000 rpm in a Beckman Optima XL-I ultracentrifuge1 (Beckman, Palo Alto, CA), using an An50 Ti eight-hole rotor. Data acquisition was conducted using the UV-Vis absorbance detection system and/or the Rayleigh interferometer on the ultracentrifuge with centrifuge cells containing either double sector 3 or 12 mm charcoal-filled Epon centerpieces and sapphire windows. Sedimentation coefficient distribution information, c(s), was obtained using the SEDFIT program with a confidence level of 0.68.19,24
Isoelectric focusing
Elecrophoresis was carried out on a pH 3–10 IEF minigel from invitrogen at 100 V for 1 h, 200 V for 1 h, and 500 V for 30 min.
Differential scanning fluorimetry
Protein unfolding was monitored by differential scanning fluorimetry. Measurements were conducted on an Mx3005p real-time PCR system (Agilent Technologies) in a 96-well format using 10 μg of protein in 50 μL PBS (at pH 7.0) supplemented with SYPRO orange fluorophor (Invitrogen) at a final concentration of 10×. Samples were heated from 25°C to 95°C at 1°C/min with fluorescence intensity measured three times every 1°C. Fluorescence intensities were plotted as a function of temperature. Tm were derived from these curves by taking the negative derivative (“-R′(T)” in the Mx3005p software) and selecting the local minima of the derivative plots. For pH studies, 10 mM sodium citrate was used as the buffering agent.
Analysis of function by direct binding ELISA
ELISA plates were coated with Lingo-1-Fc, blocked, and incubated with a threefold dilution series of each test compound starting at 10 μg/mL (eight dilutions). Bound Mab was detected using AP-anti-human Fab using 4-nitrophenyl phosphate AP substrate. Plates were read at 405 nm on a Molecular Devices plate reader. EC50 values of binding were calculated from the titration curve using prism software.
Crystallization of the Li33IgG1 Fab
IgG1 Fab at 5 mg/mL was mixed at a volumetric ratio of 1:1 with a reservoir solution consisting of 2 M ammonium sulfate, 0.1 M sodium acetate pH 3.5 and 0.1 M TCEP. Football-shaped crystals were grown by vapor diffusion at 20°C and were cryoprotected by transferring them into 2 M ammonium sulfate, 0.1 M citrate pH 3.5, 20% glycerol, 10% sucrose, 10% xylitol for 2 min and then frozen by a quick transfer into liquid nitrogen. The crystals diffracted to 3.2 Å at the SGXcat beamline at the Advanced Photon Source (Argonne, IL). Data were processed with the HKL program package v.1.97.25 The crystal structure was solved by molecular replacement based on another IgG1 homology model (AQC2 mutant Fab PDBID: 2B2X) in PHASER26 with all possible arrangements of the screw access leading to a clear solution in space group P6(5)22. Model building of the single Fab and four sulfates in Coot 0.5.227 followed by refinement using Refmac528 to 3.2 Å resolution resulted in a final R-factor of 19.3% and Rfree of 28.9% with reasonable geometry. The structure has been deposited in the RCSB with PDB code:3KYK.
Crystallization of the Li33IgG2 Fab2
IgG2 Fab2 at 7.2 mg/mL was mixed at a volumetric ratio of 1:1 with a reservoir solution consisting of 12%Peg3350, 0.1 M phosphate citrate pH 4, 0.2 M NaCl. Rod-shaped crystals grown by vapor diffusion at 20°C and were cryoprotected by transferring them into 20%Peg3350, 0.1 M phosphate citrate pH 4, 0.2 M NaCl, 15% glycerol for 2 min and then frozen by a quick transfer into liquid nitrogen. The crystals of the IgG2 Fab2 diffracted to 2.8 Å at the SGXcat beamline at the Advanced Photon Source. Data were processed with the HKL program package v.1.97.25 The crystal structure was solved by molecular replacement based on another IgG2 homology model (3GIZ) in PHASER.26 Model building of the eight Fabs in Coot 0.5.227 followed by refinement using Refmac528 to 2.6 Å resolution resulted in a final R-factor of 26% and R free of 34% with reasonable geometry. The structure has been deposited in the RCSB with PDB code:3KYM.
Acknowledgments
We wish to thank Greg Thill, Joe Amatucci, Courtney Kirouac, and Shelly Martin for cell line selection and generating conditioned medium; Sheng Gu, Dingyi Wen, Craig Wildes, and Monika Vecchi for analytical biochemistry; and Brian Miller and Scott Glaser for initial screening for W94, I57, W104, and P53 point mutations.
Glossary
Abbreviations:
- AUC
analytical ultracentrifugation
- CDR
complementarity determining region
- HC
heavy chain
- Ig
immunoglobulin
- LC
light chain
- Mab
monoclonal antibody
- SEC
size exclusion chromatography
- TCEP
Tris(2-carboxyethyl)phosphine
- Tm
melting temperature.
References
- 1.Aires da Silva F, Corte-Real S, Goncalves J. Recombinant antibodies as therapeutic agents: pathways for modeling new biodrugs. BioDrugs. 2008;22:301–314. doi: 10.2165/00063030-200822050-00003. [DOI] [PubMed] [Google Scholar]
- 2.Salfeld JG. Isotype selection in antibody engineering. Nat Biotechnol. 2007;25:1369–1372. doi: 10.1038/nbt1207-1369. [DOI] [PubMed] [Google Scholar]
- 3.Labrijn AF, Aalberse RC, Schuurman J. When binding is enough: nonactivating antibody formats. Curr Opin Immunol. 2008;20:479–485. doi: 10.1016/j.coi.2008.05.010. [DOI] [PubMed] [Google Scholar]
- 4.Allen MJ, Guo A, Martinez T, Han M, Flynn GC, Wypych J, Liu YD, Shen WD, Dillon TM, Vezina C, Balland A. Interchain disulfide bonding in human IgG2 antibodies probed by site-directed mutagenesis. Biochemistry. 2009;48:3755–3766. doi: 10.1021/bi8022174. [DOI] [PubMed] [Google Scholar]
- 5.Dillon TM, Ricci MS, Vezina C, Flynn GC, Liu YD, Rehder DS, Plant M, Henkle B, Li Y, Deechongkit S, Varnum B, Wypych J, Balland A, Bondarenko PV. Structural and functional characterization of disulfide isoforms of the human IgG2 subclass. J Biol Chem. 2008;283:16206–16215. doi: 10.1074/jbc.M709988200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrant JL, Benjamin CD, Cutler AH, Kalled SL, Hsu YM, Garber EA, Hess DM, Shapiro RI, Kenyon NS, Harlan DM, Kirk AD, Burkly LC, Taylor FR. The contribution of Fc effector mechanisms in the efficacy of anti-CD154 immunotherapy depends on the nature of the immune challenge. Int Immunol. 2004;16:1583–1594. doi: 10.1093/intimm/dxh162. [DOI] [PubMed] [Google Scholar]
- 7.Van der Neut Kolfschoten M, Schuurman J, Losen M, Bleeker WK, Martínez-Martínez P, Vermeulen E, den Bleker TH, Wiegman L, Vink T, Aarden LA, De Baets MH, van de Winkel JG, Aalberse RC, Parren PW. Anti-inflammatory activity of human IgG4 antibodies by dynamic Fab arm exchange. Science. 2007;317:1554–1557. doi: 10.1126/science.1144603. [DOI] [PubMed] [Google Scholar]
- 8.Aalberse RC, Schuurman J. IgG4 breaking the rules. Immunology. 2002;105:9–19. doi: 10.1046/j.0019-2805.2001.01341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garber E, Demarest SJ. A broad range of Fab stabilities within a host of therapeutic IgGs. Biochem Biophys Res Commun. 2007;355:751–757. doi: 10.1016/j.bbrc.2007.02.042. [DOI] [PubMed] [Google Scholar]
- 10.Shire SJ, Shahrokh Z, Liu J. Challenges in the development of high protein concentration formulations. J Pharm Sci. 2004;93:1390–1402. doi: 10.1002/jps.20079. [DOI] [PubMed] [Google Scholar]
- 11.Chennamsetty N, Voynov V, Kayser V, Helk B, Trout BL. Design of therapeutic proteins with enhanced stability. Proc Natl Acad Sci USA. 2009;106:11937–11942. doi: 10.1073/pnas.0904191106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Philo JS. A critical review of methods for size characterization of non-particulate protein aggregates. Curr Pharm Biotechnol. 2009;10:359–372. doi: 10.2174/138920109788488815. [DOI] [PubMed] [Google Scholar]
- 13.Tessier PM, Jinkoji J, Cheng YC, Prentice JL, Lenhoff AM. Self-interaction nanoparticle spectroscopy: a nanoparticle-based protein interaction assay. J Am Chem Soc. 2008;130:3106–3112. doi: 10.1021/ja077624q. [DOI] [PubMed] [Google Scholar]
- 14.Mi S, Miller RH, Lee X, Scott ML, Shulag-Morskaya S, Shao Z, Chang J, Thill G, Levesque M, Zhang M, Hession C, Sah D, Trapp B, He Z, Jung V, McCoy JM, Pepinsky RB. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci. 2005;8:745–751. doi: 10.1038/nn1460. [DOI] [PubMed] [Google Scholar]
- 15.Mi S, Hu B, Hahm K, Luo Y, Kam Hui ES, Yuan Q, Wong WM, Wang L, Su H, Chu TH, Guo J, Zhang W, So KF, Pepinsky B, Shao Z, Graff C, Garber E, Jung V, Wu EX, Wu W. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat Med. 2007;13:1228–1233. doi: 10.1038/nm1664. [DOI] [PubMed] [Google Scholar]
- 16.Mi S, Miller RH, Tang W, Lee X, Hu B, Wu W, Zhang Y, Shields CB, Zhang Y, Miklasz S, Shea D, Mason J, Franklin RJ, Ji B, Shao Z, Chédotal A, Bernard F, Roulois A, Xu J, Jung V, Pepinsky B. Promotion of central nervous system remyelination by induced differentiation of oligodendrocyte precursor cells. Ann Neurol. 2009;65:304–315. doi: 10.1002/ana.21581. [DOI] [PubMed] [Google Scholar]
- 17.Cole MS, Anasetti C, Tso JY. Human IgG2 variants of chimeric anti-CD3 are nonmitogenic to T cells. J Immunol. 1997;159:3613–3621. [PubMed] [Google Scholar]
- 18.Taylor FR, Prentice HL, Garber EA, Fajardo HA, Vasilyeva E, Pepinsky RB. Suppression of sodium dodecyl sulfate-polyacrylamide gel electrophoresis sample preparation artifacts for analysis of IgG4 half-antibody. Anal Biochem. 2006;353:204–208. doi: 10.1016/j.ab.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 19.Dam J, Schuck P. Calculating sedimentation coefficient distributions by direct modeling of sedimentation velocity concentration profiles. Methods Enzymol. 2005;384:185–212. doi: 10.1016/S0076-6879(04)84012-6. [DOI] [PubMed] [Google Scholar]
- 20.Berkowitz SA. Role of analytical ultracentrifugation in assessing the aggregation of protein biopharmaceuticals. AAPS J. 2006;8:E590–E605. doi: 10.1208/aapsj080368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Song Y, Azakami H, Hamasu M, Kato A. In vivo glycosylation suppresses the aggregation of amyloidogenic hen egg white lysozymes expressed in yeast. FEBS Lett. 2001;491:63–66. doi: 10.1016/s0014-5793(01)02151-2. [DOI] [PubMed] [Google Scholar]
- 22.Sinclair AM, Elliott S. Glycoengineering: the effect of glycosylation on the properties of therapeutic proteins. J Pharm Sci. 2005;94:1626–1635. doi: 10.1002/jps.20319. [DOI] [PubMed] [Google Scholar]
- 23.Qu Z, Sharkey RM, Hansen HJ, Shih LB, Govindan SV, Shen J, Goldenberg DM, Leung SO. Carbohydrates engineered at antibody constant domains can be used for site-specific conjugation of drugs and chelates. J Immunol Methods. 1998;213:131–144. doi: 10.1016/s0022-1759(97)00192-0. [DOI] [PubMed] [Google Scholar]
- 24.Schuck P, Perugini MA, Gonzales NR, Howlett GJ, Schubert D. Size-distribution analysis of proteins by analytical ultracentrifugation: strategies and application to model systems. Biophysical J. 2002;82:1096–1111. doi: 10.1016/S0006-3495(02)75469-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meth Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 26.McCoy AJ. Solving structures of protein complexes by molecular replacement with phaser. Acta Crystallogr D Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. Refmac5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 28.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]