

Abstract
Function studies of many proteins are waited to develop after genome sequencing. High-throughout technology of gene cloning will strongly promote proteins' function studies. Here we describe a ligation-independent cloning (LIC) method, which is based on the amplification of target gene and linear vector by PCR using phosphorothioate-modified primers and the digestion of PCR products by λ exonuclease. The phosphorothioate inhibits the digestion and results in the generation of 3′ overhangs, which are designed to form complementary double-stranded DNA between target gene and linear vector. We compared our phosphorothioate primer cloning methods with several LIC methods, including dU primer cloning, hybridization cloning, T4 DNA polymerase cloning, and in vivo recombination cloning. The cloning efficiency of these LIC methods are as follows: phosphorothioate primer cloning > dU primer cloning > hybridization cloning > T4 DNA polymerase cloning >> in vivo recombination cloning. Our result shows that the 3′ overhangs is a better cohesive end for LIC than 5′ overhang and the existence of 5′phosphate promotes DNA repair in Escherichia coli, resulting in the improvement of cloning efficiency of LIC. We succeeded in constructing 156 expression plasmids of Aeropyrum pernix genes within a week using our method.
Keywords: ligation-independent cloning, 5′ phosphate group, phosphorothioate-modified primer, cloning efficiency, Aeropyrum pernix
Introduction
Since the invention of gene cloning, many efforts have been made to improve this technique. To date, these cloning techniques include the restriction enzyme cloning, 1 blunt-end cloning,2 TA cloning,3 classical ligation-independent cloning (LIC),4–11 hybridization cloning,12,13 and in vivo cloning.14–16 In fact, the last two methods are also LIC because they are ligase-free cloning techniques. All these methods require the specific enzymatic treatment of target gene and vector,1,5–9 the use of primers carrying modified bases,4,9–11 or some specific bacterial strains,14–16 except for hybridization cloning. Among them, the LIC being easy to carry out can be easily adopted for high-throughout cloning. LIC is based on the long 3′ or 5′ overhangs of target gene and vector. The two overhangs of insert are complementary with those of the linear vector; thus, resulting in circular recombinant plasmid containing four nicks is formed. This damaged recombinant is fully repaired to integrity by host DNA repair system after transforming into Escherichia coli competent cell.
As a potential high-throughout cloning strategy, many LIC methods were developed. The early LIC was based on the 3′ exonuclease or T4 DNA polymerase. 5–7 However, both enzymes had some limitation on the selection of inserting sites of target gene. Nisson et al. developed a new LIC method, using the dU-carrying primer, uracil-DNA glycosylase, and Taq DNA polymerase.9 This strategy makes it easy to select the inserting site of target gene. However, due to its low fidelity, the Taq DNA is not suitable for construction of expression plasmid. Subsequently, LIC methods based on the ribonucleotide-modified primer were developed.10,11 The ribonucleotide prevents DNA extension by some DNA polymerases such as Vent DNA polymerase and results in the PCR fragments with 5′ overhang. On the other hand, ribonucleotide has no effect on the DNA extension by other DNA polymerases such as Taq DNA polymerase and results in the blunt PCR fragments which can be cleaved at the position of ribonucleotide by treatment of weak alkali or ribonuclease HII. However, the ribonucleotide-carrying primer is sensitive to RNase A which leads to cloning failure. Here we report a novel LIC strategy based on phosphorothioate primer and λ exonuclease. Our method had the trait of high-cloning efficiency, simple operation, and high reproducibility.
Results
Phosphorothioate primer and λ exonuclease dependent cloning
The schematic presentation of our method was shown in Figure 1. The phosphorothioate in PCR fragments counteracts the 5′-processive digestion of DNA by λ exonuclease. This results in complementary long 3′ overhangs which are complementary between insert and vector. The annealed insert and vector (circular recombinant plasmid with four nicks) was transformed into E. coli and repaired by the bacterial DNA repair system. Theoretically, all target's PCR products should be cloneable, in the presence of at least equimolar linear vector.
Figure 1.
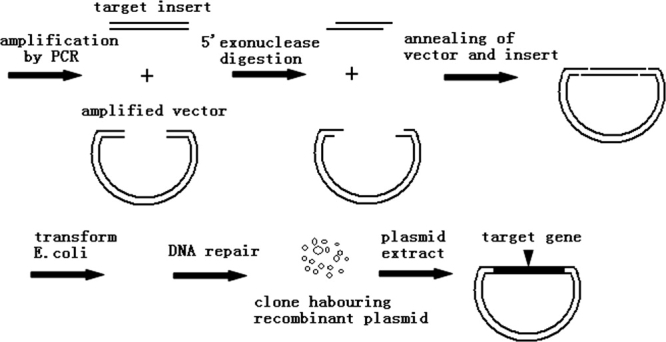
Cartoon of the phosphorothioate primer cloning of PCR fragments.
We tested the feasibility of our LIC via the insertion of a gene from Aeropyrum pernix (ape_0119) into pUC18 vector. After digestion with λ exonuclease (about 250 ng exonuclease/pmol DNA), the PCR fragments (4 ng ape_0119 target and 10 ng linear vector, molar ratio ≈ 1) amplified by the 5′-phosphorylated phosphorothioate primers (Table I) produced 82 recombinant plasmids with insert (Table II). However, the same PCR fragments without phosphorothioate did not generate any clone. The PCR fragments with phosphorothioate and 5′OH only generated two clones. As expected, the DNA mixtures without digestion by λ exonuclease did not generate clones after transformation. As shown in Table II, the transformation efficiency was up to 8.2 × 103/μg pUC18 PCR product using standard E. coli competent cells (2.3 × 105/μg circular pUC18 vector), with 95% of the screened colonies containing the correct ape_0119 insert. Four clones obtained from the transformation of the exonuclease-digested linear vector were all blank plasmid without insert. These blank plasmids could possibly be due to self-annealed linear vectors, which were repaired by the bacterial DNA repair system after transformation. Therefore, our highly efficient LIC method depends on the 5′-phosphorylated phosphorothioate primer and λ exonuclease.
Table I.
Sequences of Primers Used in This Study
| Primer | DNA sequencea |
|---|---|
| Ape_0119-F | 5′AAGCTTTAGTTACTA-TGGGTGAGGACAAGAGGGA3′ |
| Ape_0119-R | 5′AACAGCTATGACCTC-TACTCTGGATCCCTTATACC3′ |
| pUC18-F | 5′AGTAACTAAAGCTTG-GCACTGGCCGT3′ |
| pUC18-R | 5′AGGTCATAGCTGTTT-CCTGTG3′ |
| adaptor-F | 5′TCAACAAGTTTGTAC-A-T-G3′ |
| adaptor-R | 5′TACAAGAAAGCTGAA-T-T-A3′ |
| pDEST17-F | 5′TACAAACTTGTTGAT-T-C-GAGG3′ |
| pDEST17-R | 5′TCAGCTTTCTTGTAC-A-A-AGTG3′ |
Denotes the phosphorothioate modification.
Table II.
Effects of Different Parameters on Phosphorothioate Primer Cloninga
| Modification of PCR fragments | Combination of DNA | Exonuclease treatment | Number of recombinants |
|---|---|---|---|
| 5′ P + phosphorothioate | Vector + insert | + | 82 |
| 5′ P + phosphorothioate | Vector + insert | − | 0 |
| 5′ P − phosphorothioate | Vector + insert | + | 0 |
| 5′ OH + phosphorothioate | Vector + insert | + | 2 |
| 5′ OH − phosphorothioate | Vector + insert | + | 0 |
| 5′ P + phosphorothioate | Vector | + | 4 |
| 5′ P + phosphorothioate | Vector | − | 0 |
| 5′ P + phosphorothioate | Insert | + | 0 |
| 5′ P + phosphorothioate | Insert | − | 0 |
Mixtures of equimolar PCR fragments (10 ng pUC18 and 4 ng ape_0119) or respective target/vector fragments were incubated with λ exnuclease (250 ng exonuclease/pmol DNA) at 37°C for 5 min before transforming into E. coli.
Optimal parameters
Although more recombinant clones were obtained in our method, the parameter should be optimized to achieve the highest cloning efficiency. These parameters include the digestion time, the amounts of λ exonuclease, the ratio of insert to vector, the exonuclease reaction buffer, and the addition of T4 DNA ligase (Fig. 2). If the digestion time was longer than 10 min, the number of clones decreased [Fig. 2(A)]. Excess of λ exonuclease decreased the number of clones [Fig. 2(B)]. At a constant vector quantity, the number of recombinant plasmid increased with increased amount of insert [Fig. 2(C)]. This is due to the increased formation of recombinant nicked plasmid. The cloning efficiency did not depend on the reaction buffer [Fig. 2(D)]. We obtained approximately the same number of clones using PCR buffer (only adding exonuclease into new PCR products) or standard exonuclease buffer during exonuclease digestion. Moreover, the purification of PCR fragments did not result in distinct increase of the number of clones, indicating that other components present in PCR mixtures do not affect our LIC method. In addition, the inclusion of T4 DNA ligase in the reaction did not increase the cloning efficiency [Fig. 2(E)]. After optimization, we defined the best and practical protocol: directly mixing the PCR products of insert and vector in a molar ratio of 3–10; then adding λ exonuclease (250 ng/pmol DNA) and incubating for 5 min at 37°C, finally immediately transforming the mixtures into competent E. coli cell. All subsequent experiments were carried out according to the optimal protocol except specified.
Figure 2.
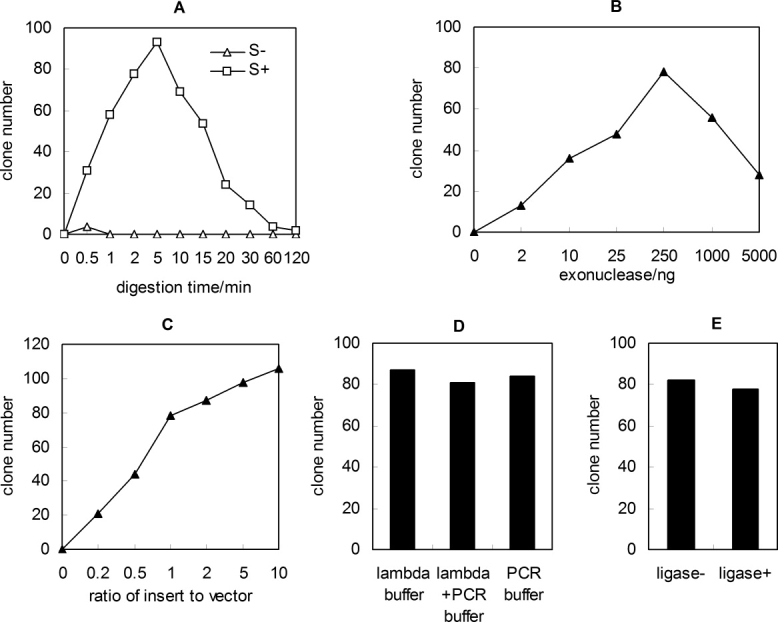
Optimization of protocol. The exonuclease reaction time (A), the amount of λ exonuclease (B), the molar ratio of insert to vector (C), the reaction buffer (D), and the addition of T4 DNA ligase (E) were studied for obtaining the best protocol. In Figure 2(C), constant amount of linear vector (10 ng) and different concentrations of insert were used. Equimolar PCR fragments (10 ng pUC18 and 4 ng ape_0119) was used in the optimization of other parameters. Except for Figure 2(B), the λ exonuclease digestion reactions contained 250 ng λ exonuclease/pmol DNA. The capital letter S indicates phosphorothioate.
Effects of overhang length and phosphorothioate number
The number of clones generated from our LIC method was dependent on the length of 30 overhang and the number of phosphorothioate (Fig. 3). Equimolar insert and vector was used for rational evaluation of the number of clones. No clone was obtained when the length of 3′ overhang was less than 8 nt. However, the length of 3′ overhang between 8 nt and 14 nt, leaded to a linear increase in the number of clones generated [Fig. 3(A)]. Above 14 nt the number of clone generated was not augmented.
Figure 3.
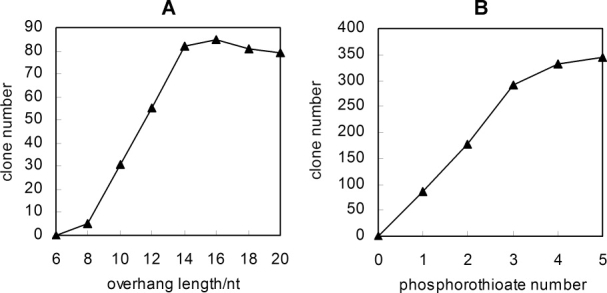
Effect of the length of 3′ overhang and the number of phosphorothioate on cloning efficiency. The mixtures of equimolar PCR fragments (10 ng pUC18 and 4 ng ape_0119) were incubated with λ exonuclease (250 ng exonuclease/pmol DNA) at 37°C for 5 min before transforming into E. coli. The primers used to generate the required overhangs are the derivatives of Ape_0119-F, Ape_0119-R, pUC18-F, and pUC18-R.
The successive increase in the number of phosphorothioate distinctly increased the number of recombinant plasmid. As shown in Figure 3(B), the number of recombinant clones increased from 87 to 345 when the number of phosphorothioate was augmented from 1 to 5. Altogether, taking into consideration of cloning efficiency and cost effectiveness, we recommended the combination of 14 nt 3′ overhang and three successive phosphorothioates.
Comparison of our method with other LIC methods
Several LIC methods, including dU primer cloning, hybridization cloning, T4 DNA polymerase cloning, and in vivo recombination cloning, were evaluated to compare their cloning efficiencies with our method. The terminal structures of DNA for various LIC are as follows: 3′ overhang (phosphorothioate primer cloning and dU primer cloning), 5′ overhang (T4 DNA polymerase cloning), blunt end (in vivo recombination cloning), 5′ and 3′ overhangs (hybridization cloning). In the phosphorothioate primer cloning and dU primer cloning, the 5′ group on DNA terminal is phosphate while that in other methods is hydroxyl group. The result showed a distinctive higher cloning efficiency was obtained with our method (Table III). The cloning efficiency of our method was at least onefold higher than that of other LIC methods. Among all tested LIC methods, the cloning efficiency of in vivo recombination cloning was the lowest. Interestingly, the cloning efficiency of 3′ overhang (phosphorothioate primer cloning and dU primer cloning) was higher than that of 5′ overhang (T4 DNA polymerase cloning) and mixture of 5′ and 3′ overhang (hybridization cloning) by at least fivefold (Table III).
Table III.
Number of Recombinant Plasmid in Various LICs
| LIC methoda | Number of recombinants |
|---|---|
| Phosphorothioate primer cloning | 287 |
| dU primer cloning9 | 138 |
| Hybridization cloning12 | 27 |
| T4 DNA polymerase cloning7 | 23 |
| In vivo recombination cloning15 | 12 |
All protocols were obtained from the respective references. In short for hybridization cloning, the mixtures of insert and vector were heated for 5 min at 95°C and slowly cooled down to room temperature; for dU primer cloning chlamydial endonuclease III was used to cleave the apyrimidinic site after treating the insert/vector mixtures with chlamydial uracil-DNA glycosylase; for T4 DNA polymerase cloning, the insert and vector were treated by T4 DNA polymerase, respectively, and then mixed to anneal their 5′ overhangs; for in vivo recombination cloning the purified insert and vector were electro-transformed into E. coli strain DY329. Except for in vivo recombination cloning (50 ng pUC18 and 20 ng ape_0119 with 40 bp homologous region), the same amount of insert and vector (10 ng pUC18 and 4 ng ape_0119 with 14 bp homogenous region) was used. The primers carrying three successive phosphorothioates were used in our method. The base sequences of overhangs of hybridization cloning and dU primer cloning were the same as our method; but in T4 DNA polymerase cloning, the little-changed base sequence was designed to obtain the 14 nt overhangs. All primers were designed as described in the references.
Because the 5′ terminal group is different for LICs methods, henceforth, we compared the cloning efficiency of various LIC methods using the two type of 5′ group, phosphate or hydroxyl, on DNA ends (Table IV). Our results showed that the phosphate group on 5′ ends improved cloning efficiency in hybrizidation cloning, T4 DNA cloning, and in vivo recombination cloning (Table IV). However, no significant effect was observed in the phosphorothioate primer cloning and dU primer cloning using 5′ phosphate or 5′ hydroxyl group (Table IV), suggesting that the efficiency of the nick repair between 3′ protruding overhang and 5′ recessed end in bacterial was not dependent on the presence of 5′ phosphate.
Table IV.
Promotion of Cloning Efficiency by 5′ Phosphate
| LIC methoda | 5′ Phosphateb | 5′ Hydroxylc |
|---|---|---|
| Phosphorothioate primer cloning | 292 | 238 |
| dU primer cloning9 | 134 | 121 |
| Hybridization cloning12 | 54 | 23 |
| T4 DNA polymerase cloning7 | 46 | 21 |
| In vivo recombination cloning15 | 34 | 11 |
The overhanged ends were generated using the protocol described in Table III. Except for in vivo recombination cloning (50 ng pUC18 and 20 ng ape_0119 with 40 bp homologous region), the same amount of insert and vector (10 ng pUC18 and 4 ng ape_0119 with 14 bp homogenous region) was used.
In hybridization cloning, T4 DNA polymerase cloning, and in vivo recombination cloning, phosphate group on the 5′ end was generated by PCR amplification using phosphorylated primers.
In phosphorothioate primer cloning and dU primer cloning, the 5′ hydroxyl group was generated by treating the 3′-overhanged DNA fragment with shrimp alkaline phosphatase.
Cloning of 192 genes from A. pernix
According to the above optimized protocol, we selected 192 genes from A. pernix and constructed their expression plasmids. The wild-type circular pDEST17 vector contains the ccdB gene, which is lethal to E. coli cells such as strain DH5α that do not carry the gyrase mutated gene; henceforth, digestion of vector parental template by Dpn I was omitted in our method.
PCR products of 161 genes from A. pernix were successfully cloned into pDEST17 vector. However, 5 of the 161 recombinant plasmids contained unspecifically amplified inserts confirmed by DNA sequencing. Therefore, 156 real recombinant expression plasmids were obtained. During cloning, 18 of 156 genes had a very low percentage of positive clones (12 genes with less than 40% of positive clones and 6 genes with about 0% of positive clones). The cause of low percentage of positive clones was the existence of plenty of primer dimer in PCR products of 18 genes. After removal of primer dimer by purification of PCR product, higher than 95% of positive clones (higher than) was obtained for all 18 genes. The length of genes cloned is up to 5.8 kb (ape_0620.1, 8 clone/ng vector), indicating that this method is highly suitable for cloning long DNA fragments.
Discussion
As a LIC approach, our method has the common advantages of LIC. First, restriction enzyme digestion and enzyme ligation reactions are not required, thus reducing the potential of cloning failure. Second, this approach is simple and rapid, as it requires minimum manipulations. Third, this procedure enables the direct cloning of PCR fragments into the final vector between any two nucleotides. Our approach also has a number of advantages over other LIC methods. First, the cost of synthesizing phosphorothioate modification primers is less than other modifications. 4,9–11 Second, the PCR amplification can be performed by high-fidelity DNA polymerase, such as KOD-plus DNA polymerase and pfu DNA polymerase, which are not suitable for dU primer cloning.9 Third, the reproducibility is very good in the presence of more successive phosphorothioate modifications. Finally, our method can also be used in site-directed mutagenesis via a pair of primers with the 5′ complementary end.
Why did our method obtain a distinctively higher cloning efficiency? We think the following factors may be involved: (i) the 3′ overhang can be repaired in E. coli with higher efficiency compared to 5′ overhang and (ii) the presence of phosphorothioate immediately after a 5′ phosophate in the insert and vector promotes DNA repair in E. coli.
Our LIC method does have some disadvantages. First, the phosphorothioate modification cannot stop the λ exonuclease digestion completely at this modification site, which may lead to complete digestion of some PCR fragments. However, this can be overcome by introduction of more than three successive phosphorothioate groups. Second, the phosphorothioate-modified primers require phosphorylation at 5′ end for efficient digestion by λ exonuclease. However, this shortcoming may be overcome by digesting the 5′OH ds DNA with T7 gene 6 protein or the 5′exonuclease domain of E. coli DNA polymerase I. 17,18 Third, like other LIC methods, our method is also dependent on the successful PCR amplification of long vector. Fortunately, many commercial DNA polymerases available allow high-fidelity amplification of long DNA fragments.
Materials and Methods
Materials
Taq DNA polymerase and T4 PNK were from Takara (Dalian, China). KOD-plus DNA polymerase and Dpn I restriction enzyme were purchased from Toyobo and Fermentas, respectively. The λ exonuclease was expressed in E. coli using pET28a recombinant plasmid (constructed by our lab) and purified via the 6×His tag. pDEST 17 vector and all primers used in this study were purchased from Invitrogen (Shanghai, China). A. pernix strain K1 was obtained from Japan Collection of Microorganisms (JCM, Japan) and its genomic DNA was extracted. E. coli strain DH5α was used in all cloning. Other reagents used were of analytical grade.
PCR amplification of target gene and linear vector
The encoding sequence (about 1kb) of ‘DNA repair and recombination protein RadA’ from A. pernix (ape_0119) was amplified from genomic DNA using primer pair Ape_0119-F and Ape_0119-R (Table I). PCR reaction (50 μL) containing 100 ng of A. pernix genomic DNA and was subjected to predenaturation of 5 min at 95°C and 20 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 60 s, followed by a 3 min extension at 72°C using the high-fidelity KOD-plus DNA polymerase (Toyobo, Japan). The linear pUC18 vector (about 2.6 kb) was amplified by PCR using primer pair pUC18-F and pUC18-R (Table I). PCR reaction (50 μL) was carried out using 1 ng of circular pUC18 DNA with the following condition: 5 min predenaturation at 95°C, 20 cycles of 94°C for 30 s, 55°C for 30 s, and 72°C for 3 min, and a final extension at 72°C for 5 min using the KOD-plus DNA polymerase. The primers were phosphorylated by T4 PNK and ATP. The PCR products of linear pUC18 vector were incubated with 10 U of Dpn I to fragment the parental vector template. PCR products were purified with the PCR purification kit (Sangon, Shanghai, China) when necessary.
Basic operation of LIC
Most of the λ exonuclease digestion reaction was performed in 10 μL of reaction buffer (66 mM glycine-NaOH, pH 9.4) at 37°C for 5 min (unless specified). After λ exonuclease digestion, the DNA mixtures were heated at 70°C for 15 min to inactivate the enzymes and slowly cooled down to room temperature for annealing of the complementary strands with the 3′ overhang. The DNA mixture was immediately transformed into 100 μL of E. coli DH5α competent cells. Or the exonuclease-treated DNA mixtures were immediately transformed into E. coli competent cells without heat inactivation of λ exonuclease. The resulting colonies were screened for the presence of the insert by colony PCR using Taq DNA polymerase. To optimize the LIC conditions for high-cloning efficiency, we tested the effects of different parameters such as the ratio of insert to linear vector, the amounts of λ exonuclease and buffers used, the digestion time, the number of phosphorothioate, and the length of complementary strand.
LIC of 192 different targets into pDEST17 vector
For constructing the expression plasmids of thermostable proteins, we chose to use the pDEST17 vector. The wild-type circular pDEST17 vector is naturally lethal to E. coli cell. Hence, permits the direct selection of recombinant clones. In this study, we selected 192 genes from A. pernix for cloning into the pDEST17 vector using our method. Briefly, two PCR reactions were carried out to amplify these target genes for our LIC method. The first PCR was carried out using gene specific primer pair that contains a common adaptor sequence at the 5′ end of the primer. A second PCR was carried out using 1/10 volume of the first PCR mixtures as template and the adaptor specific primer pair that contains three successive phosphorothioate modifications at the 3′ end. PCR, enzymatic treatment of linear pDEST17 vector and target genes, transformation of competent DH5α cell, and extraction of recombinant plasmids, were performed using 96-well microtitre plate.
References
- 1.Scharf SJ, Horn GT, Erlich HA. Direct cloning and sequence analysis of enzymatically amplified genomic sequences. Science. 1986;233:1076–1078. doi: 10.1126/science.3461561. [DOI] [PubMed] [Google Scholar]
- 2.Costa GL, Grafsky A, Weiner MP. Cloning and analysis of PCR-generated DNA fragments. PCR Methods Appl. 1994;3:338–345. doi: 10.1101/gr.3.6.338. [DOI] [PubMed] [Google Scholar]
- 3.Marchuk D, Drumm M, Saulino A, Collins FS. Construction of T-vectors, a rapid and general system for direct cloning of unmodified PCR products. Nucleic Acids Res. 1991;19:1154. doi: 10.1093/nar/19.5.1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaluz S, Flint APF. Ligation-independent cloning of PCR products with primers containing nonbase residues. Nucleic Acids Res. 1994;22:4845. doi: 10.1093/nar/22.22.4845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsiao K. Exonuclease III induced ligase-free directional subcloning of PCR products. Nucleic Acids Res. 1993;21:5528–5529. doi: 10.1093/nar/21.23.5528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aslanidis C, de Jong PJ. Ligation-independent cloning of PCR products (LIC-PCR) Nucleic Acids Res. 1990;18:6069–6074. doi: 10.1093/nar/18.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang YS, Watson WJ, Tucker PW, Capra JD. Construction of recombinant DNA by exonuclease recession. Nucleic Acids Res. 1993;21:1889–1893. doi: 10.1093/nar/21.8.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tseng H. DNA cloning without restriction enzyme and ligase. Biotechniques. 1999;27:1240–1244. doi: 10.2144/99276rr02. [DOI] [PubMed] [Google Scholar]
- 9.Nisson PE, Rashtchian A, Watkins PC. Rapid and efficient cloning of Alu-PCR products using uracil DNA glycosylase. PCR Methods Appl. 1991;1:120–123. doi: 10.1101/gr.1.2.120. [DOI] [PubMed] [Google Scholar]
- 10.Donahue WF, Turczyk BM, Jarrell KA. Rapid gene cloning using terminator primers and modular vectors. Nucleic Acids Res. 2002;30:e95. doi: 10.1093/nar/gnf094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coljee VW, Murray HL, Donahue WF, Jarrell KA. Seamless gene engineering using RNA- and DNA-overhang cloning. Nat Biotechnol. 2000;18:789–791. doi: 10.1038/77363. [DOI] [PubMed] [Google Scholar]
- 12.Tillett D, Neilan BA. Enzyme-free cloning: a rapid method to clone PCR products independent of vector restriction enzyme sites. Nucleic Acids Res. 1999;27:e26. doi: 10.1093/nar/27.19.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van den Ent F, Löwe J. RF cloning: a restriction-free method for inserting target genes into plasmids. J Biochem Biophys Methods. 2006;67:67–74. doi: 10.1016/j.jbbm.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Y, Muyrers JP, Testa G, Stewart AF. DNA cloning by homologous recombination in Escherichia coli. Nat Biotechnol. 2000;18:1314–1317. doi: 10.1038/82449. [DOI] [PubMed] [Google Scholar]
- 15.Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci USA. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iizasa E, Nagano Y. Highly efficient yeast-based in vivo DNA cloning of multiple DNA fragments and the simultaneous construction of yeast/Escherichia coli shuttle vectors. Biotechniques. 2006;40:79–83. doi: 10.2144/000112041. [DOI] [PubMed] [Google Scholar]
- 17.Nikiforov TT, Rendle RB, Kotewicz ML, Rogers YH. The use of phosphorothioate primers and exonuclease hydrolysis for the preparation of single-stranded PCR products and their detection by solid-phase hybridization. PCR Methods Appl. 1994;3:285–291. doi: 10.1101/gr.3.5.285. [DOI] [PubMed] [Google Scholar]
- 18.Zhang W, Evans DH. DNA strand transfer catalyzed by the 5′-3′ exonuclease domain of Escherichia coli DNA polymerase I. Nucleic Acids Res. 1995;23:4620–4627. doi: 10.1093/nar/23.22.4620. [DOI] [PMC free article] [PubMed] [Google Scholar]