

Abstract
Human small C-terminal domain phosphatase 1 (Scp1) modulates the phosphorylation state of the C-terminal domain (CTD) of eukaryotic RNA polymerase II (RNAP II), with preference for phosphorylated Ser5 in the tandem heptad repeats of the CTD. Additionally, Scp1 was identified as a conserved regulator of neuronal stem cell development. Scp1 is a member of haloacid dehalogenase (HAD) superfamily, whose catalysis depends on a Mg2+ ion and a DXDX(T/V) motif. The first Asp of the motif is identified as the nucleophile that is subject to phosphorylation leading to a phosphoryl-aspartate intermediate. This high-energy mixed anhydride intermediate is subsequently hydrolyzed to regenerate the enzyme. In the present study, we successfully captured the phosphoryl-aspartate intermediate in the crystal structure of a Scp1D206A mutant soaked with para-nitrophenyl phosphate (pNPP), providing strong evidence for the proposed mechanism. Furthermore, steady-state kinetic analysis of a variety of Scp1 mutants revealed the importance of Asp206 in Mg2+ coordination mediated by a water molecule. Overall, we captured the snapshots of the phosphoryl transfer reaction at each stage of Scp1-mediated catalysis. Through structural-based sequence alignment, we show that the spatial position of the D206 side chain is strictly conserved throughout HAD family. Our results strongly suggest that Asp206 and its equivalent residues in other HAD family members play important structural and possible mechanistic roles.
Keywords: HAD superfamily, phosphoryl-aspartate intermediate, small C-terminal domain phosphatase, protein phosphatese
Introduction
The CTD of RNAP II is a unique structure that not only temporally and spatially modulates the progression of RNAP II through the transcription cycle but also associates mRNA transcription to mRNA processing, DNA repair and other cellular processes.1 The biological functions of CTD are performed through controlling the recruitment of regulatory factors in accordance with the dynamic modifications and conformation changes of the CTD,2 including phosphorylation, isomerization, and possibly glycosylation.1 The CTD largely consists of tandem heptad repeats with a consensus sequence Y1S2P3T4S5P6S7 where the phosphorylation of Ser2, Ser5, and Ser7 has been shown to have important regulatory effects on the function of RNAP II.2–4 Therefore, the enzymes responsible for the phosphorylation and dephosphorylation of these residues play essential roles in the regulation of transcription and related processes.
The transcription factor IIF (TFIIF)-interacting C-terminal domain phosphatase 1 (Fcp1) is the first discovered CTD-specific phosphatase that preferentially dephosphorylates Ser2 in eukaryotes.5,6 Fcp1 has been shown to regulate the recycling of RNAP II by dephosphorylating Ser2 after each round of transcription.7 More recently, a family of small CTD phosphatases (Scps) with activities preferential for phosphoryl-Ser5 (phos.Ser5) was identified.8,9 This family includes three highly similar proteins designated Scp1, Scp2, and Scp3. They share more than 90% similarity at the catalytic domain and have essentially the same activity to dephosphorylate phos.Ser5.
In humans, Scp1 has ∼20% identity with the catalytic domain (FCPH domain) of Fcp1 but lacks the C-terminal breast cancer protein related C-terminal domain (BRCT), which is associated with CTD recognition by Fcp1. Scps show a strong preference toward Ser5 over Ser2 by 70-fold.10 The first clue to the specificity of Scps was elucidated when we determined the structures of Scp1 bound to a segment of the CTD heptad.10 These structures revealed a unique mode for CTD recognition and provided a structural explanation for the preferential binding and turnover of phos.Ser5 of the CTD. Consistent with the known specificity of Scp1 toward phos.Ser5 instead of phos.Ser2, in the complex structure the phos.Ser5 is bound to the active site, whereas phos.Ser2 flips out of the active site, making no direct interaction with the protein. Notably, Scp1 shows remarkable specificity toward the trans peptide-bond configuration of the two prolines in the CTD repeat, which can adopt both cis and trans configurations. Such configuration switching is known to modulate the structure of the CTD and its accessibility to kinases and/or phosphatases.11,12 Computational docking shows that if a similar active site conformation is adopted, the peptide with cis Pro will result in an unfavorable steric clash with the protein. However, it has yet to be established if Scps can undergo a dramatic conformational change to accommodate cis Pro.
Unlike kinases that evolved from the same ancestor, the protein phosphatases derived from separate origins which results in different structures and reaction mechanisms.13 Protein tyrosine phosphatases mediate the phosphoryl transfer via a phosphoryl-protein intermediate at a conserved Cys residue at the active site.14 On the other hand, protein serine/threonine (Ser/Thr) phosphatases normally require metal ions for their associated reactions. Ser/Thr phosphatase family is further divided into protein phosphatase P (PPP) and Mg2+- or Mn2+-dependent protein phosphatase (PPM) families with different metal requirements.15,16 Nonetheless, in both cases, the dephosphorylation reaction occurs in a single step in which a water molecule activated by metal ions targets the leaving phosphate group.17 One of the major distinctions between tyrosine phosphatases and Ser/Thr phosphatases is the involvement of metal ions (Ser/Thr phosphatases) and the generation of phosphoryl intermediate (tyrosine phosphatases). Therefore, when a group of novel human CTD phosphatases (Fcp/Scp phosphatases) were identified, they were classified as the PP2C subfamily of the PPM Ser/Thr phosphatase family.8,18 Like other PP2C phosphatases, Fcp/Scp phosphatases are Mg2+-dependent enzymes with multiple highly conserved aspartates. However, when the Scp structure was determined,19 it exhibited several fundamental distinctions from other PP2C members. First, it lacks the dimetal center which is present in other PP2C Ser/Thr phosphatases but has a single Mg2+ ion bound at the active site (Supporting Information Fig. S1). Moreover, the four strictly conserved aspartates are arranged in a different topology from PP2C active sites.
Based on the structure-based sequence alignment, Scp is similar to the HAD superfamily proteins, the majority of which are involved in phosphoryl transfer.20 Indeed, with little primary sequence identity, the active site of Scp superimposes perfectly with that of other HAD members.19,21,22 Even the Mg2+ ion position and its coordinating water molecules are conserved.10 This suggests that Scp is a unique family of Ser/Thr phosphatases that mediates phosphoryl transfer through the generation of a phosphoryl-aspartate using its DXDX(T/V) signature motif, a mechanism conserved in the HAD family. Using 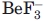 as a phosphoryl analogue, the first Asp of the motif was identified to be the nucleophile that undergoes phosphorylation leading to a phosphoryl-aspartate intermediate.19 This high-energy mixed anhydride is subsequently hydrolyzed to regenerate the enzyme.20 Although this proposed mechanism is well accepted, the phosphoryl intermediate was never directly visualized in Scp/Fcp family.
as a phosphoryl analogue, the first Asp of the motif was identified to be the nucleophile that undergoes phosphorylation leading to a phosphoryl-aspartate intermediate.19 This high-energy mixed anhydride is subsequently hydrolyzed to regenerate the enzyme.20 Although this proposed mechanism is well accepted, the phosphoryl intermediate was never directly visualized in Scp/Fcp family.
Considering the uniqueness of Scp/Fcp phosphatases in their phosphoryl reaction mechanism as a Ser/Thr phosphatase, we sought to isolate the phosphoryl intermediate combining mutagenesis and X-ray crystallography. We extended our previous structural and functional investigations to further probe the role of key residues in the dephosphorylation reaction. Steady-state kinetic analysis of a variety of Scp1 mutants verified the Mg2+-coordinating function of Asp206, a conserved residue in the HAD superfamily whose role was not highlighted before. We have successfully captured the phosphoryl-aspartate intermediate in the crystal structure of Scp1D206A mutant soaked with pNPP, providing evidence for the proposed two-step mechanism. In this study, we were able to obtain the snapshots of Scp protein at each stage of the phosphoryl-transfer reaction, which will be really valuable for the structure-function study of Scp/Fcp phosphatases.
Results and Discussion
Design of mutants based on proposed phosphoryl transfer mechanism
Based on the structures of Scp1, a two-step reaction mechanism for the dephosphorylation reaction catalyzed by Scp1 was proposed.10 In this proposed mechanism [Fig. 1(A)], the Asp96 [the first Asp in the DXDX(T/V) signature motif] acts as the attacking nucleophile in the first step of the reaction where the phosphoryl group on phos.Ser5 is transferred to Asp96, forming a phosphoryl-carboxyl mixed anhydride, which is subsequently hydrolyzed in the second step to release the phosphate and regenerate Asp96. Phosphoryl analogues have been used to understand the reaction mechanism.19 Most recently, a study conducted on Schizosaccharomyces pombe Fcp1 captured the mimic structures of the phosphoryl-aspartate intermediate and the transition state of the hydrolysis step using and  , respectively.23 In the Fcp1 structure, a water molecule occupies the position close to the Asp172 (corresponding to Asp98 in human Scp1) and can potentially be activated by Asp172 side chain for the breakdown of the phosphoryl-intermediate. This observation is consistent with the structural studies on phosphoserine phosphatase (PSP).24 Based on the similarity between Fcp1 and Scp1, it is likely that Asp98 functions as the general acid to protonate the leaving group and as the general base to activate a water molecule in the first and second steps, respectively. However, structural or biochemical data that directly test this model in Scp1 are lacking.
, respectively.23 In the Fcp1 structure, a water molecule occupies the position close to the Asp172 (corresponding to Asp98 in human Scp1) and can potentially be activated by Asp172 side chain for the breakdown of the phosphoryl-intermediate. This observation is consistent with the structural studies on phosphoserine phosphatase (PSP).24 Based on the similarity between Fcp1 and Scp1, it is likely that Asp98 functions as the general acid to protonate the leaving group and as the general base to activate a water molecule in the first and second steps, respectively. However, structural or biochemical data that directly test this model in Scp1 are lacking.
Figure 1.
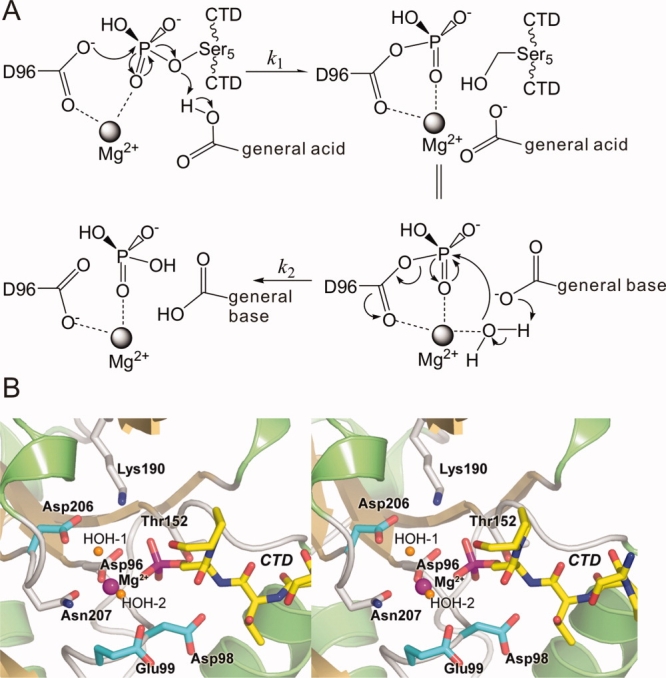
A: Proposed mechanism of phos.Ser5 dephosphorylation catalyzed by Scp1. At early steady-state, the concentration of product has not accumulated to a significant level so that the reverse reaction of the first step can be ignored. B: Stereo diagram of the CTD-bound active site of Scp1D96N. The structure of human Scp1D96N in complex with a CTD heptad repeat peptide phosphorylated at Ser5 shown as a stereo pair of ribbon diagram. The active site residues are depicted as color-coded bonds. The three residues serving as candidate general bases for hydrolysis of the phosphoryl-Asp96 intermediate are highlighted with cyan colored bonds.
One of the most efficient ways to understand the catalytic mechanism of an enzyme is to capture the intermediate of the reaction. It has been shown in an earlier study that a transient phosphoryl-cysteine intermediate was captured in the protein tyrosine phosphatase 1B crystal structure by generating an active site mutation at Gln262 to Ala.25 However, although the two-step mechanism of the dephosphorylation reaction [Fig. 1(A)] by the HAD superfamily enzymes is generally accepted,20 the phosphoryl-aspartate intermediate has rarely been shown structurally.26 In theory, there are two scenarios where the phosphoryl-aspartate intermediate may be captured in crystal structure. First, if the general acid that protonates the product [in the first step, Fig. 1(A)] is a different residue from the general base that catalyzes the hydrolysis of the phosphoryl-aspartate intermediate [the second step, Fig. 1(A)] then mutation of the latter residue would result in the accumulation of the intermediate. Second, if the general acid and the general base are the same residue, but the mutation of an auxiliary residue (e.g., a Mg2+-coordinating residue) slows down the rate of both the first and the second reactions, the overall reaction rate might be low enough so that the added substrate is not completely consumed during the process of crystallization. In this case, if the rate of the second step [k2, Fig. 1(A)] is not much higher than that of the first step [k1, Fig. 1(A)], a substantial fraction of the enzyme 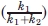 should exist as the phosphoryl-aspartate intermediate.
should exist as the phosphoryl-aspartate intermediate.
With this reasoning in mind, we designed a series of Scp1 mutants where each of the acidic residues (namely, Asp98, Asp99, and Asp206) was mutated to either the isosteric neutral residue (i.e., Asn) or Ala, examined these mutants with biochemical assays and chose suitable ones for crystallographic investigation to capture the phosphoryl intermediate.
Phosphatase activity assays for Scp1 mutants
We used two complementary methods to measure the general dephosphorylation activity of Scp1 mutants: a colorimetric assay with pNPP as the substrate and a fluorescent assay using 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) as the substrate. The pNPP assay is highly robust but shows a strong background absorbance, resulting in insensitivity with an enzyme with low activity. On the other hand, the fluorescence-based DiFMUP assay is highly responsive toward dephosphorylation and can detect very low-phosphatase activity. However, high concentrations of DiFMUP appeared to nonspecifically inhibit the activities of all tested variants of Scp1. The inhibitory effect was evident at concentrations of DiFMUP around 500 μM and was nearly complete at higher concentrations of DiFMUP (>5 mM). For this reason, the highest concentration of DiFMUP used in our assay was 150 µM which is below the Km of all active Scp1 variants, making the measurement of Km and kcat unfeasible. Nonetheless, the kcat/Km could still be measured in DiFMUP assays and were used to compare relative activities of Scp1 variants. Despite being less sensitive, the pNPP assay allowed high concentrations of substrate (up to 30 mMpNPP) and, when necessary, enabled us to obtain the kcat and Km for Scp1 variants toward pNPP. Using the two assays, significant information on the catalytic activity of these Scp1 variants was obtained.
As expected, the mutation of the active site nucleophile Asp96 to Asn (D96N) or Ala (D96A) abolished all measurable activity in both assays. Interestingly, although Asp98 was proposed to act as a general acid to protonate the leaving group of the first phosphoryl transfer and subsequently deprotonate a water to accelerate hydrolysis of the anhydride,10,19 mutation of Asp98 to Asn (D98N) retained 30% activity of the wild type Scp1 (DiFMUP assay data, Table I). Even a D98A mutant showed 10% activity of the wild type Scp1 (both assays, Table I). The kcat of D98A using pNPP as the substrate was as high as 0.5 s−1. This high kcat is noteworthy because using the model described in Figure 1(A), it can be deduced that
Table I.
Steady-State Parameters of Scp1 Variants Tested by DiFMUP and pNPP Assays
| DiFMUP assay |
pNPP assay |
|||
|---|---|---|---|---|
| kcat/Km (mM−1 min−1) | kcat (s−1) | Km (mM) | kcat/Km (mM−1 s−1) | |
| Wild type | 10.3 ± 0.8 | 2.5 ± 0.1 | 3.6 ± 0.8 | 0.7 ± 0.2 |
| D96N | N.A.a | N.A.a | N.A.a | N.A.a |
| D96A | N.A.a | N.A.a | N.A.a | N.A.a |
| D98N | 3 ± 1 | N.A.b | N.A.b | N.A.b |
| D98A | 0.8 ± 0.2 | 0.50 ± 0.02 | 7.3 ± 0.8 | 0.068 ± 0.008 |
| E99Q | 1.14 ± 0.09 | 2.0 ± 0.3 | 8 ± 4 | 0.2 ± 0.1 |
| E99A | 0.5 ± 0.1 | 0.005 ± 0.001 | N.A.c | N.A.c |
| D206Ad | 0.23 ± 0.04 | 0.022 ± 0.005 | 8 ± 4 | 0.002 ± 0.001 |
Signal below detection limit.
Although D98N showed strong activity to pNPP, the activity of this mutant appeared unstable so that during the time course required for pNPP assay the enzyme did not maintain constant activity, leading to uninterpretable data.
The Km of E99A mutant is greatly increased, and thus cannot be accurately determined from the rate versus pNPP concentration curve.
Both DiFMUP and pNPP assay were performed with 20 mM Mg2+; however, no signal can be detected for D206A mutant under this condition. Therefore, the data for D206A were obtained under 50 mM Mg2+ condition.
| (1) |
Or in other words, k2 must of necessity be greater than kcat. Therefore, the first-order rate constant for the dephosphorylation of D98A mutant (k2) must be greater than 0.5 s−1, which is at least 4 orders of magnitude higher than the reported uncatalyzed hydrolysis of phosphoryl-aspartate mixed anhydrate (10−4 s−1, Ref. 20). These results argue against the proposed function of Asp98 as the only possible general acid/base in catalysis, suggesting that complementarity by another active site residue may be applied in Scp-mediated phosphate transfer.
Similarly, mutations of Glu99 to Gln or Ala did not abolish phosphatase activity, resulting in 10% and 5% activity compared with wild type, respectively. The mutation of Asp206 to Asn resulted in greatly reduced stability and solubility of the protein. The corresponding mutation of Asp to Asn is also reported to be very disruptive for Fcp1,27 possibly due to the misfolding of the protein induced by the disruption of the salt bridge between this Asp and active site Lys. Therefore, alanine mutation was tested to investigate the functional role of D206. Under the standard assay condition with 20 mM MgCl2, the activity of D206A mutant was too low to be reliably detected in both assays. The crystal structure of Scp1 [Fig. 1(B)] shows that an active site Mg2+ is coordinated by a water molecule, which is further positioned by Asp206. Such secondary stabilization of metal ion binding via water molecules might be essential for the effective catalysis. Indeed, this role of Mg2+-coordination by residues corresponding to D206 is also observed in many other HAD family proteins in three-dimensional structure (see structural alignment later). Consistent with these structural observations, when the MgCl2 concentration was increased to 50 mM, the D206A mutant displayed a well-detectable activity toward DiFMUP. The kcat/Km was 0.2 mM−1 min−1 (∼2% of the wild type activity measured at 20 mM MgCl2). To further characterize the Mg2+-dependence of D206A mutant, we tested the catalytic activity of wild type Scp1 and the D206A mutant at various MgCl2 concentrations (Fig. 2). While the apparent Kd of wild type Scp1 with Mg2+ was 1.3 ± 0.9 mM, that of the D206A mutant was at least 100-fold higher. These results indicate that Asp206 is critical during catalysis at least in part by coordinating the catalytic Mg2+.
Figure 2.
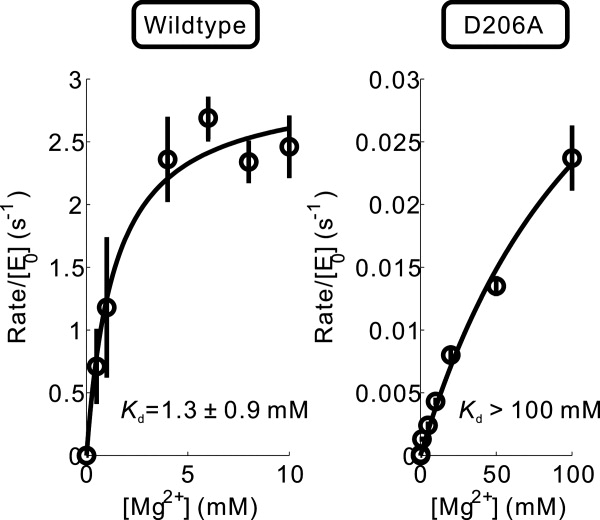
Mg2+-dependence of enzyme activity of wild type Scp1 and Scp1D206A mutant.
Phosphoryl intermediate of Scp1 obtained by incubating Scp1D206A mutant with pNPP at low Mg2+ concentration
On the basis of the kinetic characterization above, we crystallized D98A, E99A, and D206A and soaked the crystals in the buffer containing a high concentration of pNPP. X-ray diffraction data were collected for each mutant at resolutions from 2.35 to 2.45 Å (Table II). In the structure of the Scp1D98A mutant, a hydrogen bond originally observed between the side chain of Asp98 and that of Tyr158 found both in the apo protein structure (Supporting Information Fig. S2) and our previous complex structures [Fig. 3(A)] are interrupted due to mutation of Asp98. This hydrogen bond appears to anchor Tyr158 to facilitate the formation of part of the CTD binding groove.10 Without this hydrogen bond anchoring the side chain of Tyr158 by Asp98 side chain, an alternative conformation of Tyr is adopted [Fig. 3(B)], which places the phenolic side chain of Tyr in the hydrophobic pocket, previously observed to specifically sequester Pro3 of the CTD repeat sequence [Fig. 3(C)]. This hydrophobic pocket prefers binding to hydrophobic molecules, and the adoption of Pro3 of the CTD repeat is essential for CTD recognition by Scp1. The structure of Scp1E99A exhibits an identical metal center with wild type Scp1 and Scp1D96N structures (Supporting Information Fig. S3), with the only difference being the coexistence of both conformations of Tyr158 due to a minor change of the Asp98 position. Apparently, the orientation of the Tyr158 is directly affected by its hydrogen bonding to Asp98 carboxyl side chain.
Table II.
Data Collection and Refinement Statistics
| Scp1D206A soaked in pNPP at low Mg2+ | Scp1D206A soaked in pNPP at high Mg2+ | Scp1D98A soaked in pNPP at low Mg2+ | |
|---|---|---|---|
| Data collection | |||
| Space group | C2 | C2 | C2 |
| Cell dimensions | |||
| a, b, c (Å) | 124.7, 78.2, 62.6 | 125.5, 77.3, 63.1 | 124.8, 79.0, 62.9 |
| α, β, γ (°) | 90.0, 112.3, 90.0 | 90.0, 112.5, 90.0 | 90.0, 112.8, 90.0 |
| Resolution (Å) | 49.1−2.35 (2.43−2.35)a | 48.6−2.45 (2.54−2.45) | 48−2.30 (2.38−2.30) |
| No. of observation | 39,564 | 32,699 | 39,941 |
| No. of unique reflections | 23,379 | 20,421 | 24,660 |
| Rsym or Rmerge (%) | 7.1 (20.4) | 7.5 (22.7) | 7.2 (20.5) |
| I/σ(I) | 16.4 (2.6) | 13.4 (2.1) | 17.5 (3.4) |
| Completeness (%) | 91.3 (59.1) | 90.7 (53.2) | 90.2 (57.2) |
| Redundancy | 2.4 (1.7) | 3.3 (2.1) | 3.4 (2.6) |
| Refinement | |||
| Resolution (Å) | 49.1−2.35 | 48.6−2.45 | 32.2−2.35 |
| No. reflections (test set) | 23286 (1109) | 17693 (864) | 21099 (1131) |
| Rwork/Rfree (%)b | 19.5 (25.9)/24.5 (34.5) | 20.4 (25.6)/25.4 (37.7) | 19.2 (24.0)/23.1 (30.6) |
| No. atoms | |||
| Protein | 2930 | 2908 | 2928 |
| Ion | 2 | 12 | 2 |
| Water | 95 | 32 | 99 |
| B-factors (Å2) | |||
| Protein | 36.2 | 37.8 | 37.4 |
| Mg2+ | 20.5 | 39.3 | 24 |
| Water | 37.2 | 38.2 | 39.1 |
| R.m.s deviations | |||
| Bond lengths (Å) | 0.013 | 0.014 | 0.011 |
| Bond angles (°) | 1.48 | 1.60 | 1.36 |
| Ramachandran plot (%) | |||
| Most favored | 87.8 | 84.4 | 90.5 |
| Additionally allowed | 11.3 | 15.3 | 9.2 |
| Generally allowed | 0.9 | 0.3 | 0.3 |
Highest resolution shell is shown in parenthesis.
Rfree is calculated with 5% of the data randomly omitted from refinement.
Figure 3.
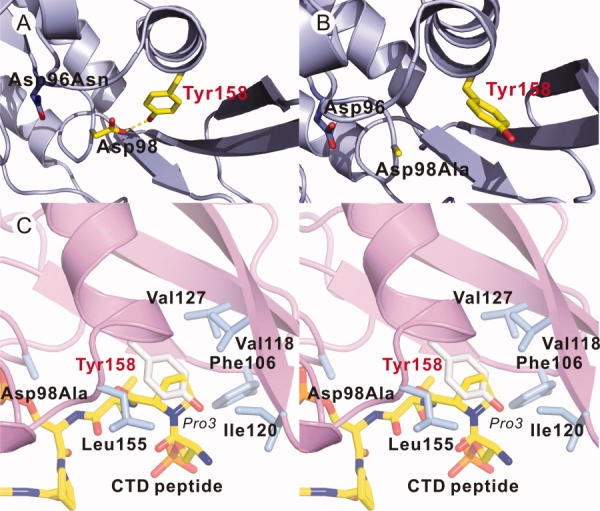
Two different conformations of Tyr158 observed in Scp1D96N structure (A) and Scp1D98A structure (B). In (A), the side chain of Tyr158 forms a hydrogen bond (yellow dashed line) with the side chain of Asp98 which is lost in (B). C: Stereo diagram of superimposition of Scp1D98A structure with Scp1D96N-CTD complex (PDB ID: 2ghq). The CTD peptide is shown in yellow stick.
Unlike D98A and E99A structures, the Scp1D206A mutant exhibited strong positive electron density directly contiguous with the electron density associated with the Asp96 residue (molecule A). The shape and high signal-to-noise ratio of the contiguous density is consistent with a covalently trapped phosphate group, which was built and refined as a phosphoryl-Asp96 intermediate [Fig. 4(A,B)]. The remaining parts of the overall Scp1D206A structure are virtually identical to the wild type and D96N Scp1 structures. Additional density resided in the Pro3 binding pocket that was refined well as a part of PEG molecule in one of the two Scp1D206A monomers in the asymmetric unit. This is consistent with previous observation that this pocket highly favors hydrophobic molecules and might nonspecifically bind to chemical molecules in crystallization solution.19
Figure 4.
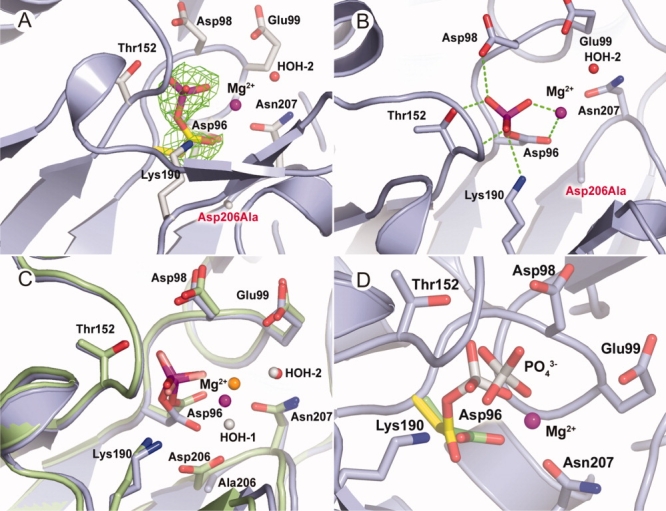
Structural studies of Scp1D206A. A: Phosphorylation of Asp96 (molecule A). The mutation of Asp206 (labeled in red) stabilizes the trapped phosphoryl-aspartate intermediate of the enzyme. B: The hydrogen bonding formed between phosphate group and the enzyme. C: Superimposition of phosphate-trapping Scp1D206A (light blue) with Scp1 bound with (1ta0) (lime). The water molecules are colored red and Mg2+ ion colored magenta for the former structure, whereas water as white and Mg2+ as orange for the latter. D: Superimposition of Scp1D206A soaked in substrate pNPP at different Mg2+ concentrations. The active site residues are shown in stick. The Asp96 covalently linked to phosphate group is shown in yellow, and the phosphoryl-free Asp96 is shown in green.
Apparently, in the crystalline environment of the Scp1D206A mutant, the high-energy phosphoryl-enzyme intermediate bound to Asp96 reaches appreciable levels and remains stable over time to allow visualization by protein X-ray crystallography. Based on the peak height from initial Fo–Fc electron density maps as well as thermal factors from active sites, we estimated that the occupancy of the phosphate group covalently attached to Asp96 reached ∼50% (molecule A). In the crystalline environment of the Scp1D206A mutant, the electron density of Mg2+ (Supporting Information Fig. S4) is significantly weaker than that obtained in the Scp1D96N-CTD peptide complex structure, suggesting the Mg2+ occupancy is not 100% (refined as 50% occupancy with B factor around 20 Å2). This is again consistent with the kinetic study that the binding affinity between Mg2+ and Scp1D206A is much lower. Therefore, using lower Mg2+ concentration in the crystallization mother liquor could be one of the major reasons why we captured this phosphoryl-enzyme intermediate in Scp1D206A crystal. The crystallization state also sheltered the phosphoryl intermediate from bulk water in the D206A mutant and slowed down the hydrolysis of phosphoryl-Asp96.
A previously reported structure of in complex with Scp1 suggested the existence of a Scp1 phosphoryl-enzyme intermediate.19 Because of the presumed chemical instability of phosphoryl-aspartate intermediates of HAD family proteins, has been used as a phosphate mimic.28 By comparing the structure of our trapped phosphoryl-aspartate intermediate and that of bound to Scp1 (PDB ID: 1ta0), we conclude that the complex structure encompassing bound to Asp96 is a reasonably acceptable mimic for the covalent phosphoryl-aspartate intermediate [Fig. 4(C)].
Product-trapping structure of Scp1 obtained by incubating Scp1D206A mutant with pNPP at high Mg2+ concentration and high pH
On the basis of the proposed mechanism that a general base is involved in the second step and our kinetic data, we hypothesized that increased Mg2+ availability and higher pH would facilitate the hydrolysis of the phosphoryl-aspartate. To evaluate these effects on the activity of Scp1D206A crystallographically, we used the same Scp1D206A crystal form but soaked the samples in 25 mMpNPP with 0.2M MgCl2 at slightly higher pH (pH 6.5–7.0). Clear and unmistakable electron density indicates a bound phosphate group in the Scp1D206A active site. However, unlike the electron density associated with phosphate for the soaked crystals at low Mg2+ concentration, this electron density clearly showed a complete loss in covalent attachment to the enzyme [Fig. 4(D)]. The phosphate group in the structure was refined to 100% occupancy with the same thermal factors as the surrounding protein residues. The position of the phosphate group moves by ∼1.2 Å away from its original position in the complex structure at low magnesium concentration, whereas the side chain of the Asp96 nucleophile rotates away from the phosphate [Fig. 4(D)]. Together with previously reported structures, these newly determined Scp1 crystal structures provide static snapshots along the reaction pathway catalyzed by Scp1 (Fig. 5). Our complex structure of Scp1D96N with CTD phosphoryl-peptide presents the prereaction binding mode. The D206A structure soaked with pNPP at low Mg2+ concentration is the capture of phosphoryl intermediate, whereas the same mutant at high Mg2+ concentration is a product trapped image with inorganic phosphate group at the active site.
Figure 5.
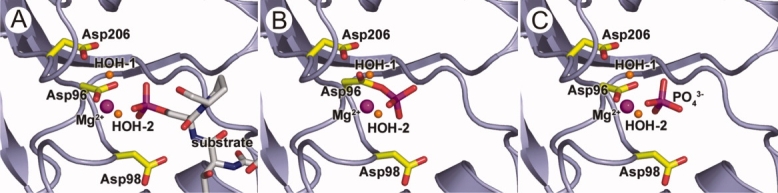
Snapshots of dephosphorylation reaction of human Scp1. This is the presumed model for the reaction catalyzed by Scp1 in substrate recognition (A), phosphoryl transfer (B), and phosphate release (C) stages. To capture them in crystallization state, certain residues were mutated (see below). Scp1 is shown in ribbon diagram in light blue, and Asp96, Asp98, and Asp206 residues are shown in stick in yellow. Mg2+ ion and the two water molecules are shown in sphere in magenta and orange, respectively. A: Scp1 bound to phosphorylated CTD (carbon chain shown in white). Asp96 was mutated to Asn to capture this complex structure. B: Phosphate group of phos.Ser5 is transferred to Asp96 and generates the phosphoryl-aspartate intermediate. Asp206 was mutated to Ala in this structure. C: Hydrolysis of the phosphoryl-aspartate intermediate and release of phosphate group from Asp96 side chain. Again, Asp206 was mutated to Ala in this structure.
Capture of phosphoryl intermediate for HAD superfamily
The formation of phosphoryl-enzyme intermediates is a crucial component of various enzymatic mechanisms involving phosphoryl transfer.25 Even though phosphoryl-aspartate is not very stable, it has been captured in other proteins containing a DXDX motif,20,29 mostly using phosphoryl analogues. In addition to the direct visualization of phosphoryl intermediate in our X-ray structure for Scp1, β-phosphoglucomutase (β-PGM) is found to be phosphorylated at the active site aspartate (Asp8) in the crystalline state (PDB ID: 1lvh).26 β-PGM is a subclass I HAD family protein, which catalyzes the transfer of a phosphate group between the 1 and the 6 positions of the cyclic glucose ring. As the enzyme needs to transfer the phosphate group from position 1 on the hexose to position 6, the phosphoryl-enzyme intermediate is sufficiently stable to allow reorientation of the acceptor in the active site as preventing water-mediated loss of phosphate. Other phosphoryl-aspartate intermediate was investigated where various phosphate analogs (, 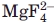 , and ) were used to mimic the different stages of phosphoryl transfer. For instance, in Ca2+-ATPase, the ATP-dependent phosphorylation of its catalytic Asp is a critical step in transducing chemical energy into conformational energy for Ca2+ ion transport.30,31 Subsequent to ATP-dependent phosphorylation of the ATPase, the dephosphorylation reaction of the phosphoryl-enzyme intermediate triggers a conformational change that closes the lumenal gate of the Ca2+ channel though the movement of a domain capping the transmembrane portal.30,31 Furthermore, the phosphoryl-aspartate intermediate was observed in S. pombe Fcp1 structure with mimicking the phosphate group (PDB ID: 3ef0).23 In the crystal structure, a covalent bond was clearly observed between the and Asp170 of Fcp1 (the first Asp in the DXDX motif). Human eyes absent (Eya) proteins, which also belong to the HAD family, play dual roles as a protein tyrosine phosphatase and a transcription factor in organ formation during development. Again, using phosphate analog and AlF3, a phosphoryl-aspartate intermediate and a transition sate of phosphoryl-enzyme were captured in the catalytic domain of Eya2 phosphatase, respectively.32 Additionally, phosphoryl-aspartate intermediates have been detected by radioactivity measurements in DNA 3′ phosphatase Tpp1,33 and in a recent study, Ca2+ ions were shown to have an inhibitory effect and induce the accumulation of a phosphoryl-aspartate intermediate detected by mass spectrometry in histidinol phosphate phosphatase.34
, and ) were used to mimic the different stages of phosphoryl transfer. For instance, in Ca2+-ATPase, the ATP-dependent phosphorylation of its catalytic Asp is a critical step in transducing chemical energy into conformational energy for Ca2+ ion transport.30,31 Subsequent to ATP-dependent phosphorylation of the ATPase, the dephosphorylation reaction of the phosphoryl-enzyme intermediate triggers a conformational change that closes the lumenal gate of the Ca2+ channel though the movement of a domain capping the transmembrane portal.30,31 Furthermore, the phosphoryl-aspartate intermediate was observed in S. pombe Fcp1 structure with mimicking the phosphate group (PDB ID: 3ef0).23 In the crystal structure, a covalent bond was clearly observed between the and Asp170 of Fcp1 (the first Asp in the DXDX motif). Human eyes absent (Eya) proteins, which also belong to the HAD family, play dual roles as a protein tyrosine phosphatase and a transcription factor in organ formation during development. Again, using phosphate analog and AlF3, a phosphoryl-aspartate intermediate and a transition sate of phosphoryl-enzyme were captured in the catalytic domain of Eya2 phosphatase, respectively.32 Additionally, phosphoryl-aspartate intermediates have been detected by radioactivity measurements in DNA 3′ phosphatase Tpp1,33 and in a recent study, Ca2+ ions were shown to have an inhibitory effect and induce the accumulation of a phosphoryl-aspartate intermediate detected by mass spectrometry in histidinol phosphate phosphatase.34
Conservation of D206 in HAD family
Although most HAD family proteins vary considerably in primary sequence, they all share the DXDX(T/V) motif (motif I) (Asp96 and Asp98 in human Scp1) as well as two other motifs at similar positions in their 3D structures [Fig. 6(A)]. Motif II is a single residue, either Thr (Thr152 in human Scp1) or Ser, which hydrogen bonds through its side chain with the phosphate moiety of the substrate. These two motifs are highly conserved and easily identified using structure-based alignment. Motif III is a DD or DN pair with a Lys residue located a short distance away on the N-terminal side of the acidic pair. The first Asp of the DD (DN) pair (Asp206 in human Scp1) aligns a water molecule, which coordinates the catalytic Mg2+ ion in the active site. This residue also forms a salt bridge to the side chain amino group of the upstream Lys (Lys190 in human Scp1) residue, which in turn forms a second intermolecular bridge to the phosphate moiety of the substrate. The second Asp or Asn (Asn207 in human Scp1) forms an axial coordination bond with the octahedrally coordinated Mg2+ ion [Fig. 1(B)]. Interestingly, the conservation of the second of the acidic stretch (Asn207) extends throughout HAD family, whereas Asp206 is found to be replaced by Gly in a lot of HAD proteins (protein serine phosphatase, P-type ATPase, phosphomannomutase, histidinol phosphate phosphatase, and sucrose phosphatase). This is very puzzling at first because our kinetic study strongly supports an essential structural function of Asp206 (or corresponding residues) in ion coordination and possibly a functional role in catalysis due to its mutation leads to capture of phosphoryl intermediate. A more careful examination of structure-based alignment shows that, in HAD proteins, where the first Asp of Motif III is replaced by Gly, the position corresponding to the side chain of Asp206 in Scp1 is always occupied by an Asp side chain extended from an amino acid 4–5 residues downstream of the second residue of Motif III [Fig. 6(B)]. Therefore, Asp206 side chain function is conserved spatially with similar functions: Mg2+ coordination and salt bridging to Lys (Table III).
Figure 6.
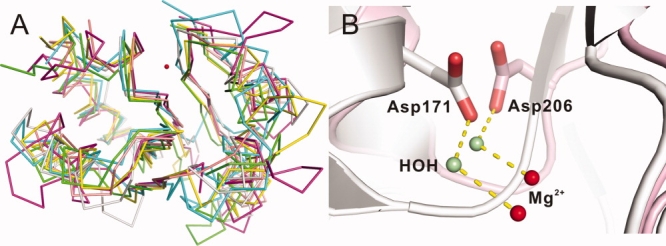
A: Structural alignment of alpha-carbon backbone of Scp1 (yellow, 2ght), PSP (green, 1l7m), β-PGM (cyan, 1lvh), PMM (magenta, 2fuc), P-Type ATPase (pink, 2zbd), and Eya2 (gray, 3geb). Red sphere is the Mg2+ in Scp1, indicating the active site. visual clarity, only the conserved portion is shown in each structure (Scp1 shows residues 89–222 without residues 104–130; PSP shows residues 4–184 without residues 19–73 and 120–137; β-PGM shows residues 1–188 without residues 15–91; PMM shows residues 12–240 without residues 82–190; P-Type ATPase shows residues 346–719 without residues 359–602 and 641–673; and Eya2 shows residues 268–519 without residues 281–424). B: Structural alignment of the active site of human Scp1 (light pink, 2ght) and MjPSP (white, 1l7m). The Asp206 in Scp1 and corresponding residue Asp171 in MjPSP are shown in stick. The water molecule hydrogen-bonding with Asp206 (or Asp171) is shown in pale green, and Mg2+ is shown in red. Hydrogen bonds are indicated by yellow dashed lines.
Table III.
Conserved Residues in the HAD Family in Comparison with Scp1
| Corres. a.a. for D96 | Corres. a.a. for D98 | Corres. a.a. for T152 | Corres. a.a. for D206 | Corres. a.a. for K190 | Corres. a.a. for N207 | |||
|---|---|---|---|---|---|---|---|---|
| Protein name | Pdb code | Seq. iden. (%) | Motif I | Motif II | Motif III | |||
| Pseudomonas sp. YL HAD | 1jud | 14 | D10 | Y12 | S118 | T175 | K151 | S176 |
| Schizosaccharomyces pombe Fcp1 | 3ef0 | 24 | D170 | D172 | T243 | D297 | K280 | D298 |
| Lactococcus lactis β-PGM | 1lvh | 14 | D8 | D10 | S114 | E169 | K145 | D170 |
| T4 polynucleotide kinase | 1ltq | 16 | D165 | D167 | S211 | D277 | K258 | D278 |
| Leishmania mexicana α-phosphomannomutase (PMM) | 2i54 | 12 | D10 | D12 | S46 | D215 | K188 | D207 |
| Human PMM | 2fuc | 16 | D19 | D21 | G52 | D226 | K198 | N218 |
| Methanocaldococcus jannaschii Phosphoserine phosphatase (MjPSP) | 1l7m | 10 | D11 | D13 | S99 | D171 | K144 | D167 |
| Human PSP | 1l8l | 15 | D20 | D22 | S109 | D183 | K158 | D179 |
| Oryctolagus cuniculus P-Type ATPase | 2zbd | 13 | D351 | T353 | T625 | D707 | K684 | D703 |
| E. coli histidinol phosphate phosphatase | 2fpr | 12 | D10 | D12 | T55 | D135 | K106 | D131 |
| Synechocystis sp. PCC6803 Sucrose phosphatase | 1tj3 | 15 | D9 | D11 | T41 | D190 | K163 | D186 |
| E. coli NagD | 2c4n | 14 | D9 | D11 | T42 | D206 | K176 | D201 |
| E. coli Class B acid phosphatase (AphA) | 2b82 | 12 | D44 | D46 | T112 | D171 | K152 | D167 |
| Human eyes absent phosphatase 2 (Eya2) | 3geb | 13 | D274 | D276 | T448 | E506 | K480 | D502 |
Possible additional role of D206 in catalysis
Our studies have established that even though the coordination of Mg2+ by D206 is mediated through a water molecule, its mutation can reduce Mg2+ binding by at least 100-fold. When considering the reaction mechanism of phosphoryl transfer reaction, the most logical candidate for general acid/based is Asp98. As shown in a recent paper about a Scp family member, S. pombe Fcp1, a water molecule is occupying at an ideal position to be activated by Asp98 and nucleophically attacks phosphorus.23 Interestingly, our mutants of Asp98 still retain a significant percentage of phosphoryl activity, which prompt us to speculate if alternative general acid/base might exist especially when Asp98 is replaced. Considering the conservation of Asp206 throughout HAD family and our capture of phosphoryl intermediate upon mutation, it is possible that this residue may also play a catalytic role. With our current experimental method for phosphatase assays, Mg2+ concentration higher than 100 mM causes precipitation of protein; therefore, it is hard to determine whether saturating concentration of Mg2+ would fully rescue the activity of D206A mutant. However, it would be interesting to investigate the structural and functional roles of this residue in other HAD family. For example, it was reported that mutation at this residue in P-type ATPase (Asp707) totally abolishes its activity.35
In summary, our current study provided definitive evidence for the existence of the phosphoryl-aspartate intermediate in the reaction catalyzed by Fcp/Scp phosphatase family. We identified Asp206 as a key residue for catalysis and metal binding, whose mutation can result in product trapping. Our results strongly suggest that Asp206 and its equivalent residues in other HAD family members play a structural and possible mechanistic role through divalent cation coordination. We believe these results will direct future studies on the mechanism of Scp1 and other HAD family enzymes.
Materials and Methods
Cloning, mutagenesis, and purification
Wild type human Scp1, residues 77–256, was sub-cloned and expressed in Escherichia coli BL21(DE3) strain using pHIS8 vector encoding a thrombin cleavable N-terminal octa-histidine tag.36 Mutant genes were generated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene, CA). The expression of Scp1 wild type or mutants was induced in E. coli at low temperature (16°C) with 250 µM IPTG and allowed to accumulate at high levels in overnight. Cells were harvested and resuspended in 0.5M NaCl, 15 mM imidazole, 50 mM Tris-HCl pH 8.0, 10% (v/v) glycerol, and 0.1% (v/v) Triton X-100. After sonication, cell debris was removed by centrifugation. The supernatant was loaded onto a Ni2+-NTA affinity column, washed extensively with sonication buffer and then with sonication buffer minus Triton X-100. Nearly homogeneous Scp1 was eluted with sonication buffer minus Triton X-100 but including 150 mM imidazole. Eluted protein was dialyzed into 20 mM Tris-HCl pH 8.0, 100 mM NaCl, 5 mM β-mercaptoethanol, and stored at a concentration of 2 mg/mL at 4°C. Samples for protein X-ray crystallography were subjected to thrombin processing to remove the octa-histidine tag followed by size exclusion chromatography for final cleanup. These samples were stored in 20 mM HEPES-Na+ pH 7.5, 100 mM NaCl, 3 mM dithiothreitol at a concentration of 12–16 mg/mL at −80°C.
pNPP assay
The steady-state parameters of D98A/N, E99Q/A, and D206A were measured using pNPP as a substrate. The reaction mixtures contained 50 mM Tris-acetate pH 5.5, 20 or 50 mM MgCl2, 0.5–50 mM of pNPP, and suitable amount of mutant proteins. The reactions were incubated at 37°C for 15 min and then quenched by adding 80 µL of 0.25N NaOH. Release of pNP was determined by measuring absorbance at 410 nm. The absorbance was converted to product concentration by a pNP standard curve. The data were analyzed by nonlinear regression performed in Matlab (The MathWorks, MA). The Mg2+ dependence of wild type Scp1 or Scp1D206A was measured by the same assay at constant substrate concentration with 0–100 mM Mg2+.
DiFMUP assay
DiFMUP (Invitrogen, CA) was dissolved in 100% DMSO to make 100 mM stock. Before each measurement, the DiFMUP was freshly diluted in 1× reaction buffer containing 50 mM Tris-acetate pH 5.5, 20 mM MgCl2 to desired concentrations. The substrate mixture and protein mixture were preincubated at 37°C for 10 min prior to mixing. The final reaction mixture contained 0–150 µM of DiFMUP and wild type Scp1 or Scp1 mutants. The generation of the fluorescent DiFMU was monitored continuously at excitation/emission maxima ∼358/450 nm for 2 h. The data were analyzed by the same method as described for pNPP assay.
Crystallization and structure determination
Scp1 mutants were crystallized in 0.5–0.8M ammonium sulfate, 100 mM HEPES-Na+ pH 7.0, with 0.2M lithium sulfate. Crystals were then transferred to a stabilizer consisting of 100 mM sodium citrate, pH 5.5 (or MES-Na+, pH 6.5), 30% (w/v) PEG 8000, 5–25 mMpNPP, and 10 mM MgCl2 or 0.2M MgCl2. The pH of the crystal stabilizer drop was retested and confirmed using pH paper. After soaking for 24 h, crystals were transferred to a cryoprotecting stabilizer containing 30% (v/v) glycerol, 30% (w/v) PEG8000, and 100 mM of the respective buffer of the original stabilizer. After a brief period of equilibration, crystals were frozen in nylon loops in liquid nitrogen and stored in liquid nitrogen before data collection. Data were collected at 100 K on beam-line 8.2.2 of the Advanced light source (ALS). Diffraction data were processed with HKL2000.37 The data collection statistics are summarized in Table II.
The crystal structures of Scp1 mutants were determined by molecular replacement (MR) using the Scp1D96N structure as a search model (PDB ID: 2ghq) using the program AmoRe38 available in the CCP4 software package.39 MR solutions were refined using CNS40 and REFMAC, reserving 5% of the measured and reduced structure factor amplitudes as an unbiased test set for cross validation (Rfree).41 SigmaA-weighted electron density maps (2Fo–Fc and Fo–Fc) were calculated after each cycle of refinement and carefully inspected to guide model rebuilding using O.42 The final models were evaluated by PROCHECK.43
Accession Numbers
Coordinates for structural studies in the paper have been deposited in the Protein Data Bank with human Scp1 D206A phosphoryl-Asp structure as 3l0b, D206A product trapped structure as 3l0c and D98A mutant as 3l0y.
Acknowledgments
The authors thank the staff of the Advanced Light Source (ALS), Berkeley, California for assistance during data collection at beam lines 8.2.2 and 5.0.1. They also thank Drs. Jack Kyte, Felicia A. Etzkorn, Xi Chen, Shuangluo Xia, Jianmin Gao, and Tony Hunter for helpful discussions. J. P. Noel and S. L. Pfaff are investigators of Howard Hughes Medical Institute.
References
- 1.Egloff S, Murphy S. Cracking the RNA polymerase II CTD code. Trends Genet. 2008;24:280–288. doi: 10.1016/j.tig.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 2.Meinhart A, Kamenski T, Hoeppner S, Baumli S, Cramer P. A structural perspective of CTD function. Genes Dev. 2005;19:1401–1415. doi: 10.1101/gad.1318105. [DOI] [PubMed] [Google Scholar]
- 3.Chapman RD, Heidemann M, Albert TK, Mailhammer R, Flatley A, Meisterernst M, Kremmer E, Eick D. Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science. 2007;318:1780–1782. doi: 10.1126/science.1145977. [DOI] [PubMed] [Google Scholar]
- 4.Egloff S, O'Reilly D, Chapman RD, Taylor A, Tanzhaus K, Pitts L, Eick D, Murphy S. Serine-7 of the RNA polymerase II CTD is specifically required for snRNA gene expression. Science. 2007;318:1777–1779. doi: 10.1126/science.1145989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chambers RS, Dahmus ME. Purification and characterization of a phosphatase from HeLa cells which dephosphorylates the C-terminal domain of RNA polymerase II. J Biol Chem. 1994;269:26243–26248. [PubMed] [Google Scholar]
- 6.Cho EJ, Kobor MS, Kim M, Greenblatt J, Buratowski S. Opposing effects of Ctk1 kinase and Fcp1 phosphatase at Ser 2 of the RNA polymerase II C-terminal domain. Genes Dev. 2001;15:3319–3329. doi: 10.1101/gad.935901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho H, Kim TK, Mancebo H, Lane WS, Flores O, Reinberg D. A protein phosphatase functions to recycle RNA polymerase II. Genes Dev. 1999;13:1540–1552. doi: 10.1101/gad.13.12.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yeo M, Lin PS, Dahmus ME, Gill GN. A novel RNA polymerase II C-terminal domain phosphatase that preferentially dephosphorylates serine 5. J Biol Chem. 2003;278:26078–26085. doi: 10.1074/jbc.M301791200. [DOI] [PubMed] [Google Scholar]
- 9.Yeo M, Lee SK, Lee B, Ruiz EC, Pfaff SL, Gill GN. Small CTD phosphatases function in silencing neuronal gene expression. Science. 2005;307:596–600. doi: 10.1126/science.1100801. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y, Kim Y, Genoud N, Gao J, Kelly JW, Pfaff SL, Gill GN, Dixon JE, Noel JP. Determinants for dephosphorylation of the RNA polymerase II C-terminal domain by Scp1. Mol Cell. 2006;24:759–770. doi: 10.1016/j.molcel.2006.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morris DP, Phatnani HP, Greenleaf AL. Phospho-carboxyl-terminal domain binding and the role of a prolyl isomerase in pre-mRNA 3′-end formation. J Biol Chem. 1999;274:31583–31587. doi: 10.1074/jbc.274.44.31583. [DOI] [PubMed] [Google Scholar]
- 12.Verdecia MA, Bowman ME, Lu KP, Hunter T, Noel JP. Structural basis for phosphoserine-proline recognition by group IV WW domains. Nat Struct Biol. 2000;7:639–643. doi: 10.1038/77929. [DOI] [PubMed] [Google Scholar]
- 13.Tonks NK. Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol. 2006;7:833–846. doi: 10.1038/nrm2039. [DOI] [PubMed] [Google Scholar]
- 14.Zhang ZY. Mechanistic studies on protein tyrosine phosphatases. Prog Nucleic Acid Res Mol Biol. 2003;73:171–220. doi: 10.1016/s0079-6603(03)01006-7. [DOI] [PubMed] [Google Scholar]
- 15.Cohen PT. Novel protein serine/threonine phosphatases: variety is the spice of life. Trends Biochem Sci. 1997;22:245–251. doi: 10.1016/s0968-0004(97)01060-8. [DOI] [PubMed] [Google Scholar]
- 16.Barford D, Das AK, Egloff MP. The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu Rev Biophys Biomol Struct. 1998;27:133–164. doi: 10.1146/annurev.biophys.27.1.133. [DOI] [PubMed] [Google Scholar]
- 17.Shi Y. Serine/threonine phosphatases: mechanism through structure. Cell. 2009;139:468–484. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 18.Chambers RS, Kane CM. Purification and characterization of an RNA polymerase II phosphatase from yeast. J Biol Chem. 1996;271:24498–24504. doi: 10.1074/jbc.271.40.24498. [DOI] [PubMed] [Google Scholar]
- 19.Kamenski T, Heilmeier S, Meinhart A, Cramer P. Structure and mechanism of RNA polymerase II CTD phosphatases. Mol Cell. 2004;15:399–407. doi: 10.1016/j.molcel.2004.06.035. [DOI] [PubMed] [Google Scholar]
- 20.Allen KN, Dunaway-Mariano D. Phosphoryl group transfer: evolution of a catalytic scaffold. Trends Biochem Sci. 2004;29:495–503. doi: 10.1016/j.tibs.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 21.Lahiri SD, Zhang G, Dunaway-Mariano D, Allen KN. The pentacovalent phosphorus intermediate of a phosphoryl transfer reaction. Science. 2003;299:2067–2071. doi: 10.1126/science.1082710. [DOI] [PubMed] [Google Scholar]
- 22.Wang W, Kim R, Jancarik J, Yokota H, Kim SH. Crystal structure of phosphoserine phosphatase from Methanococcus jannaschii, a hyperthermophile, at 1.8 A resolution. Structure. 2001;9:65–71. doi: 10.1016/s0969-2126(00)00558-x. [DOI] [PubMed] [Google Scholar]
- 23.Ghosh A, Shuman S, Lima CD. The structure of Fcp1, an essential RNA polymerase II CTD phosphatase. Mol Cell. 2008;32:478–490. doi: 10.1016/j.molcel.2008.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang W, Cho HS, Kim R, Jancarik J, Yokota H, Nguyen HH, Grigoriev IV, Wemmer DE, Kim SH. Structural characterization of the reaction pathway in phosphoserine phosphatase: crystallographic “snapshots” of intermediate states. J Mol Biol. 2002;319:421–431. doi: 10.1016/S0022-2836(02)00324-8. [DOI] [PubMed] [Google Scholar]
- 25.Pannifer AD, Flint AJ, Tonks NK, Barford D. Visualization of the cysteinyl-phosphate intermediate of a protein-tyrosine phosphatase by X-ray crystallography. J Biol Chem. 1998;273:10454–10462. doi: 10.1074/jbc.273.17.10454. [DOI] [PubMed] [Google Scholar]
- 26.Lahiri SD, Zhang G, Dunaway-Mariano D, Allen KN. Caught in the act: the structure of phosphorylated beta-phosphoglucomutase from Lactococcus lactis. Biochemistry. 2002;41:8351–8359. doi: 10.1021/bi0202373. [DOI] [PubMed] [Google Scholar]
- 27.Hausmann S, Shuman S. Defining the active site of Schizosaccharomyces pombe C-terminal domain phosphatase Fcp1. J Biol Chem. 2003;278:13627–13632. doi: 10.1074/jbc.M213191200. [DOI] [PubMed] [Google Scholar]
- 28.Cho H, Wang W, Kim R, Yokota H, Damo S, Kim SH, Wemmer D, Kustu S, Yan D. BeF(3)(-) acts as a phosphate analog in proteins phosphorylated on aspartate: structure of a BeF(3)(-) complex with phosphoserine phosphatase. Proc Natl Acad Sci USA. 2001;98:8525–8530. doi: 10.1073/pnas.131213698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diaz AR, Stephenson S, Green JM, Levdikov VM, Wilkinson AJ, Perego M. Functional role for a conserved aspartate in the Spo0E signature motif involved in the dephosphorylation of the Bacillus subtilis sporulation regulator Spo0A. J Biol Chem. 2008;283:2962–2972. doi: 10.1074/jbc.M709032200. [DOI] [PubMed] [Google Scholar]
- 30.Toyoshima C, Nomura H, Tsuda T. Lumenal gating mechanism revealed in calcium pump crystal structures with phosphate analogues. Nature. 2004;432:361–368. doi: 10.1038/nature02981. [DOI] [PubMed] [Google Scholar]
- 31.Toyoshima C, Norimatsu Y, Iwasawa S, Tsuda T, Ogawa H. How processing of aspartylphosphate is coupled to lumenal gating of the ion pathway in the calcium pump. Proc Natl Acad Sci USA. 2007;104:19831–19836. doi: 10.1073/pnas.0709978104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jung SK, Jeong DG, Chung SJ, Kim JH, Park BC, Tonks NK, Ryu SE, Kim SJ. Crystal structure of ED-Eya2: insight into dual roles as a protein tyrosine phosphatase and a transcription factor. FASEB J. 2010;24:560–569. doi: 10.1096/fj.09-143891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deshpande RA, Wilson TE. Identification of DNA 3′-phosphatase active site residues and their differential role in DNA binding, Mg2+ coordination, and catalysis. Biochemistry. 2004;43:8579–8589. doi: 10.1021/bi049434n. [DOI] [PubMed] [Google Scholar]
- 34.Rangarajan ES, Proteau A, Wagner J, Hung MN, Matte A, Cygler M. Structural snapshots of Escherichia coli histidinol phosphate phosphatase along the reaction pathway. J Biol Chem. 2006;281:37930–37941. doi: 10.1074/jbc.M604916200. [DOI] [PubMed] [Google Scholar]
- 35.McIntosh DB, Clausen JD, Woolley DG, MacLennan DH, Vilsen B, Andersen JP. Roles of conserved P domain residues and Mg2+ in ATP binding in the ground and Ca2+-activated states of sarcoplasmic reticulum Ca2+-ATPase. J Biol Chem. 2004;279:32515–32523. doi: 10.1074/jbc.M403242200. [DOI] [PubMed] [Google Scholar]
- 36.Jez JM, Ferrer JL, Bowman ME, Dixon RA, Noel JP. Dissection of malonyl-coenzyme A decarboxylation from polyketide formation in the reaction mechanism of a plant polyketide synthase. Biochemistry. 2000;39:890–902. doi: 10.1021/bi991489f. [DOI] [PubMed] [Google Scholar]
- 37.Otwinowski Z, Minor W. HKL: processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 38.Navaza J. AMoRe: an automated package for molecular replacement. Acta Crystallogr. 1994;A50:157–163. [Google Scholar]
- 39.Collaborative Computational Project, Number 4. CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr Sect D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 40.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr Sect D. 1998;54(Part 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 41.Brunger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature. 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- 42.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr Sect A. 1991;47(Part 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 43.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]