

Abstract
The antiphospholipid syndrome (APS) is a severe autoimmune disease associated with recurrent thrombosis and fetal loss and characterized by the presence of circulating autoantibodies (aAbs) mainly recognizing the N-terminal domain (DmI) of β2-glycoprotein I (β2GpI). To possibly block anti-β2GpI Abs activity, we synthesized the entire DmI comprising residues 1–64 of β2GpI by chemical methods. Oxidative disulfide renaturation of DmI was achieved in the presence of reduced and oxidized glutathione. The folded DmI (N-DmI) was purified by RP-HPLC, and its chemical identity and correct disulfide pairing (Cys4-Cys47 and Cys32-Cys60) were established by enzymatic peptide mass fingerprint analysis. The results of the conformational characterization, conducted by far- and near-UV CD and fluorescence spectroscopy, provided strong evidence for the native-like structure of DmI, which is also quite resistant to both Gdn-HCl and thermal denaturation. However, the thermodynamic stability of N-DmI at 37°C was remarkably low, in agreement with the unfolding energetics of small proteins. Of note, aAbs failed to bind to plates coated with N-DmI in direct binding experiments. From ELISA competition experiments with plate-immobilized β2GpI, a mean IC50 value of 8.8 μM could be estimated for N-DmI, similar to that of the full-length protein, IC50(β2GpI) = 6.4 μM, whereas the cysteine-reduced and carboxamidomethylated DmI, RC-DmI, failed to bind to anti-β2GpI Abs. The versatility of chemical synthesis was also exploited to produce an N-terminally biotin-(PEG)2-derivative of N-DmI (Biotin-N-DmI) to be possibly used as a new tool in APS diagnosis. Strikingly, Biotin-N-DmI loaded onto a streptavidin-coated plate selectively recognized aAbs from APS patients.
Keywords: antiphospholipid syndrome; β2-glycoprotein I; peptide synthesis; protein structure, biospectroscopy; autoimmune diseases
Introduction
The antiphospholipid syndrome (APS) is a severe autoimmune disease associated with a variety of clinical manifestations, including arterial and venous thrombosis, and recurrent fetal loss.1–3 APS is characterized serologically by high levels of autoantibodies (aAbs) mainly directed against β2-glycoprotein I (β2GpI),3,4 a 54-kDa plasma glycoprotein synthesized in the liver and abundantly present in normal plasma (∼0.2 mg mL−1).5 β2GpI is now recognized as the major autoantigen involved in APS,3,6 and the presence of anti-β2GpI Abs strongly correlates with the occurrence of thrombotic events in APS patients.4,7
The mature sequence of human β2GpI consists of 326 amino acids with four N-linked carbohydrate chains,8,9 localized in the third and fourth domain (see below) and accounting for about 19% of the protein mass (Fig. 1). The crystallographic structure reveals that human β2GpI is composed of four repeating units (Domains I–IV), which belong to the complement control protein family (CCP),10 and a distinctly folded C-terminal domain (Domain V) arranged like beads on a string to form an elongated J-shaped molecule.11,12 Conversely, small-angle X-ray scattering studies indicate that β2GpI in solution assumes a predominantly S-shaped conformation, resulting from a tilt between Domains II and III.13 β2GpI displays internal sequence and structural homology,8,11,12 and indeed Domains I–IV are homologous units of about 60 amino acids sharing a common elliptically β-sandwich structure, stabilized by two conserved disulfide bridges. These domains are also characterized by the presence of high proline content (i.e., 8–15%), a conserved Cys-Pro peptide bond in the N-terminal region of each domain, and a single Trp residue stacked against the disulfide bond connecting the first and third cysteine. Conversely, Domain V is aberrant because it contains 82 amino acids that fold into a central β-spiral structure flanked by two small helices. In addition, it contains three (instead of two) disulfides, has a relatively low proline content (i.e., 3.5%), and the single Trp residue is not structurally conserved. It is widely accepted that β2GpI binds to anionic phospholipid membranes using positively charged patches in Domain V [Fig. 1(A)],14 whereas it interacts with pathogenic aAbs by the N-terminal domain (DmI).4,15 Of note, high plasma levels of anti-β2GpI Abs recognizing DmI strongly correlate with thrombosis, whereas aAbs recognizing other different regions of β2GpI do not seem to be pathogenic.4,7 Mutagenesis studies indicate that the antigenic epitope of β2GpI in DmI is discontinuous in nature and comprises the amino acid residues Asp8 and Asp9, the Arg39-Arg43 segment, and the Domain I–II interlinker region.7,16,17 Whether anti-β2GpI aAbs directly bind to a constitutively expressed epitope of β2GpI in DmI or to a cryptic epitope that becomes exposed in DmI only after β2GpI binds to negatively charged surfaces has been the subject of a lengthy debate.18
Figure 1.
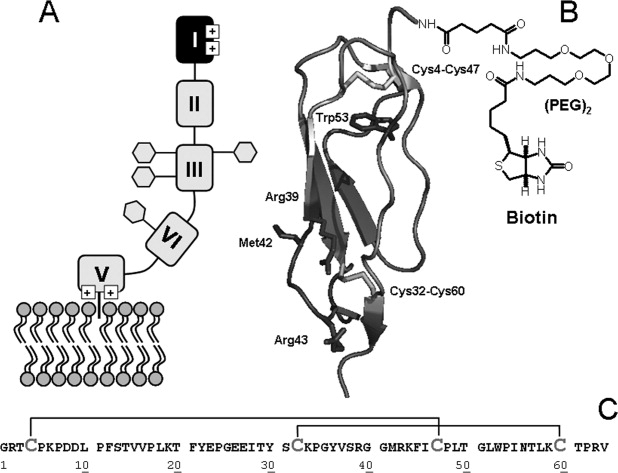
Structure and membrane binding of β2GpI. A: Schematic representation of full-length β2GpI interacting with phospholipid membranes. The four glycosylation sites in domain III and IV (i.e., Asn143, Asn164, Asn174, Asn243) are indicated by hexagons (adapted from Ref. 3). B: Schematic representation of the three-dimensional structure of N-DmI in the crystallographic structure of β2GpI (1qub).11 Disulfide bonds Cys4-Cys47 and Cys32-Cys60 and Trp53 are shown in stick together with Arg39, Met42, and Arg43 in the putatively primary antigenic epitope. Ribbon drawing of DmI was generated on the crystal structure of β2GpI using the software program ViewerPro 4.2 (Accelrys). C: Primary structure of the synthetic peptide DmI(1–64), as deduced from the amino acid sequence of full-length β2GpI.8 Cysteine residues are in gray and disulfide bonds are indicated by plain lines.
Multiple mechanisms have been proposed for explaining the clinical manifestations of APS, including (1) complement activation, (2) dysregulated activation of platelets, endothelial cells, and monocytes, (3) disruption of the interactions of anticoagulant factors (i.e., activated protein C and annexin A5), (4) inhibition of fibrinolysis, and (5) inhibition of thrombin-mediated activation of Factor XI.19–21 Even though it is still unclear which of these mechanisms is actually predominant in vivo, it is widely accepted that they are all affected by the presence of anti-β2GpI aAbs.19,22,23 Hence, the possibility to find out a molecule which is able to effectively compete with β2GpI for the binding to anti-β2GpI aAbs, without eliciting the cellular effects mediated by anti-β2GpI Abs, would be a promising approach for developing safer therapeutic strategies in APS.19 On the other hand, the presence of pathogenic and nonpathogenic aAbs recognizing several distinct regions of β2GpI in the plasma of APS patients,18 from Domain I to Domain V, has impaired so far the development of reliable immunochemical tools for the diagnosis of APS.3
In this work, we produced the entire Domain I, encompassing residues 1–64 of β2GpI, in high yields by total chemical synthesis [Fig. 1(B,C)]. The synthetic polypeptide was able to efficiently fold into a native-like structure, with the correct disulfide topology, and was remarkably stable to chemical and thermal denaturation. By ELISA competition assays, we demonstrated that synthetic N-DmI dose-dependently inhibited binding of full-length β2GpI to anti-β2GpI-Abs from APS patients, with IC50 values comparable to those of full-length β2GpI. The versatility of chemical synthesis was also exploited to produce an N-terminally biotin-(PEG)2-derivative of N-DmI (Biotin-N-DmI). When loaded onto a streptavidin-coated plate, Biotin-N-DmI selectively recognized pathogenic aAbs from APS plasma patients. Overall, our results demonstrate that large quantities of correctly folded DmI can be conveniently produced by chemical methods for potential therapeutic and diagnostic applications in APS.
Results
Purification of β2GpI
Natural β2GpI was purified from normal human plasma by means of perchloric acid precipitation,24 followed by heparin-sepharose and cation exchange chromatography. This procedure yields highly homogenous (>95%) β2GpI preparations, as obtained from RP-HPLC and SDS-PAGE (see Supporting Information Fig. S1). Mass spectrometry (MS) analysis of purified β2GpI yields an average molecular mass of 47,290 ± 10 a.m.u. (not shown). Under reducing conditions, β2GpI migrates as a single band at ∼53 kDa (Inset to Supporting Information Fig. S1), in agreement with the known lower electrophoretic mobility of glycosylated proteins.
Synthesis and chemical characterization of N-DmI
After peptide chain assembly, resin cleavage, and diethylether precipitation, the crude peptide with the Cys residues in the reduced form, R-DmI, was analyzed by RP-HPLC at 25°C [Fig. 2(A)]. The chromatogram shows two major peaks (i.e., p1 and p2) having a mass of 7163.4 ± 0.5 a.m.u., identical to the average theoretical value deduced from the primary structure of R-DmI in β2GpI (i.e., 7163.4 a.m.u.).8,9 These species are likely different cis ↔ trans proline isomers of the same polypeptide chain that expose a slightly different apolar surface to the column stationary phase and undergo isomerization on a time scale longer than that of the chromatographic separation. Notably, when the column temperature was decreased from 25 to 5°C, the intensity of p2 was much smaller, whereas p1 became predominant (see Supporting Information Fig. S2), indicating that lower temperatures favor p1 conformer(s). Similar observations on peak splitting have been reported with other Pro-rich synthetic peptides.25 Oxidative disulfide folding of R-DmI to yield the native-like species, N-DmI, was achieved by dissolving the crude R-DmI in Tris-HCl buffer, pH 8.4, and allowing the reaction to proceed for 24 h in the presence of the redox couple GSH:GSSG (1:4 mM). As shown in Figure 2(A), a single predominant peak at shorter retention time was obtained by RP-HPLC, compatible with the lower apolar surface that the folded N-DmI exposes to the RP-column. N-DmI was purified by semipreparative RP-HPLC, and its homogeneity and chemical identity were established by analytical RP-HPLC [Fig. 2(B)] and MS (Supporting Information Fig. S3), yielding a molecular mass (7159.4 ± 0.7 a.m.u.) four units lower than that of R-DmI, consistent with the formation of two disulfide bonds upon oxidative folding. The correctness of disulfide pairing was established by peptide mass fingerprint analysis with trypsin [Fig. 3(A)] and chymotrypsin [Fig. 3(B)]. MS data reported in Supporting Information Table S1 allowed us to cover the entire N-DmI sequence and to identify several proteolytic fragments each containing a single disulfide bond, Cys4-Cys47 or Cys32-Cys60 [Fig. 3(C)].
Figure 2.
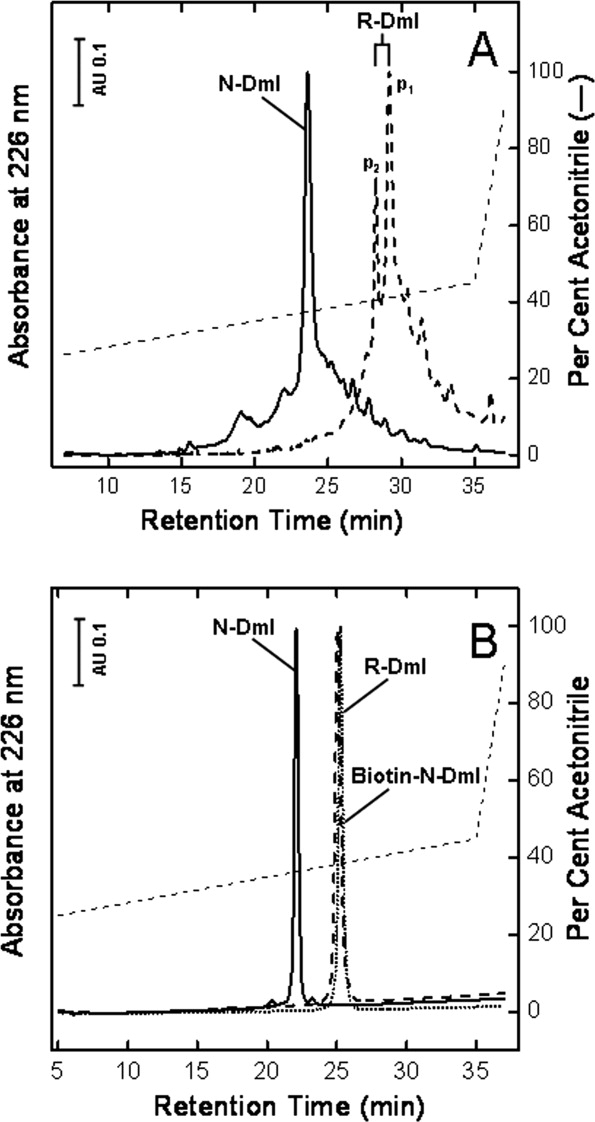
Purification of the synthetic N-DmI, R-DmI, and Biotin-N-DmI. A: RP-HPLC analysis of crude peptides. B: Purity check of HPLC-purified peptides. The column was eluted with a linear acetonitrile gradient (---) in 0.1% aqueous TFA at a flow rate of 0.8 mL min−1. The peptide material corresponding to the major peaks in the chromatograms was collected and subjected to MS analysis.
Figure 3.
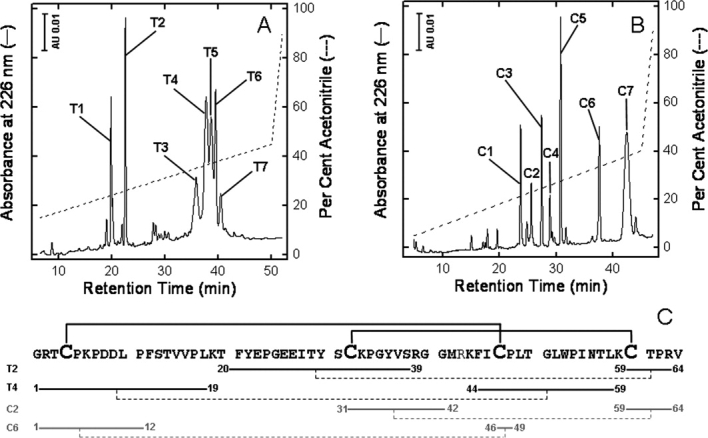
Assignment of disulfide pairing of the synthetic N-DmI by enzymatic peptide mass fingerprint analysis. A: RP-HPLC analysis of the proteolysis reaction with trypsin. B: RP-HPLC analysis of the proteolysis reaction with chymotrypsin. The chemical identity of tryptic (T) and chymotryptic (C) fragments was established by MS analysis, as reported in Supporting Information Table S1. C: Amino acid sequence and disulfide bond topology of the synthetic N-DmI, as deduced from MS data reported in Supporting Information Table S1. Only the peptide fragments containing a single disulfide bond (---) are indicated.
Folding kinetics of DmI
To improve oxidative disulfide folding, several additives were tested, such as l-arginine, β-mercaptoethanol, GSH and GSSG, and trimethylamine N-oxide. Of these, the redox couple GSH-GSSG was proven to work best in DmI renaturation, allowing us to obtain almost exclusively the correctly folded species in yields higher than 60% (Fig. 4), whereas in the absence of glutathione or with other additives (i.e., β-mercaptoethanol or arginine), the folding yields were always in the 30–40% range (see Supporting Information Table S2). At time intervals, aliquots of the refolding mixture were blocked by adding aqueous TFA and 6M Gdn-HCl to possibly solubilize all the intermediates generated during folding. Immediately after folding reaction was started, the solution became turbid (as also obtained by recording the absorbance at 350 nm), indicating the formation of some precipitate in the test tube (not shown). This was also confirmed by RP-HPLC analysis at short reaction times, showing the presence of only negligible amounts of DmI species in solution (Fig. 4). The protein pellet was centrifuged and analyzed by SDS-PAGE (Supporting Information Fig. S4). Under reducing conditions, a single intense band migrating at ∼7 kDa was present, whereas under nonreducing conditions any protein band could not be detected in the gel, suggesting that the precipitate remained undissolved in the sample loading buffer and did not even enter the gel. At longer reaction times, the solution became less turbid, and the correctly folded disulfide species appeared in the RP-HPLC plots. Altogether, these results can be rationalized according to the following Scheme 1
Figure 4.
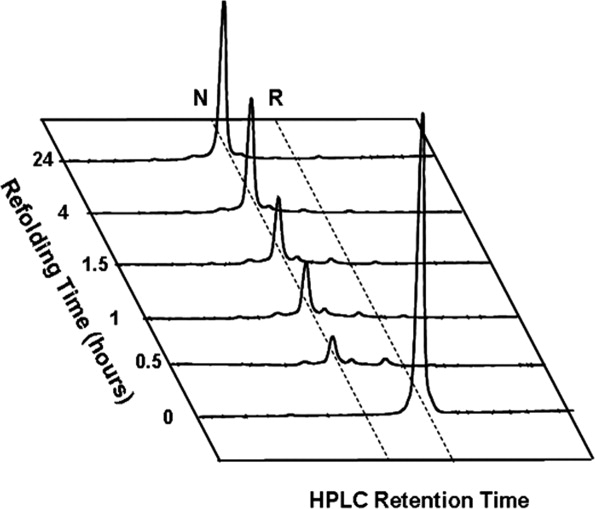
Time course RP-HPLC analysis of the oxidative disulfide folding of DmI. Fully reduced, HPLC-purified peptide R-DmI (0.25 mg) was allowed to fold at room temperature (20–22°C) in 0.1M Tris-HCl buffer, pH 8.4 (1 mL) in the presence of GSH (1 mM) and GSSG (4 mM). At time intervals, aliquots (10 μg) of the refolding mixture were acid quenched and analyzed by RP-HPLC (see Methods). R and N indicate the synthetic DmI with Cys-residues in the reduced and disulfide-bonded native state, respectively.
whereby only a low amount of soluble monomeric R-DmI exists in equilibrium with disulfide crosslinked insoluble aggregates, (DmI)n, to yield almost exclusively the natively folded product, N-DmI. Folding of DmI, in fact, is complicated by the presence of four cysteines and nine prolines. Cysteines, indeed, can combine in DmI to give seven different disulfide species, each containing one or two disulfide bonds. On the other hand, it is well known that in unfolded polypeptides, proline exists either in the cis and trans conformation, with a cis:trans ratio of 30:70, and that trans ↔ cis isomerization can remarkably slow down intramolecular protein folding,26 allowing the polypeptide chain to form intermolecular disulfide crosslinked aggregates. Of note, in native DmI, eight of the nine prolines are in the more stable trans conformation, whereas the remaining Pro17 is in the cis conformation.11
Conformational characterization of N-DmI
Fluorescence
The 280-nm emission spectrum of N-DmI recorded in sodium phosphate buffer, pH 7.4, displays a maximum centered at 347 nm [Fig. 5(A)], indicating that Trp53 is embedded in a polar environment. Nevertheless, fluorescence quenching experiments with acrylamide [see Fig. 5(B)] indicate that Trp53 in N-DmI is not exposed to the solvent, in agreement with the conformation that DmI assumes in the crystal structure of full-length β2GpI,11,12 where the indolyl moiety of Trp53 has an accessible surface area of only 5 Å2. The crystal structure also reveals that the indole N—H group is hydrogen bonded to the carbonyl oxygen of Pro5.11 Hence, we conclude that intramolecular hydrogen bonding of Trp53(N—H) with Pro5(C=O′), and not Trp exposure to the water solvent, is the major cause of the red-shifted emission of Trp53.27 In addition, the lack of tyrosine contribution indicates that an efficient Tyr-to-Trp energy transfer exists in N-DmI,28 in agreement with the crystal structure of β2GpI, showing that Tyr22 and Tyr30 are within Förster distance to Trp53. Notably, as shown in Figure 5(A), in the presence of 7M Gdn-HCl and at pH 7.4, the fluorescence λmax of N-DmI is red-shifted to 351 nm and the intensity increased by about sixfold [see also Fig. 7(A)]. These spectral changes can be well explained in the light of the three-dimensional structure of DmI in β2GpI, whereby Trp53 is stacked against the disulfide bond Cys4-Cys47.11,12 Indeed, disulfides are known to dramatically quench Trp fluorescence by an electron transfer mechanism.29 Upon guanidine-induced denaturation, the tryptophan–disulfide interaction is likely disrupted, with a resulting increase in the fluorescence intensity. For comparison, in Figure 5(A) are also shown the spectra of the disulfide-reduced species (R-DmI) in the presence or absence of denaturant and in acidic conditions (i.e., pH 2.5) to avoid disulfide formation/scrambling. Of note, the intensity of R-DmI spectrum at pH 2.5 and 7M Gdn-HCl is reduced by about 20% compared with that recorded at pH 7.4 in 7M Gdn-HCl, because of the quenching of Trp fluorescence occurring at low pH.28,29 In the absence of denaturant, and keeping the pH constant at 2.5, the emission of Trp53 in R-DmI is further reduced by 35%. This effect does not reflect any conformational change in R-DmI and is likely caused by the enhanced quantum yield of Trp fluorescence in the more dense 7M Gdn-HCl solution.30 Indeed, the intensity of the spectrum of the model compound Nα-acetyl-Trp-NH2 at pH 2.5 is about 30% lower than that taken at the same pH in 7M Gdn-HCl (data not shown).
Figure 5.
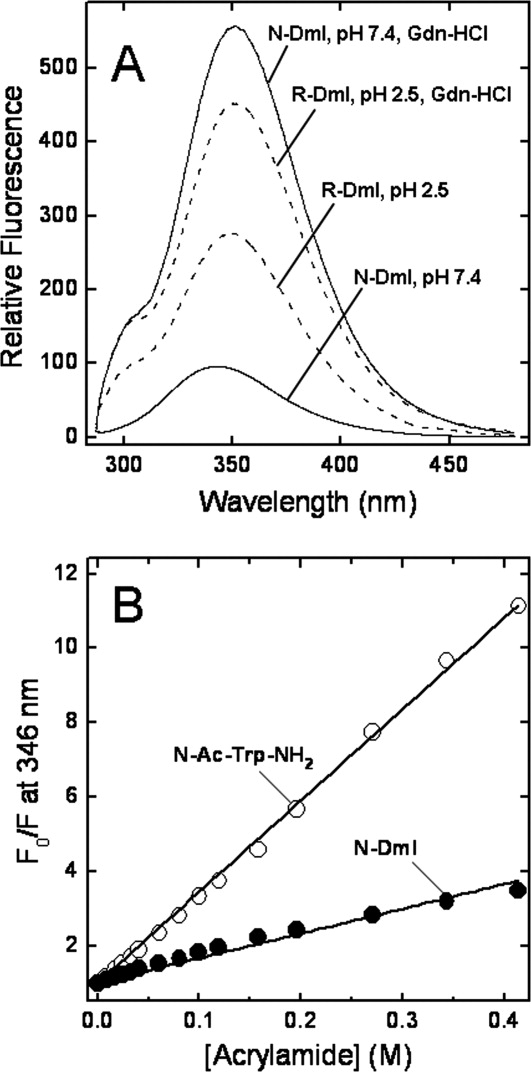
Fluorescence spectra and quenching experiments of DmI species. A: Fluorescence spectra of disulfide folded N-DmI (___) in 10 mM sodium phosphate buffer, pH 7.4, 0.15M NaCl in the presence or absence of 7M Gdn-HCl, as indicated; fluorescence spectra or the reduced species R-DmI (---) in 10 mM sodium phosphate buffer, pH 2.5, 0.15M NaCl in the presence or absence of 7M Gdn-HCl, as indicated. Protein samples (0.5 μM, 1.5 mL) were excited at 280 nm, and the spectra were recorded at 25°C ± 0.1°C. B: Acrylamide quenching of N-DmI fluorescence (•-•) and Nα-acetyl-Trp-NH2 (○-○). Fitting of data points to the Stern-Volmer equation (see Methods) yields Ksv values of 6.6 ± 0.7 and 25 ± 1.2 M−1 for N-DmI or Nα-acetyl-Trp-NH2.
Figure 7.
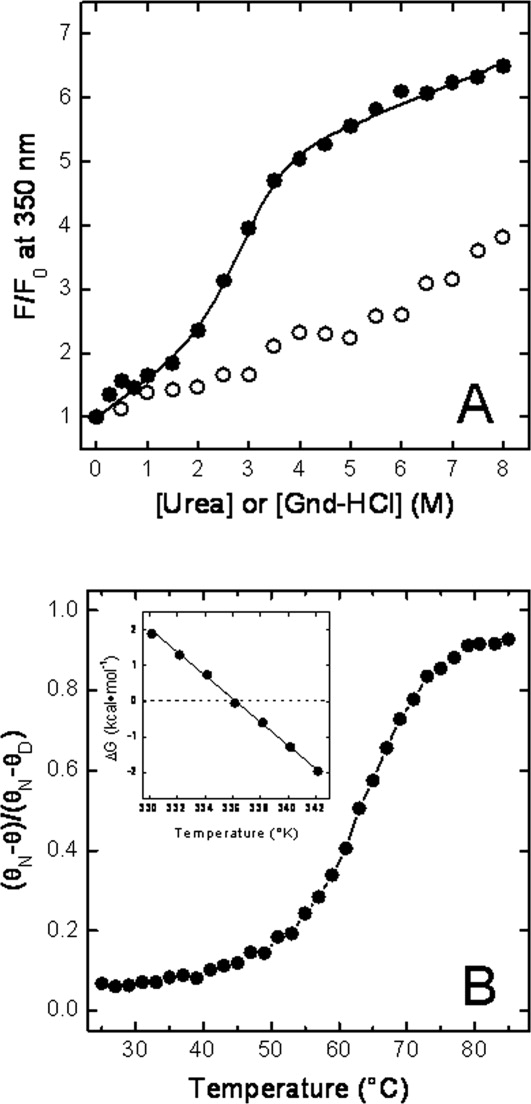
Stability of N-DmI. A: Denaturation of N-DmI induced by Gdn-HCl (•-•) and urea (○-○). Protein samples (1.5 mL, 0.5 μM) were excited at 280 nm, and the fluorescence intensity was recorded at 350 nm as a function of denaturant concentration. Fluorescence data are expressed as the ratio F/F0, where F0 and F are the fluorescence intensities of DmI in the absence or in the presence of denaturant. Continuous line represents the best fit of data points to Eq. (1), yielding a [Gdn-HCl]1/2 value of 2.8 ± 0.1M. B: Thermal denaturation of N-DmI (•-•). Denaturation was followed by recording the CD signal of the protein (2 μM) at 230 nm as a function of the sample temperature. CD signal is expressed as the ratio (θN − θ)/(θN − θD), where θ is the ellipticity at a given temperature, whereas θN and θD are the ellipticity values recorded at the lowest and highest temperature explored, respectively. Inset: Temperature dependence of the free energy change of denaturation, ΔGD, of N-DmI (•-•). All measurements were carried out at 25°C ± 0.1°C in 20 mM sodium phosphate buffer, pH 7.5, containing 0.15M NaCl.
Circular dichroism
A figure of about 53% of β-sheet secondary structure can be deduced from the X-ray structure of DmI in β2GpI.11,12 Notwithstanding, the far-UV CD spectrum of N-DmI does not conform to the features typical of a β-sheet protein, usually displaying a negative band at 210–215 nm and a positive band at 195–198 nm.31 The spectrum of N-DmI, instead, resembles that of the model compound Nα-acetyl-Trp-NH2 [Fig. 6(A) and Inset], with a negative absorption below 220 nm and a distinct positive band at 230 nm. Of note, this band disappears when the disulfide bonds are broken [Fig. 6(A)] and the spectrum of the reduced species, R-DmI, becomes that typical of an unfolded polypeptide chain.31 The unusual band at 230 nm can be assigned to the absorption of aromatic amino acids and in particular to Trp53 that in the β2GpI structure interacts with the disulfide bridge Cys4-Cys47 and with Tyr22. The contribution of aromatics to the far-UV CD is indeed most prominent in proteins displaying low CD signal (i.e., β-sheet proteins) and containing interacting aromatics.32 Of note, three aromatic pairs can be identified in the structure of DmI in β2GpI: Phe12-Tyr36, Tyr30-Phe45, and Tyr22-Trp53.11,12
Figure 6.
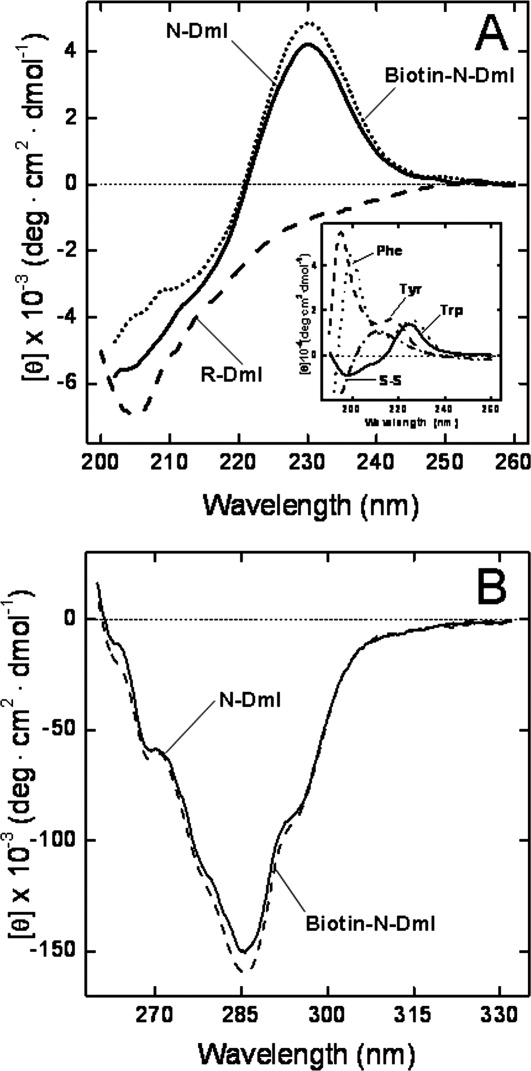
Circular dichroism spectra of N-DmI, R-DmI, and Biotin-N-DmI. A: Far-UV CD spectra. B: Near-UV CD spectra. CD spectra were recorded at a protein concentration of 20 and 140 μM in the far- and near-UV region, respectively. Inset: Far-UV CD spectra of the model compounds cysteine and Nα-acetyl-amide derivatives of Tyr, Phe, and Trp, as indicated. CD data for model compound solutions are expressed as molar ellipticity.
The near-UV CD spectrum of N-DmI [Fig. 6(B)] displays an extensive vibronic structure, demonstrating that, after in vitro folding, the synthetic peptide acquires a well-defined and compact fold.33 In particular, the 6-nm spaced bands at 263 and 269 nm can be assigned to the contribution of phenylalanines, whereas the absorption of the three tyrosines appears as a shallow band at 280 nm, superimposed to the dominant negative 1Lb band of Trp53 occurring at 285 and 293 nm.33 The presence of this band is consistent with the three-dimensional structure of β2GpI, showing that the single Trp53 is embedded in a rigid and asymmetric environment [see Fig. 1(B)].11
Thermodynamic stability
Taking advantage of the denaturant-dependent increase of fluorescence intensity, we monitored the denaturation of N-DmI as a function of urea or Gdn-HCl concentration [Fig. 7(A)]. In both cases, the fluorescence change was fully reversible. In the presence of Gdn-HCl, the denaturation curve displayed a sigmoidal shape, indicative of a cooperative unfolding transition, whereas in the presence of urea, the emission of N-DmI gradually increased without a distinct transition, in agreement with the well-known lower denaturant potency of urea compared with that of Gdn-HCl.34 Analysis of the fluorescence data was carried out within the assumption of a two-state denaturation process30,34 and allowed us to determine a [Gdn-HCl]1/2 value of 2.8 ± 0.1M, with a denaturation index m of −1.5 ± 0.1 kcal mol−1 M−1. Linear extrapolation of denaturation free energy change, ΔGD, to [Gdn-HCl] = 0 yielded a 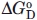 of 4.4 ± 0.1 kcal mol−1 at 25°C.
of 4.4 ± 0.1 kcal mol−1 at 25°C.
Denaturation of N-DmI was also monitored by recording the decrease of the CD signal at 230 nm as a function of temperature [Fig. 7(B)]. Even in this case, the unfolding process was highly cooperative and fully reversible. From the plot of ΔGD versus T in the transition region, a melting temperature, Tm, of 64.5°C ± 0.1°C was calculated, together with the enthalpy [ΔHm = 47 ± 2 kcal mol−1] and entropy [ΔSm = 138 ± 5 cal mol−1 K−1] change of denaturation at Tm. The values of ΔH(T) in the transition region were calculated using the van't Hoff equation. From the plot of ΔH(T) versus T, a value of ΔCp (i.e., the heat capacity change of denaturation at constant pressure) was calculated as 390 ± 50 cal mol−1 K−1. The conformational stability, ΔGD, of N-DmI at 25 or 37°C was obtained by inserting the values of Tm, ΔHm, ΔSm, and ΔCp, previously determined, into Eq. (3) and calculated as 3.3 ± 0.1 kcal mol−1 at 37°C and 4.5 ± 0.1 kcal mol−1 at 25°C. The latter value is very similar to that estimated from Gdn-HCl-induced denaturation of N-DmI at the same temperature (i.e., 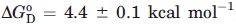 ). It is interesting to note that both [Gdn-HCl]1/2 and Tm values determined for N-DmI are very similar to those previously estimated for the full-length β2GpI (i.e., [Gdn-HCl]1/2 ∼ 2.5M and Tm = 63.5°C ± 0.1°C).8,35 These findings reflect the internal sequence and structural similarity of β2GpI and suggest that the protein domains behave independently during denaturation, in keeping with the inherent interdomain flexibility of β2GpI.13
). It is interesting to note that both [Gdn-HCl]1/2 and Tm values determined for N-DmI are very similar to those previously estimated for the full-length β2GpI (i.e., [Gdn-HCl]1/2 ∼ 2.5M and Tm = 63.5°C ± 0.1°C).8,35 These findings reflect the internal sequence and structural similarity of β2GpI and suggest that the protein domains behave independently during denaturation, in keeping with the inherent interdomain flexibility of β2GpI.13
Despite the relatively high resistance to chemical and thermal denaturation, the difference in free energy between the denatured and native state, ΔGD, of N-DmI is only 6- to 7-fold larger than the energy due to thermal motion of molecules at 37°C (i.e., RT ∼ 0.6 kcal mol−1). This behavior is quite common in small-size globular proteins and arises from the low values of the danaturation index, m, and heat capacity change of unfolding, ΔCp, that characterize their chemical or thermal denaturation.36 In the case of N-DmI, however, two additional factors contribute to protein stability: the presence of disulfide bridges and high proline content. In N-DmI structure, Cys4-Cys47 and Cys32-Cys60 crosslink the N- and C-terminal ends to the central β-sheet and thus can stabilize local interactions in the native state. On the other hand, the two disulfides can also stabilize N-DmI by reducing the conformational entropy of the polypeptide in the unfolded state, with a resulting lower entropy change of unfolding, ΔSD.37 Similar considerations apply for the presence in N-DmI of nine Pro residues, which account for a relative abundance of 14%, much higher than the frequency of Pro found in natural proteins (i.e., 4.8%). The pyrrolidine ring, in fact, imposes severe steric constrains to the polypeptide chain preferentially in the unfolded state, with a resulting decrease of ΔSD and an increase in ΔGD.38
ELISA competition experiments
The ability of N-DmI to competitively inhibit binding of anti-β2GpI Abs to natural β2GpI was investigated by absorbing purified β2GpI onto hydrophilic plates and then incubating the plates with increasing concentrations of N-DmI. Natural β2GpI and RC-DmI (i.e., Cys-reduced and carboxamidomethylated DmI) were used as a positive and negative control, respectively. A fixed volume of plasma from three patients affected by APS was added in all experiments, and the residual amount of aAbs remaining on the plate was quantified by using a secondary alkaline phosphatase-conjugated anti-human IgG antibody detection method. The selected patients (P1, P2, and P3) displayed triple positivity in specific assays for lupus anticoagulant (LAC+), anticardiolipin antibodies (IgG aCL+), and anti-β2GpI antibodies (IgG aβ2GpI+). Fitting of data points shown in Figure 8 to Eq. (4) allowed us to estimate the IC50 values reported in Table I. These data can be summarized as follows: first, free β2GpI competes with plate-bound β2GpI for binding to anti-β2GpI Abs, with a mean IC50 value of 6.4 μM, similar to that reported by others.39 This finding seems to argue against the existence of a cryptic epitope in DmI and suggests that the major antigenic epitope is constitutively expressed in the full-length protein in solution, accessible for binding to anti-β2GpI aAbs.18 Second, the synthetic N-DmI can effectively compete with immobilized β2GpI for binding to anti-β2GpI aAbs, with a mean IC50 value only 30% lower than that of full-length β2GpI. Third, the unfolded RC-DmI was unable to inhibit binding of antibodies from plasma patients to immobilized β2GpI, up to the highest concentration of RC-DmI tested (i.e., 48 μM), in agreement with previous results obtained with reduced full-length β2GpI. These results indicate that the antigenic epitope on DmI is nonlinear (i.e., comprises residues that are close in the three-dimensional structure but quite distant in the amino acid sequence) and is formed only after the protein domain folds into the native structure.
Figure 8.
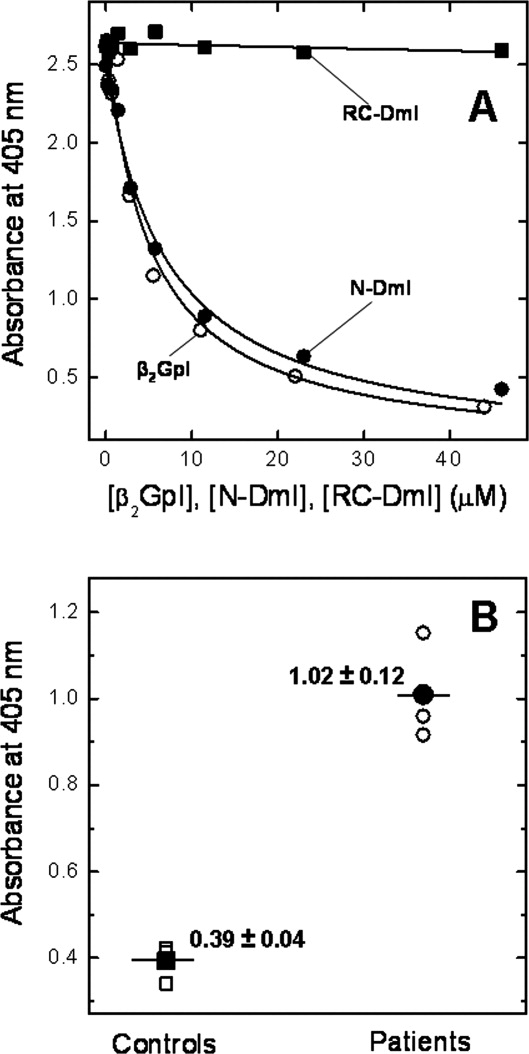
ELISA experiments. A: Competitive inhibition experiments with plate-bound β2GpI were carried out by mixing diluted plasma from APS patients with an equal volume of a solution at increasing concentrations of competitor: β2GpI (○), N-DmI (•), and RC-DmI (▪). The resulting solution was added to β2GpI-coated wells and incubated for 1 h at 37°C. After washing, quantification of anti-β2GpI Abs still bound to the microplate-absorbed β2GpI was performed by using a secondary alkaline phosphatase-conjugated anti-human IgG antibody in the presence of pNPP. The release of p-nitrophenol was determined by measuring the absorbance of the solution at 405 nm, after incubation for 30 min at 25°C. Fitting of data points to Eq. (4) yielded the IC50 values reported in Table I. B: Direct binding ELISA experiments were carried out by incubating streptavidin-coated microtiter plates with a solution of Biotin-N-DmI. Thereafter, a fixed volume of plasma from selected APS patients was added, and quantification of anti-β2GpI Abs bound to Biotin-N-DmI was carried out as detailed in A. Open symbols (○, □) refer to the experimental values, whereas filled symbols (•, ▪) are the average values together with the standard deviation.
Table I.
IC50 values (μM) obtained by ELISA competition binding experimentsa
Synthesis and characterization of N-biotinyl- NH-(PEG)2-N-DmI
To demonstrate the utility of the chemical accessibility to β2GpI domain I, we produced a biotinylated N-terminal derivative of N-DmI (Biotin-N-DmI) by solid-phase peptide synthesis [Fig. 1(B)] to be used for developing novel avidin–biotin ELISA systems in the diagnosis of APS. Biotin-N-DmI was refolded and purified as detailed for wild-type N-DmI. The chemical identity and homogeneity of Biotin-N-DmI were established by RP-HPLC [Fig. 2(B)] and MS (Supporting Information Fig. S3) that yielded a molecular mass of 7702.5 ± 0.6 a.m.u, consistent with the incorporation of the N-biotinyl-NH-(PEG)2-CO-moiety [Fig. 1(B)]. Of note, Biotin-N-DmI elutes in RP-HPLC later than N-DmI, in agreement with the apolar nature of biotin [Fig. 2(B)]. The comparative analysis of the CD spectra in the far- and near-UV region well documents that the N-terminal extension does not alter neither the secondary nor the tertiary structure of N-DmI (Fig. 6).
Finally, a direct ELISA system was developed to test whether plate-immobilized Biotin-N-DmI was able to recognize anti-β2GpI aAbs in the plasma of APS patients. With this aim, a Biotin-N-DmI solution (200 μL, 25 μg mL−1) was incubated with streptavidin-coated plates. Thereafter, a fixed volume of plasma from the triple positive P1, P2, and P3 APS patients, previously selected, was added in each well, and the residual amount of aAbs remaining on the plate was quantified by using the secondary alkaline phosphatase-conjugated anti-human IgG antibody detection method previously described. Strikingly, the data shown in Figure 8(B) clearly indicate that Biotin-N-DmI can effectively discriminate between the plasma of APS patients and that of healthy subjects, used as controls. Notably, when the synthetic wild-type N-DmI was coated onto a plastic plate, it failed to recognize anti-β2GpI aAbs in direct ELISA experiments (not shown), likely because the plate-bound N-DmI was poorly accessible for interaction with aAbs.40 In the case of Biotin-N-DmI, instead, N-DmI is ready for interaction likely because it is properly spaced by a 20-atom linker from the biotin–streptavidin complex on the plate [see Fig. 1(B)].
Discussion
Arterial and venous thrombosis are the most frequent clinical manifestations of APS,2 which strongly associate to high titers of aAbs directed against Domain I of β2GpI.6,22 To date, the only treatment proven to reduce the risk of thrombosis in APS is life-long anticoagulation, which often has severe side effects.3 Despite antithrombotic therapy, a significant proportion of patients with APS undergo rethrombosis,41 likely because anticoagulant therapy affects only the final outcome, without interfering with the early biochemical events from which thrombotic events originate, that is production of anti-β2GpI Abs and binding to β2GpI.19,22,42 Hence, the possibility to identify a molecule that is able to block anti-β2GpI Abs activities could disclose new therapeutic strategies in APS. With respect to this, the recombinant N-terminal β2GpI domain I (rDmI) has been expressed in submilligram quantities in E. coli as a C-terminally -Gly-(His)6-tagged derivative, and, more recently, it has been shown to inhibit the activity of pathogenic anti-β2GpI Abs in mice.43,44
Here, we have demonstrated that large quantities (>30 mg) of correctly folded and functionally active DmI can be produced in high yields in a fast (less than 2 weeks) and convenient way by chemical methods for future structural (i.e., NMR) and functional studies. The purity of the synthetic DmI was ascertained by RP-HPLC, SDS-PAGE, and MS, whereas its chemical identity and correctness of disulfide pairing were established by peptide mass fingerprint analysis with trypsin and chymotrypsin. All spectroscopic data herein reported are fully consistent with the crystal structure of DmI in natural β2GpI and concurrently indicate that the synthetic N-DmI has a native-like structure.11 This finding is particularly important, allowing us to interpret antibody-binding properties of N-DmI on the basis of the structure it assumes in the natural β2GpI.11 The results of ELISA competition experiments [Fig. 8(A) and Table I] indicate that the synthetic N-DmI is able to effectively inhibit binding of aAbs to plate-immobilized β2GpI with an affinity similar to that exhibited by full-length β2GpI. The versatility of solid-phase peptide synthesis will be also exploited in structure–activity relationship studies for improving the affinity of DmI for anti-β2GpI Abs by incorporating coded and noncoded amino acids with tailored side chains.45
Besides the typical clinical manifestations of APS, the diagnosis of this autoimmune disease greatly depends upon laboratory diagnostics that, however, is complicated by the limited specificity of existing assays for detecting clinically relevant antiphospholipid aAbs.19,46 Plasma of APS patients, in fact, contain pathogenic anti-β2GpI aAbs, predominantly recognizing DmI,4 and nonpathogenic anti-β2GpI aAbs, recognizing β2GpI domains different from DmI.18 Therefore, ELISA systems based on direct interaction of aAbs with immobilized β2GpI do not allow us to safely take high titers of anti-β2GpI aAbs as a reliable risk factor of thrombosis in APS patients.19,46
The data shown in Figure 8(B) clearly demonstrate that streptavidin-bound Biotin-N-DmI can selectively recognize, in direct ELISA experiments, anti-β2GpI aAbs from APS patients with a history of thrombosis and displaying triple positivity for lupus anticoagulant (LAC+), anticardiolipin antibodies (IgG aCL+), and anti-β2GpI antibodies (IgG aβ2GpI+). These results are unprecedented and disclose novel opportunities for developing more reliable diagnostic tools based on avidin/biotin ELISA systems. With respect to this, systematic experiments are undergoing in our laboratories to reduce the background signal of plasma samples in control subjects and to validate our data with a much larger set of patients displaying different clinical manifestations of APS. Preliminary results also indicate that pathogenic aAbs can be easily purified form APS plasma patients by immunoaffinity chromatography using Biotin-N-DmI bound to a streptavidin-linked column (unpublished). In conclusion, we have shown here that large quantities of correctly folded and functionally active DmI can be conveniently produced by chemical methods for potential therapeutic and diagnostic applications in APS.
Materials and Methods
Purification and chemical characterization of β2GpI
Natural β2GpI was purified from normal human plasma by means of perchloric acid precipitation, followed by affinity chromatography on a HiTrap (1 cm × 2.5 cm) heparin-Sepharose column (GE-Healthcare) and cation exchange chromatography on a Mono-S (0.6 cm × 5 cm) column (GE-Healthcare). The homogeneity of β2GpI preparations was established by Coomassie-stained polyacrylamide gel electrophoresis in the presence of SDS (4–12% acrylamide) (SDS-PAGE), under reducing and nonreducing conditions, and by RP-HPLC on a Zorbax (Agilent Technologies, Santa Clara, CA) C4 analytical column (4.6 mm × 150 mm), eluted with a linear acetonitrile–0.1% TFA gradient. The protein material eluted from the column was lyophilized and analyzed by MS. Typically, samples (10 μL, 1–10 μM) in water:acetonitrile solution (1:1 v/v), containing 1% formic acid, were loaded at a flow rate of 10 μL min−1 on a Mariner ESI-TOF instrument from PerSeptive Biosystems (Stafford, TX). Spray tip potential was set at 3.0 kV, whereas the nozzle potential and temperature were set at 200 V and 140°C, respectively. Deglycosylation of β2GpI with N-glycanase F (Roche, Mannheim, Germany) was carried out as detailed in the Supporting Information.
Synthesis and chemical characterization
Synthesis
The peptide sequence 1–64 of β2GpI was synthesized by the solid-phase method using the 9-fluorenylmethyloxycarbonyl(Fmoc) strategy on a model PS3 automated synthesizer from Protein Technologies International (Tucson, AZ).47 The peptide chain was assembled stepwise on a NovaSyn TGA resin (Novabiochem, Switzerland) derivatized with Fmoc-Val (0.24 mequiv g−1). tert-butyloxycarbonyl side-chain protecting group was used for Lys and Trp; tert-butyl for Ser, Thr, Asp, Glu, and Tyr; triphenylmethyl for Asn and Cys; and 2,2,4,6,7-pentamethyldihydrobenzofuran-5-sulfonyl group was used for Arg. Removal of Nα-Fmoc-protecting groups was achieved by treatment for 20 min with 20% piperidine in N-methylpirrolidone. Standard coupling reactions were performed with 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and 1H-hydroxy-benzotriazole (HOBt) as activating agents, with a fourfold molar excess of Nα-Fmoc-protected amino acids (Novabiochem) in the presence of diisopropylethylamine. For double couplings at peptide bonds involving Val, Ile, Leu, and Phe, the stronger activator 2-(7-aza-1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate was used (HATU). After peptide assembly was completed, the side chain-protected peptidyl resin was treated for 120 min at room temperature with a mixture of TFA/H2O/ethandithiol/triisopropylsilane (90:5:4:1 v/v/v/v). The resin was removed by filtration, and the acidic solution, containing the unprotected peptide, was precipitated with ice-cold diethylether and then lyophilized. The crude peptide with Cys residues in the reduced free thiol state, R-DmI, was fractionated by RP-HPLC on a Zorbax C18 analytical column. The peptide material eluted in correspondence of the major chromatographic peaks was collected, lyophilized, and analyzed by MS. Disulfide-mediated oxidative renaturation of the crude R-DmI, to yield the correctly folded species, N-DmI, was carried out by dissolving the crude peptide (1.8 mg mL−1) in 0.1M Tris-HCl buffer, pH 8.4, and allowing the reaction to proceed for 24 h in the presence of 1 mM GSH and 4 mM GSSG. The folding reaction was monitored by RP-HPLC, using a Zorbax C18 analytical column. For preparative purposes, aliquots (∼2 mg) of the crude N-DmI were injected onto a semipreparative Grace-Vydac (Hesperia, CA) C-18 column (1 cm × 25 cm, 5-μm particle size) eluted with a linear acetonitrile–0.1% TFA gradient from 30 to 42% in 30 min. The material corresponding to the major peak was collected, lyophilized, and used for subsequent studies.
Biotin-N-DmI was synthesized by reacting the peptidyl 1–64 resin with a twofold molar excess of N-biotinyl-NH-(PEG)2-COOH (Novabiochem; cat. 01-63-0133) in the presence of HBTU and HOBt. The resulting biotinylated DmI was purified and characterized as detailed above for the wild-type peptide.
Disulfide bonds assignment
The refolded peptide, N-DmI, (30 μg) was subjected to proteolysis with bovine trypsin (Sigma) in 50 mM NaHCO3 buffer, pH 8.4 (300 μL), or with bovine chymotrypsin (Sigma) in 50 mM Tris-HCl buffer, pH 7.8, containing 10 mM CaCl2. Reactions were allowed to proceed for 3 h at 37°C using a protease:substrate ratio of 1:25 (w/w). Thereafter, proteolysis reactions were stopped by acid quenching with 4% aqueous TFA, and immediately analyzed by RP-HPLC on a Zorbax C18 analytical column (4.6 mm × 150 mm). The chemical identity of the proteolytic fragments was established by MS analysis.
Refolding kinetics
To initiate folding, HPLC-purified R-DmI was dissolved (0.25 mg mL−1) in 0.1M Tris-HCl buffer, pH 8.4, in the absence or in the presence of 1 mM GSH and 4 mM GSSG. Before use, all buffers were extensively degassed under vacuum for at least 30 min. The folding intermediates were trapped in a time course manner by acidifying aliquots (50 μL) of the refolding mixture with an equal volume of 4% (v/v) aqueous TFA. Acid-trapped intermediates were added with 7M Gdn-HCl solution (280 μL) and analyzed by RP-HPLC. The yield of the correctly folded species was estimated by integrating the area under the chromatographic peaks.
Reduction and carboxamidomethylation of cysteines
Purified N-DmI (150 μg) was reduced for 40 min at 37°C in 0.5M Tris-HCl buffer (125 μL), pH 8.3, containing 1 mM EDTA, 6M Gdn-HCl, and 0.1M dithiothreitol. Carboxamidomethylation of Cys residues was carried out for 1 h in the dark, in the presence of 0.2M iodoacetamide, maintaining the solution pH constant at 8.3. The reduced and carboxamidomethylated peptide, RC-DmI, was purified by RP-HPLC, and its chemical identity was established by MS analysis.
Spectroscopic measurements
Peptide concentration was determined by UV absorption at 280 nm on a double-beam V-630 spectrophotometer from Jasco (Tokyo, Japan). Molar absorptivity values were calculated according to Pace et al.48 and taken as 10,200 M−1 cm−1 for N-DmI and Biotin-N-DmI and 9970 M−1 cm−1 for R-DmI and RC-DmI. Circular dichroism (CD) spectra were recorded on a Jasco J-810 spectropolarimeter equipped with a water-jacketed cell holder, connected to a NesLab RTE-111 (Newington, NH) water-circulating bath. The final spectra resulted from the average of four accumulations after base line subtraction. CD data were expressed as the mean residue ellipticity, [θ] = θobs·MRW/(10·l·c), where θobs is the observed signal in degrees, MRW is the mean residue weight, l is the cuvette pathlength in cm, and c is the protein concentration in g mL−1. Fluorescence spectra were recorded on a Jasco model FP-6500 spectrofluorimeter, equipped with a Peltier model ETC-273T temperature control system. Protein samples were excited at 280 nm, using excitation and emission slits of 10 nm. Fluorescence quenching experiments were performed by recording the decrease of fluorescence intensity of N-DmI or Nα-acetyl-tryptophanamide as a function of acrylamide concentration. Fluorescence quenching data were fitted to the Stern-Volmer equation: F0/F = 1 + Ksv·[Q], where F0 and F are the fluorescence intensities in the absence and presence of quencher, Q, and Ksv is the Stern-Volmer quenching constant.28
Stability measurements and data analysis
Chemical denaturation
Gdn-HCl and urea-induced denaturation experiments were carried out by exciting protein samples at 280 nm (excitation slit 5 nm) and recording the fluorescence intensity at 350 nm (emission slit 10 nm). Before measurements, samples were incubated for 1 h at 25°C ± 0.1°C. At each denaturant concentration, the fluorescence signal was subtracted for that of the corresponding blank. Reversibility of denaturation was estimated by measuring the recovery of the fluorescence intensity upon 20-fold dilution of a protein stock solution (10 μM) in 8M Gdn-HCl or 8M urea with nondenaturing buffer.
Thermal denaturation
The decrease of the CD signal at 230 nm was recorded as a function of the sample temperature, T. Denaturation experiments were carried out in a 1-cm pathlength cuvette heated under gentle stirring at a linear heating rate of 40°C h−1. Reversibility of the thermal unfolding was determined by measuring the recovery of the CD signal upon cooling to the starting temperature.
Data analysis
Denaturant-induced unfolding data were analyzed within the framework of a two-state process, N ⇔ D, and the data points fitted to the equation30:
| (1) |
where F is the observed fluorescence intensity, FN and FU are the intensities of the native (N) and denatured (D) state in the absence of denaturant, sN and sU are the base line slopes for the native and denatured regions, is the free energy change for the unfolding reaction in the absence of denaturant at 25°C, and m is the denaturation index (m = −dΔGU/d[D]), which is the dependence of ΔGD on denaturant concentration. Alternatively, was estimated by linear extrapolation to [Gdn-HCl] = 0 of ΔGD values calculated in the transition region, according to the equation34:
| (2) |
Thermal denaturation transition curves were analyzed according to a two-state model, as previously detailed.49 For each temperature in the transition region, it is possible to derive the equilibrium denaturation constant, KD, and the free energy change of unfolding, ΔGD = −RT·lnKD, where R is the gas constant (1.987 cal mol−1 K−1) and T is the absolute temperature. The melting temperature, Tm, defined as the temperature at which ΔGD = 0 was derived from the linear regression equation obtained by plotting ΔGD as a function of T in the transition region. Entropy, ΔSm, and enthalpy, ΔHm, change of unfolding at Tm were calculated according to the equations ΔSm = −dΔG/dT and ΔHm = Tm·ΔSm, respectively. The enthalpy change, ΔHD(T), at a given temperature in the transition region was calculated by plotting the value of −R·lnKD as a function of 1/T, using the van't Hoff equation ΔHD(T) = −[d(lnKD)/d(1/T)]R. The heat capacity change of unfolding at constant pressure, ΔCp = Cp(D) − Cp(N), was calculated as the slope (ΔCp = dΔH(T)/dT) of the straight line interpolating the data points in the plot of ΔHD(T) versus T. The conformational stability of N-DmI outside the transition region, at 25 or 37°C, was determined according to the equation:
| (3) |
Fitting of data points was performed using the computer program Origin version 7.5 (Microcal, CA).
Serological assays and ELISA experiments
Serological assays
Plasma samples from three patients (P1, P2, and P3) with primary APS, a history of thrombosis, and positive for anti-β2GpI IgG antibodies were included in this study. Ethical approval for the study was granted by the Research Ethics Committee of the University of Padova. Antibodies against β2GpI (anti-β2GpI Abs) were measured by ELISA, as described earlier, and were considered positive when the value (arbitrary units) exceeded the 99th percentile obtained using plasma from 40 healthy subjects (16 U).50 To determine LAC activity, activated partial thromboplastin time and diluted Russell Viper Venom Time (dRVVT) assays were performed as detailed elsewhere.4,51 Patients were considered LAC positive with a dRVVT mixing test ratio of more than 1.2 and a positive confirmatory test. Anticardiolipin antibodies (anti-CL Abs) were measured by ELISA as described and considered positive when IgG phospholipid (GPL) units were 40 or more.4,50
Competitive inhibition ELISA
Polyvinyl chloride 96-wells microtiter plates from Falcon (Franklin Lakes, NJ) were coated overnight at 4°C with a solution (10 μg mL−1, 100 μL/well) of β2GpI purified from human plasma in 0.1M sodium bicarbonate buffer, pH 9.5. Thereafter, the plates were washed four times (4×) with phosphate-buffered saline (PBS), containing 0.1% Tween-20, and the reactive sites blocked by treatment with 4% BSA (100 μL/well) in PBS for 2 h. Samples were prepared by mixing a fixed volume (50 μL) of diluted plasma (1:50 v/v) from selected APS patients with an equal volume of solutions containing increasing concentrations (i.e., from 0 to 46 μM) of natural β2GpI or synthetic N-DmI and RC-DmI. Each solution (100 μL) was incubated in the corresponding well for 1 h at 37°C. Anti-β2GpI Abs titers of 218, 340, and 392 U mL−1 were determined in plasma samples of patient P1, P2, and P3, respectively, by an ELISA as described elsewhere.50 Briefly, the plates were washed (4×) with PBS, containing 0.1% Tween-20, and 100 μL of alkaline phopshatase-conjugated anti-human IgG (Sigma), diluted 1:1000 (v/v) in PBS-2% BSA, was added per well and incubated at 37°C for 1 h. Then, the plates were washed (4×) with PBS-0.1% Tween-20, and p-nitrophenylphosphate (pNPP) (Sigma) (100 μL/well) was added and incubated for 30 min. The release of p-nitrophenol was monitored by recording the absorbance at 405 nm using a microplate autoreader from Tecan (Männedorf, Switzerland). The data were corrected for the corresponding background values, plotted as a function of inhibitor concentration, I (i.e., β2GpI, N-DmI, and RC-DmI) and fitted to the equation:
| (4) |
where A405 is the absorbance at the specified inhibitor concentration, [I], A0 and AI are the absorbance measured in the absence or presence of saturating [I], and IC50 is the [I] value at which 50% inhibition was observed.
Direct binding ELISA with Biotin-N-DmI
Streptavidin-coated microtiter plates (Sigma; cat. S-6940) were incubated with a solution of Biotin-N-DmI (25 μg mL−1, 200 μL/well) in PBS, pH 7.4, for 2 h at room temperature. After washing (3×) with PBS-0.1% Tween-20, each well was added with a fixed volume (100 μL) of plasma from selected APS patients, diluted (1:100 v/v) with 4% BSA in PBS. After washing (3×) with PBS-0.05% Tween-20, determination of anti-β2GpI aAbs was carried out with an alkaline phopshatase-conjugated anti-human IgG, as detailed above.
Acknowledgments
The authors thank dr. Daniele Dalzoppo for critical reading of the manuscript and dr. Marco Daniele for technical assistance with the peptide synthesizer. The support of dr. Olmetta Iadicicco is also gratefully acknowledged.
Glossary
Abbreviations:
- β2GpI
β2-glycoprotein I
- aAbs
autoantibodies
- APS
antiphospholipid syndrome
- Biotin-N-DmI
N-DmI derivatized at the N-terminus with N-biotinyl-NH-(PEG)2-
- BSA
bovine serum albumin
- CD
circular dichroism
- ELISA
enzyme-linked immunosorbent assay
- ESI
electrospray ionization
- Fmoc
9-fluorenylmethyloxycarbonyl
- Gdn-HCl
guanidine hydrochloride
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- MS
mass spectrometry
- N-DmI
natively folded synthetic N-terminal domain 1–64 of β2GpI
- PBS
phosphate-buffered saline
- R-DmI
DmI with Cys residues in the reduced state
- RC-DmI
DmI with Cys residues reduced and carboxamidomethylated
- RP
reversed phase
- SDS-PAGE
polyacrylamide gel electrophoresis in the presence of sodium dodecylsulphate
- UV
ultraviolet.
References
- 1.Wilson WA, Piette JC, Brey R, Derksen RH, Harris EN, Hughes GRV, Triplett DA, Khamashta MA. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;4:1309–1311. doi: 10.1002/1529-0131(199907)42:7<1309::AID-ANR1>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 2.Levine JS, Branch DW, Rauch J. The antiphospholipid syndrome. N Engl J Med. 2002;346:752–763. doi: 10.1056/NEJMra002974. [DOI] [PubMed] [Google Scholar]
- 3.Giannakopoulos B, Krilis SA. How I treat the antiphospholipid syndrome. Blood. 2009;114:2020–2030. doi: 10.1182/blood-2009-05-220756. [DOI] [PubMed] [Google Scholar]
- 4.de Laat B, Pengo V, Pabinger I, Musial J, Voskuyl AE, Bultink IE, Ruffatti A, Rozman B, Kveder T, de Moerloose P, Boehlen F, Rand J, Ulcova-Gallova Z, Mertens K, de Groot PG. The association between circulating antibodies against domain I of beta2-glycoprotein I and thrombosis: an international multicenter study. J Thromb Haemost. 2009;7:1767–1773. doi: 10.1111/j.1538-7836.2009.03588.x. [DOI] [PubMed] [Google Scholar]
- 5.Lee NS, Brewer HB, Jr, Osborne JC., Jr Beta 2-glycoprotein I. Molecular properties of an unusual apolipoprotein, apolipoprotein H. J Biol Chem. 1983;258:4765–4770. [PubMed] [Google Scholar]
- 6.de Laat B, Derksen RH, de Groot PG. Beta(2)-glycoprotein I, the playmaker of the antiphospholipid syndrome. Clin Immunol. 2004;112:161–168. doi: 10.1016/j.clim.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 7.de Laat B, Derksen RH, Urbanus RT, de Groot PG. IgG antibodies that recognize epitope Gly40-Arg43 in domain I of beta 2-glycoprotein I cause LAC, and their presence correlates strongly with thrombosis. Blood. 2005;105:1540–1545. doi: 10.1182/blood-2004-09-3387. [DOI] [PubMed] [Google Scholar]
- 8.Lozier J, Takahashi N, Putnam FW. Complete amino acid sequence of human plasma beta 2-glycoprotein I. Proc Natl Acad Sci USA. 1984;81:3640–3644. doi: 10.1073/pnas.81.12.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bendixen E, Halkier T, Magnusson S, Sottrup-Jensen L, Kristensen T. Complete primary structure of bovine beta 2-glycoprotein I: localization of the disulfide bridges. Biochemistry. 1992;31:3611–3617. doi: 10.1021/bi00129a009. [DOI] [PubMed] [Google Scholar]
- 10.Bork P, Downing AK, Kieffer B, Campbell ID. Structure and distribution of modules in extracellular proteins. Q Rev Biophys. 1996;29:119–167. doi: 10.1017/s0033583500005783. [DOI] [PubMed] [Google Scholar]
- 11.Bouma B, de Groot PG, van den Elsen JM, Ravelli RB, Schouten A, Simmelink MJ, Derksen RH, Kroon J, Gros P. Adhesion mechanism of human beta(2)-glycoprotein I to phospholipids based on its crystal structure. EMBO J. 1999;18:5166–5174. doi: 10.1093/emboj/18.19.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwarzenbacher R, Zeth K, Diederichs K, Gries A, Kostner GM, Laggner P, Prassl R. Crystal structure of human beta2-glycoprotein I: implications for phospholipid binding and the antiphospholipid syndrome. EMBO J. 1999;18:6228–6239. doi: 10.1093/emboj/18.22.6228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hammel M, Kriechbaum M, Gries A, Kostner GM, Laggner P, Prassl R. Solution structure of human and bovine beta(2)-glycoprotein I revealed by small-angle X-ray scattering. J Mol Biol. 2002;321:85–97. doi: 10.1016/s0022-2836(02)00621-6. [DOI] [PubMed] [Google Scholar]
- 14.Hoshino M, Hagihara Y, Nishii I, Yamazaki T, Kato H, Goto Y. Identification of phospholipid-binding site of human β2-glycoprotein I domain V by heteronuclear magnetic resonance. J Mol Biol. 1998;304:927–939. doi: 10.1006/jmbi.2000.4243. [DOI] [PubMed] [Google Scholar]
- 15.Iverson GM, Victoria EJ, Marquis DM. Anti-β2 glycoprotein I autoantibodies recognize an epitope on the first domain of β2GPI. Proc Natl Acad Sci USA. 1998;95:15542–15546. doi: 10.1073/pnas.95.26.15542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iverson GM, Reddel SW, Victoria EJ, Cockerill KA, Wang YX, Marti-Renom MA, Sali A, Marquis DM, Krilis SA, Linnik MD. Use of single point mutations in domain I of β2-glycoprotein I to determine fine antigenic specificity of antiphospholipid autoantibodies. J Immunol. 2002;169:7097–7103. doi: 10.4049/jimmunol.169.12.7097. [DOI] [PubMed] [Google Scholar]
- 17.Ioannou Y, Pericleous C, Giles I, Latchman DS, Isenberg DA, Rahman A. Binding of antiphospholipid antibodies to discontinuous epitopes on domain I of human beta(2)-glycoprotein I: mutation studies including residues R39 to R43. Arthritis Rheum. 2007;56:280–290. doi: 10.1002/art.22306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giles IP, Isenberg DA, Latchman DS, Rahman A. How do antiphospholipid antibodies bind beta2-glycoprotein I? Arthritis Rheum. 2003;48:2111–2121. doi: 10.1002/art.11101. [DOI] [PubMed] [Google Scholar]
- 19.Giannakopoulos B, Passam F, Rahgozar S, Krilis SA. Current concepts on the pathogenesis of the antiphospholipid syndrome. Blood. 2007;109:422–430. doi: 10.1182/blood-2006-04-001206. [DOI] [PubMed] [Google Scholar]
- 20.Hulstein JJ, Lenting PJ, de Laat B, Derksen RH, Fijnheer R, de Groot PG. Beta2-glycoprotein I inhibits von Willebrand factor dependent platelet adhesion and aggregation. Blood. 2007;110:1483–1491. doi: 10.1182/blood-2006-10-053199. [DOI] [PubMed] [Google Scholar]
- 21.Sikara MP, Routsias JG, Samiotaki M, Panayotou G, Moutsopoulos HM, Vlachoyiannopoulos PG. Beta2-glycoprotein I (beta-2GPI) binds platelet factor 4 (PF4): implications for the pathogenesis of antiphospholipid syndrome. Blood. 2009;115:440–441. doi: 10.1182/blood-2009-03-206367. [DOI] [PubMed] [Google Scholar]
- 22.Meroni PL. Pathogenesis of the antiphospholipid syndrome: an additional example of the mosaic of autoimmunity. J Autoimmun. 2008;30:99–103. doi: 10.1016/j.jaut.2007.11.014. [DOI] [PubMed] [Google Scholar]
- 23.Rand JH. The antiphospholipid syndrome. Hematology Am Soc Hematol Educ Program. 2007:136–142. doi: 10.1182/asheducation-2007.1.136. [DOI] [PubMed] [Google Scholar]
- 24.Wurm H. Beta2-glycoprotein-I (apolipoprotein H) interactions with phospholipid vescicles. Int J Biochem. 1984;16:511–515. doi: 10.1016/0020-711x(84)90168-x. [DOI] [PubMed] [Google Scholar]
- 25.De Filippis V, Draghi A, Frasson R, Grandi C, Musi V, Fontana A, Pastore A. o-Nitrotyrosine and p-iodophenylalanine as spectroscopic probes for structural characterization of SH3 complexes. Protein Sci. 2007;16:1257–1265. doi: 10.1110/ps.062726807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brandts JF, Halvorson HR, Brennan M. Consideration of the possibility that the slow step in protein denaturation reactions is due to cis–trans isomerism of proline residues. Biochemistry. 1975;14:4953–4963. doi: 10.1021/bi00693a026. [DOI] [PubMed] [Google Scholar]
- 27.Vivian JT, Callis PR. Mechanisms of tryptophan fluorescence shifts in proteins. Biophys J. 2001;80:2093–2109. doi: 10.1016/S0006-3495(01)76183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lakowicz JR. Principles of fluorescence spectroscopy. 2nd ed. New York: Kluwer Academic/Plenum; 1999. [Google Scholar]
- 29.Chen Y, Barkley MD. Toward understanding tryptophan fluorescence in proteins. Biochemistry. 1998;37:9976–9982. doi: 10.1021/bi980274n. [DOI] [PubMed] [Google Scholar]
- 30.Eftink MR. The use of fluorescence methods to monitor unfolding transitions in proteins. Biophys J. 1994;66(2 Part 1):482–501. doi: 10.1016/s0006-3495(94)80799-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brahms S, Brahms J. Determination of protein secondary structure in solution by vacuum ultraviolet circular dichroism. J Mol Biol. 1980;138:149–178. doi: 10.1016/0022-2836(80)90282-x. [DOI] [PubMed] [Google Scholar]
- 32.De Filippis V, De Dea E, Lucatello F, Frasson R. Effect of Na+ binding on the conformation, stability, and molecular recognition properties of thrombin. Biochem J. 2005;390:485–492. doi: 10.1042/BJ20050252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Strickland EH. Aromatic contributions to circular dichroism spectra of proteins. CRC Crit Rev Biochem. 1974;3:113–175. doi: 10.3109/10409237409105445. [DOI] [PubMed] [Google Scholar]
- 34.Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 35.Hammel M, Schwarzenbacher R, Gries A, Kostner GM, Laggner P, Prassl R. Mechanism of the interaction of beta(2)-glycoprotein I with negatively charged phospholipid membranes. Biochemistry. 2001;40:14173–14181. doi: 10.1021/bi0114372. [DOI] [PubMed] [Google Scholar]
- 36.Myers JK, Pace CN, Scholtz JM. Denaturant m values and heat capacity changes: relation to changes in accessible surface areas of protein unfolding. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrison PM, Sternberg MJ. Analysis and classification of disulphide connectivity in proteins. The entropic effect of cross-linkage. J Mol Biol. 1994;244:448–463. doi: 10.1006/jmbi.1994.1742. [DOI] [PubMed] [Google Scholar]
- 38.Matthews BW, Nicholson H, Becktel WJ. Enhanced protein thermostability from site-directed mutations that decrease the entropy of unfolding. Proc Natl Acad Sci USA. 1987;84:6663–6667. doi: 10.1073/pnas.84.19.6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tincani A, Spatola L, Prati E, Allegri F, Ferremi P, Cattaneo R, Meroni P, Balestrieri G. The anti-beta2-glycoprotein I activity in human anti-phospholipid syndrome sera is due to monoreactive low-affinity autoantibodies directed to epitopes located on native beta2-glycoprotein I and preserved during species' evolution. J Immunol. 1996;157:5732–5738. [PubMed] [Google Scholar]
- 40.Iverson GM, Matsuura E, Victoria EJ, Cockerill KA, Linnik MD. The orientation of beta2GPI on the plate is important for the binding of anti-beta2GPI autoantibodies by ELISA. J Autoimmun. 2002;18:289–297. doi: 10.1006/jaut.2002.0590. [DOI] [PubMed] [Google Scholar]
- 41.Espinosa G, Cervera R. Morbidity and mortality in the antiphospholipid syndrome. Curr Opin Pulm Med. 2009;15:413–417. doi: 10.1097/MCP.0b013e32832d0463. [DOI] [PubMed] [Google Scholar]
- 42.Shoenfeld Y, Meroni PL, Cervera R. Antiphospholipid syndrome dilemmas still to be solved: 2008 status. Ann Rheum Dis. 2008;67:438–442. doi: 10.1136/ard.2007.083873. [DOI] [PubMed] [Google Scholar]
- 43.Ioannou Y, Giles I, Lambrianides A, Richardson C, Pearl LH, Latchman DS, Isenberg DA, Rahman A. A novel expression system of domain I of human beta2 glycoprotein I in Escherichia coli. BMC Biotechnol. 2006;6:8. doi: 10.1186/1472-6750-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ioannou Y, Romay-Penabad Z, Pericleous C, Giles I, Papalardo E, Vargas G, Shilagard T, Latchman DS, Isenberg DA, Rahman A, Pierangeli S. In vivo inhibition of antiphospholipid antibody-induced pathogenicity utilizing the antigenic target peptide domain I of beta2-glycoprotein I: proof of concept. J Thromb Haemost. 2009;7:833–842. doi: 10.1111/j.1538-7836.2009.03316.x. [DOI] [PubMed] [Google Scholar]
- 45.De Filippis V, Colombo G, Russo I, Spadari B, Fontana A. Probing hirudin-thrombin interaction by incorporation of noncoded amino acids. Biochemistry. 2002;41:13556–13569. doi: 10.1021/bi0203482. [DOI] [PubMed] [Google Scholar]
- 46.Urbanus RT, Derksen RH, de Groot PG. Current insight into diagnostics and pathophysiology of the antiphospolipid syndrome. Blood Rev. 2008;22:93–105. doi: 10.1016/j.blre.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 47.Atherton E, Sheppard RC. Solid phase peptide synthesis. Oxford, UK: IRL Press; 1989. [DOI] [PubMed] [Google Scholar]
- 48.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;11:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.De Filippis V, De Antoni F, Frigo M, Polverino de Laureto P, Fontana A. Enhanced protein thermostability by Ala→Aib replacement. Biochemistry. 1998;37:1686–1696. doi: 10.1021/bi971937o. [DOI] [PubMed] [Google Scholar]
- 50.Pengo V, Biasiolo A, Fior MG. Autoimmune antiphospholipid antibodies are directed against a cryptic epitope expressed when beta 2-glycoprotein I is bound to a suitable surface. Thromb Haemost. 1995;73:29–34. [PubMed] [Google Scholar]
- 51.Pengo V, Tripodi A, Reber G, Rand JH, Ortel TL, Galli M, de Groot PG. Update of the guidelines for lupus anticoagulant detection. J Thromb Haemost. 2009;7:1737–1740. doi: 10.1111/j.1538-7836.2009.03555.x. [DOI] [PubMed] [Google Scholar]