

Abstract
An array of genetic screens and selections has been developed for reporting protein folding and solubility in the cytoplasm of living cells. However, there are currently no analogous folding assays for the bacterial periplasm, despite the significance of this compartment for the expression of recombinant proteins, especially those requiring important posttranslational modifications (e.g., disulfide bond formation). Here, we describe an engineered genetic selection for monitoring protein folding in the periplasmic compartment of Escherichia coli cells. In this approach, target proteins are sandwiched between an N-terminal signal recognition particle (SRP)-dependent signal peptide and a C-terminal selectable marker, TEM-1 β-lactamase. The resulting chimeras are localized to the periplasmic space via the cotranslational SRP pathway. Using a panel of native and heterologous proteins, we demonstrate that the folding efficiency of various target proteins correlates directly with in vivo β-lactamase activity and thus resistance to ampicillin. We also show that this reporter is useful for the discovery of extrinsic periplasmic factors (e.g., chaperones) that affect protein folding and for obtaining folding-enhanced proteins via directed evolution. Collectively, these data demonstrate that our periplasmic folding reporter is a powerful tool for screening and engineering protein folding in a manner that does not require any structural or functional information about the target protein.
Keywords: cotranslational SRP pathway, directed evolution, disulfide bond formation, folding and solubility reporter, molecular chaperones, periplasm, protein engineering
Introduction
Ever since the inception of recombinant DNA technology, maximizing the solubility of heterologous proteins in Escherichia coli has been a principal goal of modern biotechnology.1–4 Despite numerous advances in this area over the past 3 decades, high-level expression of correctly folded, soluble proteins for laboratory and preparative purposes remains a significant challenge. Indeed, following their expression in the cytoplasm of E. coli, many recombinant proteins of prokaryotic and eukaryotic origin are prone to misfolding5–7 and are subsequently degraded by cellular proteases8,9 or deposited into inactive inclusion bodies.10,11 Protein misfolding in the cytoplasm can be a consequence of the relative crowdedness of this compartment, where macromolecule concentration can reach 300–400 mg/mL.12 In addition, posttranslational processing steps such as disulfide bond formation13 or N-linked glycosylation,14 which are often required for correct folding, are absent from the cytoplasm of wild-type E. coli cells.
The periplasm in E. coli, on the other hand, is often the preferred compartment for protein expression because it: (1) contains significantly fewer proteins, particularly proteases, compared to the cytoplasm; (2) houses a network of redox enzymes that catalyze the formation and isomerization of disulfide bonds; and (3) permits N-linked glycosylation in Campylobacter jejuni14 and E. coli.15 Consequently, periplasmic proteins are often easier to isolate, less prone to crowding-induced aggregation and/or proteolytic degradation, and more efficiently folded compared with their cytoplasmic counterparts. However, even though the significance of the periplasm for protein expression is firmly established,3,16 there are currently no genetic reporter assays for monitoring protein folding in this subcellular compartment. In stark contrast, numerous folding reporter systems have been developed for detecting correctly folded, soluble proteins in the cytoplasm of living E. coli cells.17–23 These approaches commonly rely on a genetic fusion between a protein-of-interest (POI) and a reporter protein whose specific phenotype is independent of the POI's function.24 In this scenario, when the POI folds into a soluble conformation, the reporter to which it is fused is functional. In contrast, when the POI misfolds or aggregates, the fused reporter is inactive and a null phenotype is observed. Another typical feature of these approaches is that they can be used even when structural or functional information about the target is lacking. Perhaps the most useful aspect of these assays is that they can be combined with well-established methods for creating protein diversity libraries to screen or select for soluble variants of recalcitrant proteins19,22,23,25–27 with only one exception reported so far.28 Hence, a screen or selection for monitoring protein folding in the bacterial periplasm would be a desirable accomplishment, but to date has been met with technical difficulties.29
In this study, we developed an activity-independent selection strategy that reliably reports the “folding robustness” of POIs expressed in the periplasm. The term folding robustness is used here to denote both the chemical solubility related to correct folding of the POI and the avoidance of aggregation or degradation. The assay is based on a tripartite fusion between: (1) an N-terminal signal peptide from E. coli DsbA (ssDsbA), which has previously been shown to direct proteins through the bacterial signal recognition particle (SRP)-dependent translocation pathway,30 (2) the POI, and (3) a C-terminal fusion of mature TEM-1 β-lactamase (Bla). We chose the ssDsbA signal peptide because it is known to direct cotranslational export of heterologous proteins through the SRP pathway30,31 and thus should effectively partition ssDsbA-POI-Bla fusions to the periplasm. Using this mode of export, we observed that the antibiotic resistance of cells expressing engineered ssDsbA-POI-Bla chimeras correlated with the periplasmic folding behavior of the POI. Thus, simple selection on β-lactam antibiotics such as ampicillin (Amp) enabled discrimination between folded and misfolded conformations of POIs, even when (i) structural and functional information and (ii) activity assays for the POIs were lacking. The method was also capable of evaluating trans-acting factors that influence protein folding in the periplasm such as disulfide bond formation enzymes and molecular chaperones. Finally, the potential of the assay for improving periplasmic protein folding and solubility was demonstrated using a directed evolution strategy. Taken together, these results reveal our periplasmic protein folding assay to be a powerful new experimental tool for elucidating the factors that affect the folding of proteins in this important biological compartment.
Results
An engineered assay for folding and solubility in the periplasm
To create a rapid protein folding assay for the periplasm, we constructed a plasmid called pDMB that permitted expression of any POI as a sandwich fusion between an N-terminal export signal (ssDsbA) and a C-terminal selectable marker (Bla). Consistent with the logic underlying fusion-based folding reporters for the cytoplasm,18–21 we reasoned that productive folding of the downstream Bla protein domain and consequent resistance to β-lactam antibiotics is directly related to the folding robustness (avoidance of aggregation and inclusion body formation) of the upstream POI. Thus, correctly folded, soluble proteins would be expected to confer Amp resistance to E. coli cells and provide a selectable phenotype for periplasmic protein folding (Fig. 1). Here, more than 40 different POIs were cloned into pDMB and characterized based on their ability to confer Amp resistance to E. coli cells as described below.
Figure 1.
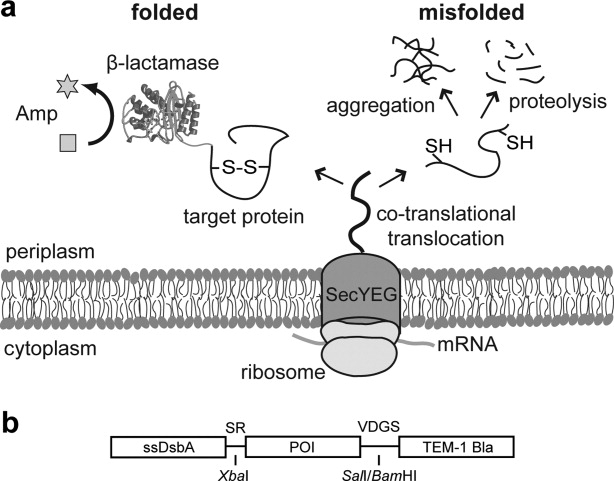
Schematic of SRP-mediated periplasmic folding reporter. (a) The DsbA signal peptide enables cotranslational translocation across the cytoplasmic membrane via the SRP-dependent pathway. Nascent polypeptides enter the periplasm where they fold properly, conferring Amp resistance to cells, or misfold, leading to eventual aggregation and/or proteolysis. (b) Fusion proteins created in this study contained an N-terminal DsbA signal peptide (ssDsbA) followed by a POI and a C-terminal selectable marker (TEM-1 Bla). Restrictions sites (XbaI and SalI/BamHI) resulted in short linker sequences (SR and VDGS) at the junctions.
Monitoring DsbA-dependent folding of enzymes and antibodies in the periplasm
As proof-of-concept, we first investigated whether our assay could report the folding behavior of E. coli alkaline phosphatase (PhoA). We chose PhoA because its correctly folded, catalytically active conformation is dependent upon two disulfide bonds, one between Cys168 and Cys178 and the other between Cys286 and Cys336, that are formed by the primary periplasmic oxidant DsbA.32 Expression of ssDsbA-PhoA-Bla in wild-type DHB4 cells conferred strong resistance to 100 μg/mL Amp as evidenced by spot plating [Fig. 2(a)]. When the same construct was expressed in DHA cells, which lack DsbA, resistance to this concentration of Amp was no longer observed. To quantify this difference in Amp resistance, single colonies of DHB4 and DHA were challenged on increasing levels of Amp, and values for the minimum bacteriocidal concentration (MBC) were determined. The MBC for DHB4 cells expressing ssDsbA-PhoA-Bla was eightfold higher than that measured for DHA cells expressing the same fusion (Table I). Plating on LB agar lacking Amp revealed no measurable growth difference between the DHB4 and DHA cells [Fig. 2(a)], confirming that the difference in Amp resistance was due to DsbA-dependent oxidation of protein thiols required for PhoA folding. For comparison, we evaluated cytoplasmic folding of PhoA in DHB4 cells using a previously developed assay based on the twin-arginine translocation (Tat) protein export pathway.22 In this system, POI-Bla fusions are targeted by an N-terminal ssTorA signal peptide to the Tat pathway, which requires that protein substrates fold correctly in the cytoplasm before transport across the inner membrane.33 Thus, the efficiency of ssTorA-POI-Bla export, and the corresponding Amp resistance phenotype, is regulated by the folding behavior of the POI in the cytoplasm.22,25 As expected, we observed that DHB4 cells expressing ssTorA-PhoA-Bla were sensitive to 100 μg/mL Amp [Fig. 2(b) and Table I]. This Amp sensitivity was due to the fact that the cytoplasm of DHB4 cells is a reducing environment that renders PhoA misfolded and incompetent for export via the Tat pathway. To confirm that these results were not attributable to redox-dependent changes in the folding of the Bla moiety itself, cells expressing ssDsbA fused directly to Bla were similarly plated on Amp. Consistent with earlier findings,34 the periplasmic redox state had no measurable effect on the catalytic activity of Bla as evidenced by the identical resistance phenotypes for DHB4 and DHA cells expressing ssDsbA-Bla [Fig. 2(b) and Table I]. Western blot analysis of the cytoplasmic and periplasmic fractions derived from DHB4 and DHA cells expressing the ssDsbA-PhoA-Bla fusions confirmed that soluble expression of PhoA-Bla in the periplasm corresponded with the respective Amp phenotypes [Fig. 2(c)]. Proteolytic degradation of the fusion was observed in the cytoplasm and periplasm, but as discussed below, it does appear to affect the assay readout for PhoA and does not occur for most of the other proteins tested.
Figure 2.
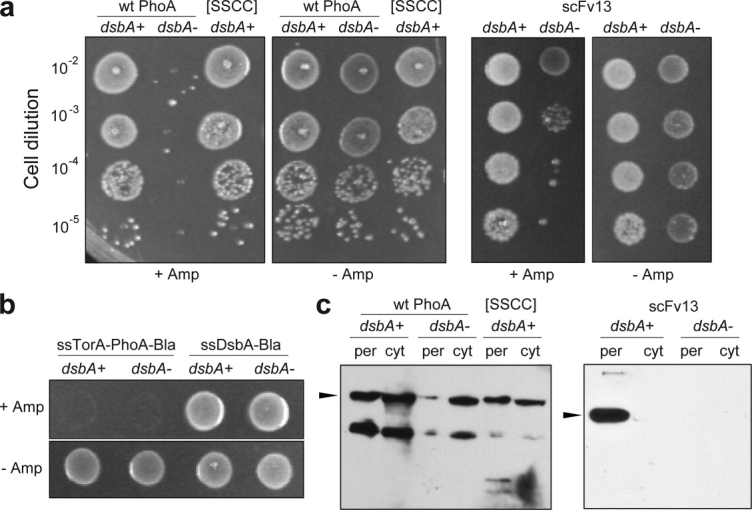
Redox-dependent folding reported via SRP selection. (a) An equivalent number of DHB4 (dsbA+) and DHA (dsbA−) cells expressing either ssDsbA-PhoA-Bla, ssDsbA-PhoA[SSCC]-Bla, or ssDsbA-scFv13-Bla were spotted on LB/agar containing either 50 μg/mL Cm (−Amp) or 100 μg/mL Amp (+Amp). Cell dilutions from 10−2 to 10−5 were spotted on LB plates containing different concentrations of Amp. (b) Cells expressing ssTorA-PhoA-Bla or ssDsbA-Bla spotted on LB/agar containing either 50 μg/mL Cm (−Amp) or 100 μg/mL Amp (+Amp) at a dilution of 10−2 from overnight cultures. (c) Western blot analysis of periplasmic (per) and cytoplasmic (cyt) fractions derived from DHB4 (dsbA+) and DHA (dsbA−) cells expressing either ssDsbA-PhoA-Bla, ssDsbA-PhoA[SSCC]-Bla, or ssDsbA-scFv13-Bla. Samples were blotted with Bla-specific antibodies. Arrow indicates expected size of the fusion proteins.
Table I.
Amp Resistance Conferred by Different Target Proteins
| Protein-of-interesta | Strain | MBC (μg/mL)b | MIC (μg/mL)b |
|---|---|---|---|
| Alkaline phosphatase | |||
| PhoA | DHB4 | 200 | 200 |
| PhoA | DHA | 25 | 25 |
| PhoA[SSCC] | DHB4 | 400 | 200 |
| ssTorA-PhoA-Bla | DHB4 | 6 | 6 |
| ssTorA-PhoA-Bla | DHA | 12 | 6 |
| Bla | DHB4 | 3200 | 1600 |
| Bla | DHA | 3200 | 1600 |
| Anti-β-gal scFv13 | |||
| scFv13 (wild type) | DHB4 | 400 | 200 |
| scFv13 (wild type) | DHA | 50 | 50 |
| scFv13-R4 | DHB4 | 400 | 400 |
| scFv13-R4 | DHA | 100 | 50 |
| Maltose-binding proteins | |||
| MalE (wild type) | DH5α | 250 | 200 |
| G32D | DH5α | 200 | 175 |
| I33P | DH5α | 200 | 175 |
| MalE31 | DH5α | 175 | 175 |
| Other proteins | |||
| GFP | DH5α | 200 | 175 |
| GST | DH5α | 50 | 25 |
| MetF | DH5α | 25 | 25 |
| MetK | DH5α | 6 | 3 |
| TrxA | DH5α | 100 | 100 |
| Amyloid-β peptides | |||
| Aβ42 (wild type) | DH5α | 12 | 12 |
| A2 | DH5α | 25 | 25 |
| A4 | DH5α | 25 | 25 |
| B9 | DH5α | 25 | 25 |
| B12 | DH5α | 25 | 25 |
| H2 | DH5α | 50 | 25 |
| GM6 | DH5α | 100 | 100 |
| GM7 | DH5α | 100 | 50 |
| GM11 | DH5α | 100 | 50 |
Unless otherwise indicated, protein-of-interest is a fusion of ssDsbA-POI-Bla.
Approximately 500 CFUs were plated overnight at 37°C.
Recently, Kadokura and Beckwith35 reported that, in contrast to the findings of Sone et al.,32 introduction of cysteine to alanine mutations into each of the two cysteine pairs of PhoA (Cys168-Cys178 and Cys286-Cys336) did not destabilize the protein in vivo. They suggested this discrepancy could be due to the differences of the conditions between the two studies: Kadokura and Beckwith replaced the cysteine residues of PhoA with alanine, whereas Sone et al. substituted serine; the temperature for culture was 30°C in the Kadokura and Beckwith study, but 37°C in the Sone et al. report. Here, we attempted to resolve this discrepancy by replacing the Cys168-Cys178 pair in PhoA with serine and testing this mutant in our folding reporter at 22 and 37°C. Consistent with the observations of Kadokura and Beckwith, PhoA[SSCC] conferred resistance that was indistinguishable from wild-type (wt) PhoA at both 22°C (data not shown) and 37°C [Fig. 2(a) and Table I].
To determine whether the ability to report DsbA-dependent folding was a general feature of our assay, we next analyzed the behavior of a single-chain antibody fragment specific for β-galactosidase (scFv13).36 Like PhoA, scFv13 folding is dependent on the formation of two intradomain disulfide bonds, one in the VH and one in the VL domain. In the absence of these bonds, the scFv is only expressed at very low levels.36 Similar to what was seen above, DHB4 and DHA cells expressing ssDsbA-scFv13-Bla exhibited distinctly different Amp resistance phenotypes [Fig. 2(a)]. The MBC was eightfold greater for DHB4 cells compared with DHA cells expressing this construct (Table I). Western blot analysis revealed significant accumulation of soluble ssDsbA-scFv13-Bla in the periplasmic fraction of DHB4 cells, whereas virtually no cross-reacting bands were detected in any fraction (cytoplasmic or periplasmic) isolated from DHA cells [Fig. 2(c)]. This result confirmed that scFv13 folding and solubility was dependent on disulfide bond formation. The fact that scFv13-Bla accumulated predominantly in the periplasm provided additional support for our hypothesis that many proteins targeted to the SRP pathway are efficiently partitioned in the periplasm. It is also noteworthy that we did not observe any proteolytic degradation of the scFv13-Bla fusion. We also tested scFv13-R4, a variant of scFv13 engineered previously to have greater solubility under reducing conditions.36 In DHA cells, where the periplasm does not support disulfide bond formation, scFv13-R4 showed a twofold increase in MBC over wild-type scFv13 (Table I), consistent with the improved ability of scFv13-R4 to fold correctly in the absence of disulfide bonds.36 Interestingly, expression of ssDsbA-scFv13-R4-Bla in DHB4 cells resulted in an eightfold higher MBC relative to expression in DHA cells. This is likely due to the fact that oxidation of scFv13-R4 in the periplasm further increases the protein's stability by the addition of at least one disulfide bond.36 Like its parent, scFv13-R4 accumulated stably in cells with no apparent degradation [Fig. 3(a)].
Figure 3.
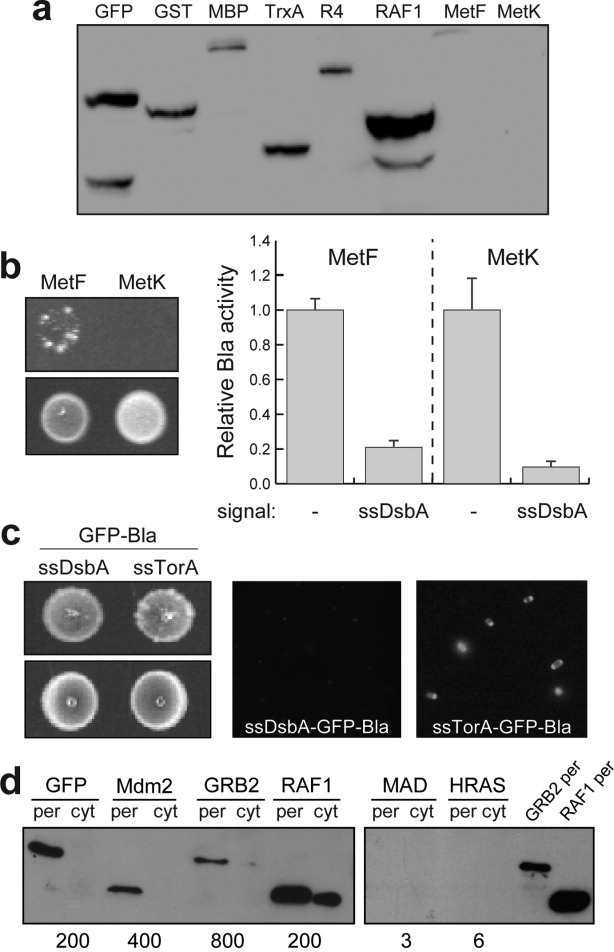
A universal folding reporter. (a) Western blot analysis of whole cell lysates generated from an equivalent number of DH5α cells expressing the ssDsbA-POI-Bla constructs as indicated. Samples were blotted with Bla-specific antibodies. (b) An equivalent number of DH5α cells expressing ssDsbA-MetF-Bla or ssDsbA-MetK-Bla were spotted on LB/agar containing either 20 μg/mL Cm (−Amp) or 100 μg/mL Amp (+Amp) at a dilution of 10−2 from overnight cultures. Nitrocefin hydrolysis activity of whole-cell lysates from cells expressing MetF-Bla and MetK-Bla with or without the ssDsbA signal peptide. Activity was measured as the initial velocity of absorbance change at 486 nm. Relative Bla activity was obtained by normalizing to the activity of the fusion without the signal peptide. Bla activity was measured in triplicate, and error bars represent standard error of the mean. (c) DH5α cells expressing ssDsbA-GFP-Bla or ssTorA-GFP-Bla were spotted as in (b). Fluorescence microscopy analysis (×100) of DH5α cells expressing ssDsbA-GFP-Bla or ssTorA-GFP-Bla as indicated. (d) Western blot analysis of periplasmic (per) and cytoplasmic (cyt) fractions generated from an equivalent number of DH5α cells expressing ssDsbA-POI-FLAG fusions lacking the Bla moiety for the clones indicated. Blots were probed by anti-FLAG antibodies. GroEL was used as a fractionation marker by probing with anti-GroEL antibodies (data not shown). GRB2 per and RAF1 per samples were included as positive loading controls for the blot on the right. MBC values (μg/mL Amp) corresponding to each protein are indicated below the panels.
A universal assay of periplasmic protein folding
We next tested whether folding behavior unrelated to disulfide bond formation could be discriminated using this selection strategy. Specifically, we tested several proteins that are physiologically expressed in the E. coli cytoplasm (e.g., GST, MetF, MetK, and TrxA) as well as GFP. We reasoned that some of these would fold correctly in the periplasm. Indeed, selective plating of cells expressing these constructs revealed that GST, TrxA, and GFP are each correctly folded in the periplasm as evidenced by MBC values of 50, 100, and 200 μg/mL Amp, respectively (Table I). For comparison, expression of Bla fused to the maltose-binding protein (MBP), a native periplasmic protein that is known to be extremely soluble in E. coli,37 resulted in an MBC of 250 μg/mL Amp (Table I). Importantly, each of these was found to stably accumulate in cells with little to no proteolytic degradation [Fig. 3(a)]. We further reasoned that other proteins such as MetF and MetK would misfold and/or aggregate outside of their native environment. This is because MetF folding involves the binding of a flavin cofactor, which would likely not be present in the periplasm.38 Similarly, MetK has redox-sensitive Cys residues that could become misoxidized in the periplasm. In addition, both proteins are strictly dependent on the cytoplasmic chaperone GroEL for correct folding.39 As expected, the MetF and MetK fusions conferred little to no resistance to cells plated on 100 μg/mL Amp [Fig. 3(b)] and resulted in MBC values of only 25 and 6 μg/mL, respectively (Table I). The weak Amp resistance conferred by MetF and MetK was accompanied by the complete absence of each fusion protein following Western blot analysis [Fig. 3(a)], indicating that incorrect folding of each protein in the periplasm was followed by proteolytic degradation. To rule out the possibility that MetF and MetK are incapable of folding when fused to Bla, we expressed MetF-Bla and MetK-Bla that lacked the ssDsbA export signals. Each of these folded very efficiently in the cytoplasm as evidenced by the relatively high Bla activity measured in the lysates of cells expressing these constructs [Fig. 3(b)]. For comparison, very little Bla activity was measured in the lysates of cells expressing ssDsbA-MetF-Bla and ssDsbA-MetK-Bla. Thus, C-terminal fusions of Bla to MetF and MetK are indeed capable of correct folding when expressed in the cytoplasm but become misfolded and inactive when localized in the periplasm.
The observation that GFP folded correctly in the periplasm was surprising based on earlier findings that it could not attain a fluorescent conformation when localized in the periplasm via the Sec pathway.40,41 Indeed, cells expressing the ssDsbA-GFP-Bla fusion were nonfluorescent [Fig. 3(c)]. However, the absence of cell fluorescence could not be attributed to global misfolding or aggregation of GFP in the periplasm because ssDsbA-GFP-Bla conferred a strong Amp-resistant phenotype to cells [Table I and Fig. 3(c)]. For comparison, cells expressing GFP-Bla via the Tat export pathway resulted in highly fluorescent cells that were similarly Amp resistant [Table II and Fig. 3(c)], consistent with earlier findings.22,42 From these data, it is clear that GFP is soluble in both the cytoplasmic and periplasmic compartments; hence, the inability of GFP to fluoresce in the periplasm is not attributable to its overall folding and stability in this compartment.
Table II.
Periplasmic and Cytoplasmic Folding of Mammalian Target Proteins
| Protein (organism)a | Domainb | Subcellular location | MBC (μg/mL) periplasmc | MBC (μg/mL) cytoplasmc | STE ratiod |
|---|---|---|---|---|---|
| CASP2 (Hs) | FL | Cytoplasm | 50 | 50 | 0.00 |
| CCND2 (Hs) | FL | Cytoplasm | 100 | 9 | 0.12 |
| CD44 (Hs) | FL | Extracellular | 6 | 6 | 0.13 |
| CDK4 (Hs) | FL | Cytoplasm | 50 | 12 | 0.25 |
| CDKN1B (Hs) | FL | Cytoplasm | 400 | 50 | 0.10 |
| Efna1 (Mm) | EC | Extracellular | 200 | 25 | 0.36 |
| Efnb2 (Mm) | EC1 | Extracellular | 200 | 9 | 0.06 |
| Efnb2 (Mm) | EC2 | Extracellular | 800 | 9 | 0.32 |
| Epha2 (Mm) | LB | Extracellular | 6 | 6 | 0.03 |
| Ephb2 (Mm) | SAM | Cytoplasm | 800 | 100 | 0.16 |
| Ephb2 (Mm) | TK | Cytoplasm | 100 | 3 | 0.07 |
| FOS (Hs) | FL | Nuclear | 100 | 25 | 0.01 |
| GATA2 (Hs) | FL | Nuclear | 3 | 500 | 0.09 |
| GFP (Av) | FL | Cytoplasm | 200 | 400 | 0.69 |
| GRB2 (Hs) | FL | Cytoplasm | 800 | 30 | 0.31 |
| HRAS (Hs) | FL | Cytoplasm | 3 | 25 | 0.52 |
| JUN (Hs) | FL | Nuclear | 400 | 25 | 0.05 |
| MAD (Hs) | FL | Nuclear | 6 | 25 | 0.52 |
| Mdm2 (Mm) | p53-bd | Nuclear/Cytoplasm | 400 | 12 | 0.42 |
| MMPI (Hs) | FL | Extracellular | 200 | 50 | 0.09 |
| RAF1 (Hs) | Ras-bd | Cytoplasm | 200 | 400 | 0.52 |
Organism: Hs, Homo sapiens; Mm, Mus musculus; Av, Aequoria victoria.
Domain: FL, full length; EC, extracellular; TK, tyrosine kinase; LB, ligand binding, SAM, sterile alpha motif; bd, binding domain.
Approximately 500 CFUs were plated overnight at 37°C.
Soluble versus total expression (STE) ratios were obtained for each protein by normalizing the soluble expression data by the total expression data reported in Tables II and III of Ref. 5. Values reported are the average of over all 6× and 10× his-tagged constructs.
To further test the limits of our selection strategy, we attempted to identify mammalian proteins that fold correctly in the periplasm. For this, a total of 20 mammalian proteins and protein domains were evaluated for SRP-mediated expression and selection. These proteins were of human or murine origin and represented several diverse protein families with extracellular, cytoplasmic, and nuclear cell locations (Table II). We tested a collection of full-length and truncated proteins, which are described elsewhere,5 by cloning each into the pDMB selection vector as ssDsbA-POI-Bla fusions and determining the MBC for each. For 15 of the proteins tested, the MBC was ≥50 μg/mL Amp (Table II), suggesting that these were all relatively well folded and soluble in the periplasm. Of these, Efnb2(EC2), Ephb2(SAM), and GRB2 conferred a level of resistance (800 μg/mL Amp) that was only fourfold lower than the MBC measured for cells expressing ssDsbA-Bla with no target protein, indicating that these were extremely soluble when expressed in the periplasm. The resistance conferred by these 15 clones correlated with soluble expression of the fusion proteins, as exemplified by the RAF1-Bla fusion [Fig. 3(a)]. Only five of the clones (CD44, Epha2(LB), GATA2, HRAS, and MAD) exhibited MBCs of ≤6 μg/mL Amp that were comparable to the MBC reported for plasmid-free wt MC4100 cells43 and indicative of poor folding and solubility. It is informative to compare the MBC results obtained here for periplasmic folding versus the MBC values for cytoplasmic solubility determined previously using a Tat-mediated folding reporter that also used TEM-1 Bla.44 For some of the target proteins (e.g., CCND2, Efnb2(EC1), Efnb2(EC2), GRB2, and Mdm2-p53-bd), strong resistance was only conferred by periplasmic expression but not by cytoplasmic expression, whereas GATA2, HRAS, and MAD conferred greater resistance following cytoplasmic expression compared with periplasmic expression (Table II). We also observed that some of the proteins (e.g., EphB2(SAM), GFP, and RAF1) were soluble in both the periplasmic and cytoplasmic compartments, whereas certain others (e.g., CD44 and Epha2(LB)) were not very soluble in either of these subcellular locations (Table II).
To independently confirm soluble periplasmic expression, we expressed a subset of the mammalian clones as ssDsbA-POI-FLAG chimeras in which the C-terminal Bla moiety was replaced by a nine-residue FLAG epitope tag. Subcellular fractionation analysis of several representative positive clones (i.e., those conferring resistance to ≥50 μg/mL Amp) including GRB2, Mdm2(p53-bd), RAF1, and GFP revealed that each of these accumulated in the soluble periplasmic fraction of wt cells [Fig. 3(d)]. Conversely, we observed no soluble accumulation in either the periplasmic or cytoplasmic fractions for two negative clones, namely HRAS and MAD, that conferred resistance to ≤6 μg/mL Amp. These results confirm that soluble expression in the periplasm correlates well with the resistance conferred by each of these clones.
cis- and trans-acting factors that affect protein folding in the periplasm
We next determined whether the assay could report changes in folding robustness caused by cis-acting factors such as sequence mutations or trans-acting factors such as molecular chaperones. For these studies, we used E. coli MBP and a collection of MBP variants that become kinetically trapped in off-pathway intermediates that are prone to aggregation.29 One of these, a double mutant called MalE31 (G32D/I33P), reportedly forms inclusion bodies in the periplasmic space.45 We cloned four different versions of MBP (wt, G32D, I33P, and MalE31) into pDMB and evaluated their ability to confer Amp resistance to cells. Spot plating revealed that cells expressing wt MBP but not MalE31 were resistant to 200 μg/mL Amp [Fig. 4(a)]. Western blot analysis revealed that the resistance phenotypes conferred by ssDsbA-MBP-Bla and ssDsbA-MalE31-Bla correlated with the intracellular accumulation of each protein [Fig. 3(b) for MBP and Fig. 4(b) for MalE31]. Plating of the same cells on LB lacking Amp showed no difference in growth phenotype for wt MBP versus MalE31 [Fig. 4(a)]. The measured MBC for the wt MBP fusion was 42% greater than its MalE31 counterpart (Table I). It is noteworthy that although the MalE31 conferred significantly less Amp resistance than MBP, these Bla fusions were not completely inactive. This is in agreement with recent findings of Betton and coworkers who showed that despite its poor folding efficiency in the periplasm, a fusion of MalE31-Bla retained some catalytic activity.29 Our previous findings showed that MalE31 expressed in the cytoplasm is somewhat soluble, albeit to a lesser extent than wt MBP.22
Figure 4.
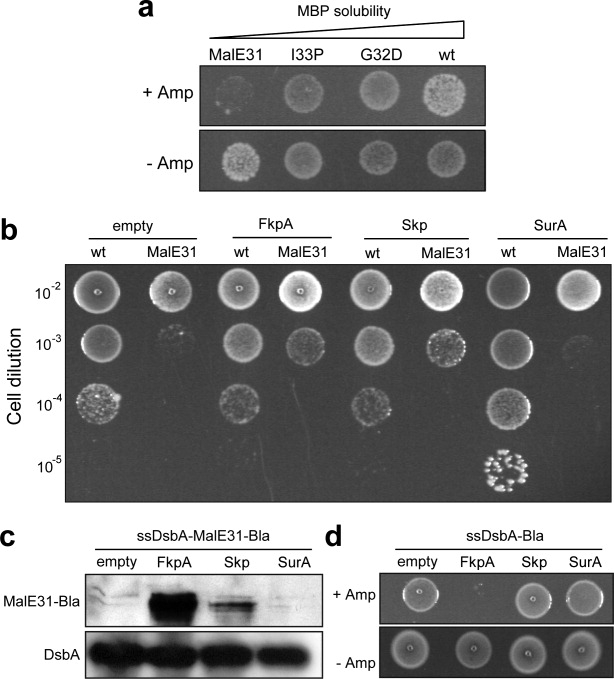
Effect of cis- and trans-acting factors on periplasmic protein folding. (a) DH5α cells expressing wt MBP and its variants MalE31, I33P, and G32D as sandwich fusions between ssDsbA and Bla. An equivalent number of cells were spotted on 100 μg/mL Amp (+Amp) or 20 μg/mL Cm (−Amp) at a dilution of 10−3 from overnight cultures. Open triangle indicates increasing solubility of MBP. (b) Growth of an equivalent number of DH5α cells coexpressing periplasmic chaperones FkpA, Skp, and SurA (or with empty pBAD18-Kan) along with wt MBP or MalE31 fusions. Cell dilutions from 10−2 to 10−5 were spotted on LB plates supplemented with 200 μg/mL Amp and 50 μg/mL Kan. (c) Western blot analysis of periplasmic fractions from cells coexpressing FkpA, Skp, and SurA chaperones as indicated along with ssDsbA-MalE31-Bla. Samples were probed with Bla-specific antibodies. Detection of DsbA using anti-DsbA serum was performed as a loading control. (d) An equivalent number of cells coexpressing ssDsbA-Bla with either FkpA, Skp, or SurA chaperones as indicated were spotted on LB plates containing 50 μg/mL Kan and either 800 μg/mL Amp (+Amp) or 20 μg/mL Cm (−Amp) at a dilution of 10−2 from overnight cultures.
It has been shown that the periplasmic chaperone FkpA can decrease aggregation of MalE31, whereas another periplasmic chaperone SurA did not affect MalE31 aggregation.46 In addition, a periplasmic chaperone called Skp is well known for its ability to interact with a broad range of substrates47 and has been used to improve expression of phage-displayed proteins.48 On the basis of these data, we reasoned that a chaperone-mediated decrease in MalE31 aggregation would lead to increased Bla activity in our assay. To test this, cells coexpressing FkpA, SurA, or Skp with ssDsbA-MalE31-Bla were plated on Amp. FkpA and Skp coexpression with ssDsbA-MalE31-Bla resulted in a measurable increase in Amp resistance compared with the control case where ssDsbA-MalE31-Bla was expressed alone [Fig. 4(b)]. In contrast, SurA coexpression conferred no measurable growth difference compared with the control [Fig. 4(b)]. Cells grown on control plates lacking Amp showed no differences in growth phenotype (data not shown). Western blot analysis of periplasmic fractions confirmed that coexpression of FkpA and Skp, but not SurA, increased the solubility of ssDsbA-MalE31-Bla [Fig. 4(c)]. Finally, to show that improved Amp resistance resulted from an increase in MalE31 solubility and not by improved activity of the Bla domain itself, we coexpressed each of the periplasmic chaperones with ssDsbA-Bla and found that Bla activity did not increase in the presence of any of the periplasmic chaperones [Fig. 4(d)]. In fact, coexpression of FkpA caused a decrease in Bla activity as evidenced by lack of cell growth on 800 μg/mL Amp. Interestingly, we found that coexpression of SurA increased the solubility of the wt MBP fusion [Fig. 4(b)], suggesting that SurA promotes folding of wt MBP but not MalE31.
Enhancing the solubility of aggregation-prone proteins
To demonstrate the utility of this system, we next attempted to isolate solubility-enhanced protein variants in the periplasm using a directed evolution approach. For this purpose, we inserted the 42-residue amyloid-β peptide (Aβ42) in the POI position of our folding reporter. Aβ42 is the primary constituent in dense amyloid fibrils that accumulate in the brains of patients with Alzheimer's disease.49 As in humans, Aβ42 aggregates extensively when expressed in E. coli.22,50 We and others have isolated variants of Aβ42 that were significantly more soluble in the bacterial cytoplasm compared with wt Aβ42.22,50 When expressed in the periplasm as a ssDsbA-Aβ42-Bla fusion, these solubility-enhanced variants conferred increased Amp resistance to cells relative to wt Aβ42 [Fig. 5(a)]. The most soluble variant, clone GM6,50 displayed an eightfold higher MBC relative to wt Aβ42 (Table I).
Figure 5.
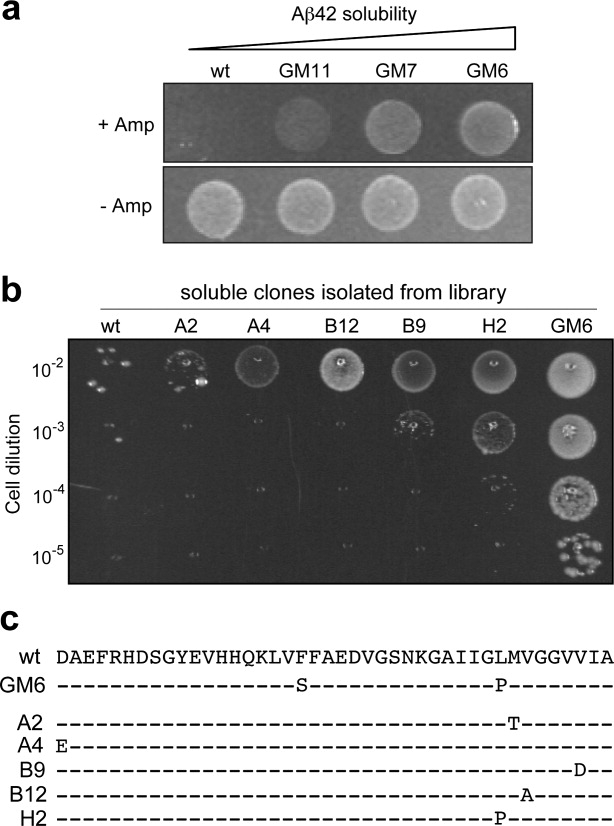
Directed evolution of the Aβ42 peptide. (a) An equivalent number of cells expressing fusions wild-type Aβ42 and solubility-enhanced variants GM11, GM7, and GM6 spotted on 100 μg/mL Amp (+Amp) or 20 μg/mL Cm (−Amp) at dilution of 10−2 from overnight cultures. The variants are derivatives of Aβ42 with the following mutations: GM11 (H6Q/V12A/V24A/132M/V36G), GM7 (V12A/I32T/L34P), and GM6 (F19S/L34P). (b) Growth of an equivalent number of cells expressing evolved variants of Aβ42 with greater solubility than wild-type Aβ42. Cell dilutions from 10−2 to 10−5 were spotted on LB plates supplemented with 100 μg/mL Amp. For comparison, wild-type Aβ42 and soluble variant GM6 are shown. (c) Amino acid sequences of evolved Aβ42 peptides, along with Aβ42 and GM6 for comparison.
Encouraged by the observation that growth selection could easily distinguish more soluble Aβ42 variants, we next attempted to isolate solubility-enhanced Aβ42 variants from a combinatorial library of Aβ42 sequences. An error-prone library of Aβ42 in pDMB was created that contained ∼5 × 104 members. Plating of ∼104 cells from this library on 25 μg/mL Amp resulted in five clones exhibiting an Amp-resistant phenotype above wt Aβ42 [Fig. 5(b)]. Each of these variants showed at least a twofold improvement in MBC over wt Aβ42, with clone H2 exhibiting a fourfold increase in MBC compared with wt (Table I). Interestingly, clone H2 contains the L34P mutation [Fig. 5(c)], which is known to render Aβ42 less prone to aggregation.50 The other isolated clones had point mutations located primarily within hydrophobic stretches in the central and C-terminal parts of Aβ42 that have been implicated by others as key determinants in aggregation and fibril formation50,51: A2 (M35T), B9 (V40D), and B12 (V37A) [Fig. 5(c)]. It is noteworthy that this improvement in Aβ42 solubility only required a single round of mutagenesis and selection, confirming the robustness of our assay for searching large volumes of sequence space for proteins with altered structural characteristics.
Discussion
To date, several reporters of protein folding have been developed in E. coli.17–21 Such methods include: transcriptional fusion reporters that signal the cell's genetic response to misfolding and aggregation of the POI,52 and translational fusion reporters between the POI and a peptide (e.g., tetracysteine motif binding site17), protein (e.g., chloramphenicol acteyltransferase,18 GFP19), or split protein fragment (e.g., β-galactosidase,20 GFP21) whose activity is modulated by the folding behavior of the POI. Along similar lines, we recently developed a protein solubility reporter that exploits the authentic protein folding quality control mechanism of the E. coli Tat pathway.22 Surprisingly, there are currently no reports of an analogous reporter system for the periplasmic folding environment despite the significance of this compartment for the expression and engineering of heterologous proteins.1–3 This is due in part to the fact that GFP, one of the most useful cellular reporter proteins that has been used to successfully assay cytoplasmic folding,19 fails to reach a fluorescent conformation when routed to the periplasm via the Sec export mechanism.40 To remedy this, we developed a genetic reporter system comprising a POI fused between an N-terminal cotranslational export signal (ssDsbA) and a C-terminal selectable marker (Bla) that enables intimate coupling between periplasmic protein folding and antibiotic resistance. It should be noted that Bla gene fusions have been used previously in E. coli to: (1) isolate genes encoding exported and membrane proteins of prokaryotic53 and eukaryotic54 origin; (2) increase secretion efficiency of exported and membrane proteins55; and (3) dissect membrane protein topology and assembly.56 However, no studies have used Bla for reporting the folding robustness of target proteins in the periplasmic space. Another unique aspect of our system is its use of the cotranslational SRP pathway such that all folding events are relegated exclusively to the domain of the periplasm. One concern that arises when using Bla fusions is that proteolytic degradation can release active Bla. Indeed, our experiments revealed some degradation of the PhoA-Bla fusion. However, the degradation does not appear to have released functional Bla because the MBC of these cells (100 μg/mL) was notably lower than that seen for other cases where the ssDsbA-POI-Bla fusion was not proteolyzed (e.g., MBP-Bla, scFv13-Bla, and scFv13-R4-Bla). Even though we cannot rule out the possibility in the future that certain protein fusions may be degraded to release functional Bla, we see no evidence in the cases that were tested here. In fact, most of the other Bla fusions tested in this study experienced little to no proteolytic degradation. Further, although the correctly folded ssDsbA-PhoA-Bla fusion was not completely stable, the misfolded counterpart was even less stable (i.e., completely degraded) as evidenced by the total absence of cross-reacting bands on the Western blot. Thus, the assay was still able to reliably report the folding robustness of the PhoA protein. It turns out that this was not a unique phenomenon as many of the incorrectly folded proteins (e.g., MetF and MetK) were efficiently degraded. In light of these results, we contend that any Bla activity that might arise because of fusion instability should not impact the usefulness of the assay provided that proper negative controls are applied, as was done here.
As in many cases protein expression in the periplasm is often advantageous compared with expression in the cytoplasm,1–3 this assay should be useful in the development and optimization of numerous biotechnological applications. For instance, we demonstrated that the assay is useful for the isolation of solubility-enhanced variants from a combinatorial library of protein sequences. These results indicate that this method could be used as a platform for (1) engineering proteins with superior solubility, that is, “superfolder” proteins57 and (2) preselection of large combinatorial libraries to eliminate incorrectly folded proteins as soluble structure is a prerequisite for function. In the future, it might be possible to use this system in combination with our recently reported Tat-based cytoplasmic folding reporter,22 which also relies on Bla activity, to comparatively and comprehensively explore the intracellular protein folding landscape. It might also be possible to use this selection in conjunction with fluorescence-activated cell sorting to isolate GFP variants that fold and function in the periplasm. In addition to sequence-related determinants of protein folding, we have also shown that this assay can be used to probe extrinsic factors (e.g., molecular chaperones) that contribute to protein solubility in the periplasm. The ability to characterize or discover factors that affect folding in the periplasm and are orthogonal to protein sequence is intriguing. A more comprehensive model of the periplasmic space could have great implications for expression of proteins whose primary structure is constrained for reasons of, for example, therapeutic activity and immunogenicity. Besides chaperones, this assay can also be used to analyze how periplasmic posttranslational modifications such as disulfide bond formation, as demonstrated here, or even N-linked glycosylation58 affect folding and solubility. The stability conferred by disulfide bonds is often paramount to achieving soluble expression of many therapeutic proteins in E. coli, many of which are currently produced in E. coli by inclusion body formation and refolding procedures due to disulfide bond–related instability in the reducing cytoplasm. It should be noted that for those proteins whose proper folding depends on posttranslational processing, the periplasmic folding reporter becomes a genetic reporter of that posttranslational processing pathway. Finally, the fact that our assay can be used to evaluate the contribution of these processes to protein folding should help illuminate the various ways in which disulfide bonds and glycosylation reactions, or even their interplay,59 affects the structure, function, and stability of proteins.
Materials and Methods
Strains and plasmids
DHB4 E. coli cells (F′ lacIqpro/λ- ΔlacX74galE galK thi rpsL phoR ΔphoA(PvuII) ΔmalF3) or an isogenic derivative of DHB4, namely DHA, that carries the dsbA::kan allele,33 were used for experiments where cellular redox state was investigated. All other experiments were performed in DH5α or MC4100 E. coli cells as indicated. Cloning was performed using standard molecular biological techniques and protocols.60 Plasmid pDMB was constructed by inserting DNA for the DsbA signal peptide (ssDsbA; DNA nucleotides 1–57 of the E. colidsbA gene) between SacI and XbaI sites of pTrc99A-Cm.22 Next, the gene encoding TEM-1 Bla was inserted between the BamHI and HindIII sites. Finally, genes encoding different POIs were inserted between the XbaI and SalI or BamHI sites resulting in a sandwich fusion between ssDsbA and Bla. The resulting plasmids contained very short linker sequences, Ser-Arg and Val-Asp-Gly-Ser, at the junctions of ssDsbA-POI and POI-Bla [see Fig. 1(a)], corresponding to the translated portions of the XbaI and SalI-BamHI sites, respectively. The POIs included: MetF, MetK, GST, PhoA, and TrxA, all of which were amplified from the E. coli genome using colony PCR; PhoA[SSCC], which was amplified from pMS104 lacking the fifth aberrant cysteine,61 was kindly provided by M. Berkman; MalE, MalE31, MalE-G32D and MalE-I33P,45 and scFv13 and scFv13-R436 were kindly provided by J.-M. Betton; wild-type Aβ42 peptide and solubility-enhanced Aβ42 variants GM7, GM11, and GM650 were kindly provided by M.H. Hecht; and GFP, which was PCR-amplified from pTMB-GFP.22 Plasmid pTMB is identical to pDMB except that it contains the signal peptide of E. coli trimethylamine N-oxide reductase (ssTorA; DNA bases 1–126 of the E. coli torA gene) between SacI and XbaI sites. Genes encoding the periplasmic chaperones SurA, FkpA, and Skp were PCR-amplified from E. coli genomic DNA and inserted between the NcoI and SalI sites of pBAD18-Kan.62 For mammalian protein expression (and GFP), a vector called pDSALK was created. This plasmid was created from a kanamycin-resistant version of pSALect44 and contains the DsbA signal sequence between the NotI and XbaI restriction sites. Mammalian proteins were cloned between the XbaI and either SalI or BamHI sites of pDSALK and then transformed into MC4100 cells for plating on Amp. For Western blot analysis of mammalian proteins expressed without the Bla moiety, proteins or protein domains with N-terminal ssDsbA were PCR-amplified from pDSALK constructs and cloned between the NcoI and HindIII sites of pTrc99A. A FLAG affinity tag was added to the C-termini of each protein or protein domain by PCR. Sequences of all plasmids constructed in this study were confirmed by DNA sequencing.
Expression of fusion proteins and cell growth assays
Cells carrying a folding reporter plasmid were grown overnight at 37°C in LB medium containing 50 μg/mL chloramphenicol (Cm). Screening of cells on LB agar was performed by first normalizing overnight cultures by OD600 and then spotting 5 μL of serially diluted (10- to 105-fold) cells on LB agar plates containing 100 μg/mL Amp or 20 μg/mL Cm. LB agar plates used to analyze coexpression of periplasmic chaperones were supplemented with 50 μg/mL kanamycin (Kan), along with either 20 μg/mL Cm or 200 μg/mL Amp and either 0.2% arabinose or 0.2% glucose. The trc promoter allows for leaky expression, and therefore, no IPTG was used to induce cultures. In all cases, the plates were incubated for 16 h at 37°C and then imaged using a ChemiDoc System (BioRad). For MBC/MIC determination, ∼200 colony-forming units (CFUs) of each clone were plated on LB agar plates containing 0, 3, 6, 12, 25, 50, 100, 200, 400, 800, or 1600 μg/mL Amp or 20 μg/mL Cm. The MBC was determined as the minimum Amp concentration at which no colonies appeared on the plates. Minimum inhibitory concentration (MIC) was determined as the minimum concentration of Amp on which colony size or number of colonies was significantly smaller than control.
Protein analysis
Cells were grown overnight at 37°C in flasks containing 50-mL LB media with appropriate antibiotics. As above for the plating experiments, no IPTG was used to induce cultures because the trc promoter allows for sufficient leaky expression. Subcellular fractionation using the ice-cold osmotic shock procedure33 was performed on an equivalent number of cells to generate soluble cytoplasmic and periplasmic fractions. Cytoplasmic fractions were obtained by sonication of resuspended spheroplasts following release of periplasmic proteins. Western blotting of these fractions was performed as previously described33 using 10 μg/mL anti-β-lactamase, anti-FLAG M2 (Stratagene) at 1:500 dilution, anti-alkaline phosphatase antibodies (Sigma) at 1:20,000 dilution, or anti-DsbA serum diluted 1:5000 (kindly provided by Dr. James Bardwell) as the primary antibody and anti-mouse or anti-rabbit horseradish peroxidase conjugate diluted 1:2500 (Promega, Madison, WI) as the secondary antibody. Bands were visualized via chemiluminescent substrate (Bio-Rad) on Kodak film. The quality of all fractionations was determined by immunodetection of the cytoplasmic GroEL protein33 or the periplasmic protein DsbA. Fractions from GFP-expressing cells were assayed for fluorescence by loading 100-μL portions into 96-well plates and quantifying the GFP activity (ex: 488 nm; em: 509 nm) using a microplate reader (Synergy HT, BioTek Instruments). Finally, soluble fractions were assayed for Bla activity based on nitrocefin (50 μM) hydrolysis in 96-well format as described.63 All fluorescence and Bla activity measurements were performed in triplicate.
Fluorescence microscopy
Cells expressing ssDsbA-GFP-Bla and ssTorA-GFP-Bla were visualized as described previously64 using a Zeiss Axioskop 40 fluorescent microscope with Spotflex color digital camera and filter sets for GFP (485 nm for excitation and 505 nm for emission) and rhodamine (540 nm for excitation and 600 nm for emission).
Library creation and selection of clones
A library of Aβ42 sequences was created according to Fisher et al.,22 except that the plasmid backbone was pDMB. Briefly, error-prone PCR was performed on the gene encoding the Aβ42 peptide. The gene library was cloned between the XbaI and SalI sites of pDMB and estimated to contain ∼50,000 members. Selection was performed by plating ∼2000 CFUs per plate on 25 μg/mL Amp. To eliminate false positives (e.g., small in-frame fragments that confer higher-than-expected resistance to Amp), clones growing on 25 μg/mL Amp were inoculated in 96-well cultures and replica spotted as above at 10−3 dilution on 25 and 100 μg/mL Amp. Only those clones that grew on 25 μg/mL Amp but failed to grow on 100 μg/mL Amp were sequenced and characterized by MBC determination.
Acknowledgments
The authors thank Dr. Jean-Michel Betton, Dr. Mehmet Berkman and Dr. Michael Hecht for plasmid DNA and Dr. James Bardwell for anti-DsbA serum used in these studies. They also thank Dr. Michael Dyson for providing plasmid DNA for the mammalian proteins.
References
- 1.Baneyx F, Mujacic M. Recombinant protein folding and misfolding in Escherichia coli. Nat Biotechnol. 2004;22:1399–1408. doi: 10.1038/nbt1029. [DOI] [PubMed] [Google Scholar]
- 2.Swartz JR. Advances in Escherichia coli production of therapeutic proteins. Curr Opin Biotechnol. 2001;12:195–201. doi: 10.1016/s0958-1669(00)00199-3. [DOI] [PubMed] [Google Scholar]
- 3.Georgiou G, Segatori L. Preparative expression of secreted proteins in bacteria: status report and future prospects. Curr Opin Biotechnol. 2005;16:538–545. doi: 10.1016/j.copbio.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Sorensen HP, Mortensen KK. Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microb Cell Fact. 2005;4:1. doi: 10.1186/1475-2859-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dyson MR, Shadbolt SP, Vincent KJ, Perera RL, McCafferty J. Production of soluble mammalian proteins in Escherichia coli: identification of protein features that correlate with successful expression. BMC Biotechnol. 2004;4:32. doi: 10.1186/1472-6750-4-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luan CH, Qiu S, Finley JB, Carson M, Gray RJ, Huang W, Johnson D, Tsao J, Reboul J, Vaglio P, Hill DE, Vidal M, Delucas LJ, Luo M. High-throughput expression of C. elegans proteins. Genome Res. 2004;14:2102–2110. doi: 10.1101/gr.2520504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braun P, Hu Y, Shen B, Halleck A, Koundinya M, Harlow E, LaBaer J. Proteome-scale purification of human proteins from bacteria. Proc Natl Acad Sci USA. 2002;99:2654–2659. doi: 10.1073/pnas.042684199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker TA, Sauer RT. ATP-dependent proteases of bacteria: recognition logic and operating principles. Trends Biochem Sci. 2006;31:647–653. doi: 10.1016/j.tibs.2006.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomoyasu T, Mogk A, Langen H, Goloubinoff P, Bukau B. Genetic dissection of the roles of chaperones and proteases in protein folding and degradation in the Escherichia coli cytosol. Mol Microbiol. 2001;40:397–413. doi: 10.1046/j.1365-2958.2001.02383.x. [DOI] [PubMed] [Google Scholar]
- 10.Bowden GA, Paredes AM, Georgiou G. Structure and morphology of protein inclusion bodies in Escherichia coli. Biotechnology. 1991;9:725–730. doi: 10.1038/nbt0891-725. [DOI] [PubMed] [Google Scholar]
- 11.Villaverde A, Carrio MM. Protein aggregation in recombinant bacteria: biological role of inclusion bodies. Biotechnol Lett. 2003;25:1385–1395. doi: 10.1023/a:1025024104862. [DOI] [PubMed] [Google Scholar]
- 12.Ellis RJ, Minton AP. Cell biology: join the crowd. Nature. 2003;425:27–28. doi: 10.1038/425027a. [DOI] [PubMed] [Google Scholar]
- 13.Kadokura H, Katzen F, Beckwith J. Protein disulfide bond formation in prokaryotes. Annu Rev Biochem. 2003;72:111–35. doi: 10.1146/annurev.biochem.72.121801.161459. [DOI] [PubMed] [Google Scholar]
- 14.Weerapana E, Imperiali B. Asparagine-linked protein glycosylation: from eukaryotic to prokaryotic systems. Glycobiology. 2006;16:91R–101R. doi: 10.1093/glycob/cwj099. [DOI] [PubMed] [Google Scholar]
- 15.Wacker M, Linton D, Hitchen PG, Nita-Lazar M, Haslam SM, North SJ, Panico M, Morris HR, Dell A, Wren BW, Aebi M. N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science. 2002;298:1790–1793. doi: 10.1126/science.298.5599.1790. [DOI] [PubMed] [Google Scholar]
- 16.Missiakas D, Raina S. Protein folding in the bacterial periplasm. J Bacteriol. 1997;179:2465–2471. doi: 10.1128/jb.179.8.2465-2471.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ignatova Z, Gierasch LM. Monitoring protein stability and aggregation in vivo by real-time fluorescent labeling. Proc Natl Acad Sci USA. 2004;101:523–528. doi: 10.1073/pnas.0304533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maxwell KL, Mittermaier AK, Forman-Kay JD, Davidson AR. A simple in vivo assay for increased protein solubility. Protein Sci. 1999;8:1908–1911. doi: 10.1110/ps.8.9.1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waldo GS, Standish BM, Berendzen J, Terwilliger TC. Rapid protein-folding assay using green fluorescent protein. Nat Biotechnol. 1999;17:691–695. doi: 10.1038/10904. [DOI] [PubMed] [Google Scholar]
- 20.Wigley WC, Stidham RD, Smith NM, Hunt JF, Thomas PJ. Protein solubility and folding monitored in vivo by structural complementation of a genetic marker protein. Nat Biotechnol. 2001;19:131–136. doi: 10.1038/84389. [DOI] [PubMed] [Google Scholar]
- 21.Cabantous S, Terwilliger TC, Waldo GS. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat Biotechnol. 2005;23:102–107. doi: 10.1038/nbt1044. [DOI] [PubMed] [Google Scholar]
- 22.Fisher AC, Kim W, DeLisa MP. Genetic selection for protein solubility enabled by the folding quality control feature of the twin-arginine translocation pathway. Protein Sci. 2006;15:449–458. doi: 10.1110/ps.051902606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chautard H, Blas-Galindo E, Menguy T, Grand'Moursel L, Cava F, Berenguer J, Delcourt M. An activity-independent selection system of thermostable protein variants. Nat Methods. 2007;4:919–921. doi: 10.1038/nmeth1090. [DOI] [PubMed] [Google Scholar]
- 24.Waldo GS. Genetic screens and directed evolution for protein solubility. Curr Opin Chem Biol. 2003;7:33–38. doi: 10.1016/s1367-5931(02)00017-0. [DOI] [PubMed] [Google Scholar]
- 25.Fisher AC, DeLisa MP. Efficient isolation of soluble intracellular single-chain antibodies using the twin-arginine translocation machinery. J Mol Biol. 2009;385:299–311. doi: 10.1016/j.jmb.2008.10.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pedelacq JD, Piltch E, Liong EC, Berendzen J, Kim CY, Rho BS, Park MS, Terwilliger TC, Waldo GS. Engineering soluble proteins for structural genomics. Nat Biotechnol. 2002;20:927–932. doi: 10.1038/nbt732. [DOI] [PubMed] [Google Scholar]
- 27.Cabantous S, Waldo GS. In vivo and in vitro protein solubility assays using split GFP. Nat Methods. 2006;3:845–854. doi: 10.1038/nmeth932. [DOI] [PubMed] [Google Scholar]
- 28.Philibert P, Martineau P. Directed evolution of single-chain Fv for cytoplasmic expression using the beta-galactosidase complementation assay results in proteins highly susceptible to protease degradation and aggregation. Microb Cell Fact. 2004;3:16. doi: 10.1186/1475-2859-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arie JP, Miot M, Sassoon N, Betton JM. Formation of active inclusion bodies in the periplasm of Escherichia coli. Mol Microbiol. 2006;62:427–437. doi: 10.1111/j.1365-2958.2006.05394.x. [DOI] [PubMed] [Google Scholar]
- 30.Schierle CF, Berkmen M, Huber D, Kumamoto C, Boyd D, Beckwith J. The DsbA signal sequence directs efficient, cotranslational export of passenger proteins to the Escherichia coli periplasm via the signal recognition particle pathway. J Bacteriol. 2003;185:5706–5713. doi: 10.1128/JB.185.19.5706-5713.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steiner D, Forrer P, Stumpp MT, Pluckthun A. Signal sequences directing cotranslational translocation expand the range of proteins amenable to phage display. Nat Biotechnol. 2006;24:823–831. doi: 10.1038/nbt1218. [DOI] [PubMed] [Google Scholar]
- 32.Sone M, Kishigami S, Yoshihisa T, Ito K. Roles of disulfide bonds in bacterial alkaline phosphatase. J Biol Chem. 1997;272:6174–6178. doi: 10.1074/jbc.272.10.6174. [DOI] [PubMed] [Google Scholar]
- 33.DeLisa MP, Tullman D, Georgiou G. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci USA. 2003;100:6115–6120. doi: 10.1073/pnas.0937838100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frech C, Wunderlich M, Glockshuber R, Schmid FX. Competition between DsbA-mediated oxidation and conformational folding of RTEM1 beta-lactamase. Biochemistry. 1996;35:11386–11395. doi: 10.1021/bi9608525. [DOI] [PubMed] [Google Scholar]
- 35.Kadokura H, Beckwith J. Detecting folding intermediates of a protein as it passes through the bacterial translocation channel. Cell. 2009;138:1164–1173. doi: 10.1016/j.cell.2009.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martineau P, Jones P, Winter G. Expression of an antibody fragment at high levels in the bacterial cytoplasm. J Mol Biol. 1998;280:117–127. doi: 10.1006/jmbi.1998.1840. [DOI] [PubMed] [Google Scholar]
- 37.Kapust RB, Waugh DS. Escherichia coli maltose-binding protein is uncommonly effective at promoting the solubility of polypeptides to which it is fused. Protein Sci. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sheppard CA, Trimmer EE, Matthews RG. Purification and properties of NADH-dependent 5,10-methylenetetrahydrofolate reductase (MetF) from Escherichia coli. J Bacteriol. 1999;181:718–725. doi: 10.1128/jb.181.3.718-725.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kerner MJ, Naylor DJ, Ishihama Y, Maier T, Chang HC, Stines AP, Georgopoulos C, Frishman D, Hayer-Hartl M, Mann M, Hartl FU. Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli. Cell. 2005;122:209–220. doi: 10.1016/j.cell.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 40.Feilmeier BJ, Iseminger G, Schroeder D, Webber H, Phillips GJ. Green fluorescent protein functions as a reporter for protein localization in Escherichia coli. J Bacteriol. 2000;182:4068–4076. doi: 10.1128/jb.182.14.4068-4076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fisher AC, DeLisa MP. Laboratory evolution of fast-folding green fluorescent protein using secretory pathway quality control. PLoS ONE. 2008;3:e2351. doi: 10.1371/journal.pone.0002351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lim HK, Mansell TJ, Linderman SW, Fisher AC, Dyson MR, DeLisa MP. Mining mammalian genomes for folding competent proteins using Tat-dependent genetic selection in Escherichia coli. Protein Sci. 2009;18:2537–2549. doi: 10.1002/pro.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liebscher M, Jahreis G, Lucke C, Grabley S, Raina S, Schiene-Fischer C. Fatty acyl benzamido antibacterials based on inhibition of DnaK-catalyzed protein folding. J Biol Chem. 2007;282:4437–4446. doi: 10.1074/jbc.M607667200. [DOI] [PubMed] [Google Scholar]
- 44.Lim HK, Mansell TJ, Linderman S, Fisher AC, Dyson MR, DeLisa MP. Mining mammalian genomes for folding competent proteins using Tat-dependent genetic selection in Escherichia coli. Protein Sci. 2009;18:2537–2549. doi: 10.1002/pro.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Betton JM, Hofnung M. Folding of a mutant maltose-binding protein of Escherichia coli which forms inclusion bodies. J Biol Chem. 1996;271:8046–8052. doi: 10.1074/jbc.271.14.8046. [DOI] [PubMed] [Google Scholar]
- 46.Arie JP, Sassoon N, Betton JM. Chaperone function of FkpA, a heat shock prolyl isomerase, in the periplasm of Escherichia coli. Mol Microbiol. 2001;39:199–210. doi: 10.1046/j.1365-2958.2001.02250.x. [DOI] [PubMed] [Google Scholar]
- 47.Missiakas D, Betton JM, Raina S. New components of protein folding in extracytoplasmic compartments of Escherichia coli SurA, FkpA and Skp/OmpH. Mol Microbiol. 1996;21:871–884. doi: 10.1046/j.1365-2958.1996.561412.x. [DOI] [PubMed] [Google Scholar]
- 48.Bothmann H, Pluckthun A. Selection for a periplasmic factor improving phage display and functional periplasmic expression. Nat Biotechnol. 1998;16:376–380. doi: 10.1038/nbt0498-376. [DOI] [PubMed] [Google Scholar]
- 49.Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 50.Wurth C, Guimard NK, Hecht MH. Mutations that reduce aggregation of the Alzheimer's Abeta42 peptide: an unbiased search for the sequence determinants of Abeta amyloidogenesis. J Mol Biol. 2002;319:1279–1290. doi: 10.1016/S0022-2836(02)00399-6. [DOI] [PubMed] [Google Scholar]
- 51.Kheterpal I, Williams A, Murphy C, Bledsoe B, Wetzel R. Structural features of the Abeta amyloid fibril elucidated by limited proteolysis. Biochemistry. 2001;40:11757–11767. doi: 10.1021/bi010805z. [DOI] [PubMed] [Google Scholar]
- 52.Kraft M, Knupfer U, Wenderoth R, Pietschmann P, Hock B, Horn U. An online monitoring system based on a synthetic sigma32-dependent tandem promoter for visualization of insoluble proteins in the cytoplasm of Escherichia coli. Appl Microbiol Biotechnol. 2007;75:397–406. doi: 10.1007/s00253-006-0815-6. [DOI] [PubMed] [Google Scholar]
- 53.Smith H, de Jong A, Bron S, Venema G. Characterization of signal-sequence-coding regions selected from the Bacillus subtilis chromosome. Gene. 1988;70:351–361. doi: 10.1016/0378-1119(88)90207-7. [DOI] [PubMed] [Google Scholar]
- 54.Tan R, Jiang X, Jackson A, Jin P, Yang J, Lee E, Duggan B, Stuve LL, Fu GK. E. coli selection of human genes encoding secreted and membrane proteins based on cDNA fusions to a leaderless beta-lactamase reporter. Genome Res. 2003;13:1938–1943. doi: 10.1101/gr.1000903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Broome-Smith JK, Bowler LD, Spratt BG. A simple method for maximizing the yields of membrane and exported proteins expressed in Escherichia coli. Mol Microbiol. 1989;3:1813–1817. doi: 10.1111/j.1365-2958.1989.tb00167.x. [DOI] [PubMed] [Google Scholar]
- 56.Edelman A, Bowler L, Broome-Smith JK, Spratt BG. Use of a beta-lactamase fusion vector to investigate the organization of penicillin-binding protein 1B in the cytoplasmic membrane of Escherichia coli. Mol Microbiol. 1987;1:101–106. doi: 10.1111/j.1365-2958.1987.tb00533.x. [DOI] [PubMed] [Google Scholar]
- 57.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Engineering and characterization of a superfolder green fluorescent protein. Nat Biotechnol. 2006;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 58.Szymanski CM, Wren BW. Protein glycosylation in bacterial mucosal pathogens. Nat Rev Microbiol. 2005;3:225–237. doi: 10.1038/nrmicro1100. [DOI] [PubMed] [Google Scholar]
- 59.Bosques CJ, Imperiali B. The interplay of glycosylation and disulfide formation influences fibrillization in a prion protein fragment. Proc Natl Acad Sci USA. 2003;100:7593–7598. doi: 10.1073/pnas.1232504100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sambrook J, Russell DW. Molecular cloning : a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- 61.Sone M, Akiyama Y, Ito K. Additions and corrections to differential in vivo roles played by DsbA and DsbC in the formation of protein disulfide bonds. J Biol Chem. 1998;273:27756. doi: 10.1074/jbc.272.16.10349. [DOI] [PubMed] [Google Scholar]
- 62.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Galarneau A, Primeau M, Trudeau LE, Michnick SW. Beta-lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein protein interactions. Nat Biotechnol. 2002;20:619–622. doi: 10.1038/nbt0602-619. [DOI] [PubMed] [Google Scholar]
- 64.Kim JY, Doody AM, Chen DJ, Cremona GH, Shuler ML, Putnam D, DeLisa MP. Engineered bacterial outer membrane vesicles with enhanced functionality. J Mol Biol. 2008;380:51–66. doi: 10.1016/j.jmb.2008.03.076. [DOI] [PMC free article] [PubMed] [Google Scholar]