

Abstract
InvA is a prominent inner-membrane component of the Salmonella type III secretion system (T3SS) apparatus, which is responsible for regulating virulence protein export in pathogenic bacteria. InvA is made up of an N-terminal integral membrane domain and a C-terminal cytoplasmic domain that is proposed to form part of a docking platform for the soluble export apparatus proteins notably the T3SS ATPase InvC. Here, we report the novel crystal structure of the C-terminal domain of Salmonella InvA which shows a compact structure composed of four subdomains. The overall structure is unique although the first and second subdomains exhibit structural similarity to the peripheral stalk of the A/V-type ATPase and a ring building motif found in other T3SS proteins respectively.
Keywords: type III secretion, Salmonella, crystal structure, InvA
Introduction
The bacterial injectisome, or nonflagellar type III secretion system (NF-T3SS), is a specialized needle-like protein-export system utilized by many pathogenic or symbiotic Gram-negative bacteria for the injection of virulence inducing proteins (effectors) into host cells. It is essential for the virulence of many medically relevant bacteria including Pseudomonas, Yersinia, Salmonella, Shigella and enteropathogenic E. coli (EPEC). Bacterial proteins destined for both needle assembly and host cell targeting are translocated via the injectisome in a process known as type III secretion. This process has characteristics in common with the assembly of the flagellar motility machinery (F-T3SS) of bacteria and there exists extensive homology between these two systems particularly within the inner membrane components.
Indispensable for secretion are a set of highly conserved inner membrane proteins believed to be located in the pore at the base of the injectisome (SpaPQRS+InvA, Salmonella nomenclature; YscRSTUV, Yersinia nomenclature; FliO(no NF-T3SS homologue)PQR+FlhBA, flagellar nomenclature) and several membrane associated proteins most notably the type III ATPase InvC/YscN/FliI (Salmonella/Yersinia/flagellar). The membrane components are predicted to form an export channel with the cytoplasmic domains of SpaS/YscU/FlhB and InvA/YscV/FlhA acting as a docking platform for the substrate bound InvC/YscN/FliI complex.1 Upon binding, InvC/YscN/FliI is proposed to use the energy derived from ATP hydrolysis to dissociate substrate-chaperone complexes2 before translocation through the T3SS in a process driven by the proton motive force.3,4
InvA/YscV/FlhA, the largest member of the inner membrane export apparatus, consists of a highly conserved ∼35 kDa N-terminal integral membrane region connected by a 20–30 residue flexible linker to a more variable ∼40 kDa C-terminal soluble domain proposed to be localized to the bacterial cytoplasm. The paralogous C-terminal domain of Salmonella flagellar FlhA [∼22% sequence identity with the C-terminal domain of InvA; Fig. 1(a)] has been studied using a variety of biochemical and cellular approaches. It has been shown to interact with several soluble flagellar apparatus proteins including the ATPase FliI (InvC/YscN), its regulator FliH (OrgB/YscL), and FliJ (InvI/YscO); and is associated with a dominant-negative multicopy effect, inhibiting flagellar action by sequestering soluble components of the assembly apparatus in non-productive complexes.1,5-8 It is proposed that the C-terminal domain of FlhA provides one of the main sites of interaction at the base of the F-T3SS for the soluble assembly complex before subsequent substrate insertion into the inner membrane portal of the F-T3SS.7
Figure 1.
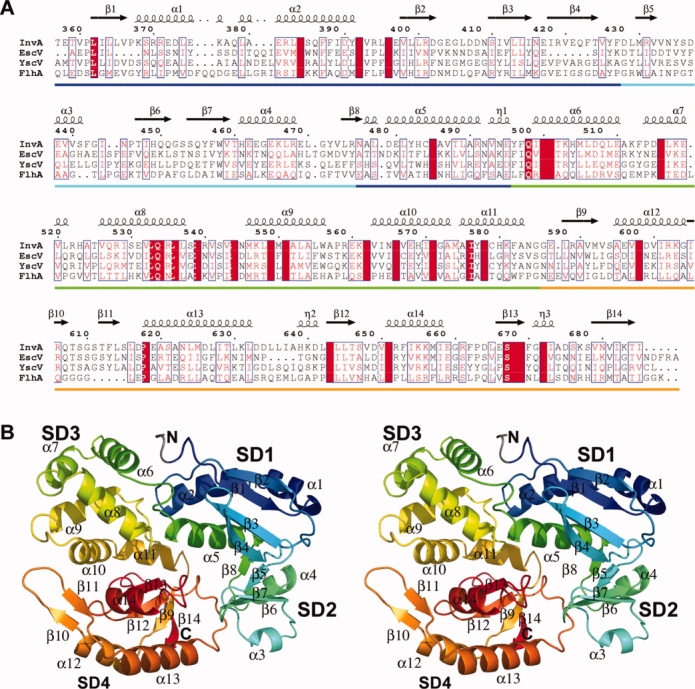
Structure of InvA357–685. (a) Sequence alignment of InvA357–685 and homologues from EPEC (EscV), Yersinia (YscV) and the Salmonella F-T3SS (FlhA). Secondary structure from InvA structure depicted above. Subdomains indicated along bottom (SD1–blue; SD2–cyan; SD3–green; SD4–orange). (b) Stereo diagram of overall structure of InvA357–685. Colored from N-terminal (blue) to C-terminal (red) (N-terminal tag gray). All structural figures were prepared with PyMOL.9
Structural data for the conserved core inner membrane components of the T3SS export apparatus is limited to the C-terminal cytoplasmic domain of SpaS/YscU/FlhB from several NF-T3SS homologues10-13 and the ATPase from both NF-T3SS14 and F-T3SS15 homologues. To gain insight into the function of InvA/YscV/FlhA we have solved the first representative structure of this family, the C-terminal domain of NF-T3SS Salmonella enterica serovar Typhimurium InvA.
Results and Discussion
Structure determination
The structure of the C-terminal domain of S. enterica serovar Typhimurium InvA (InvA357–685) from the Salmonella pathogenicity island 1 (SPI-1) was determined to 1.5 Å resolution. The protein crystallized in two crystal forms with space groups P212121 and P3221, both with one molecule in the asymmetric unit (for data collection and refinement statistics see Table I). Phases were experimentally determined from form 1 crystals using multiple wavelength anomalous dispersion with an ethyl mercury phosphate derivative. Three heavy-atom positions were located bound to residues His461, Cys474, and Cys579. The phases for the native data collected from form 2 crystals were solved using molecular replacement with the form 1 structure as a search model. The refined structures have good overall stereochemistry and are in good agreement with a backbone RMSD of 0.8 Å. In addition to the heavy atoms, two calcium atoms and a PEG molecule have been modelled in the form 1 data and two phosphates and one CAPS molecule included in the form 2 model.
Table I.
Data Collection and Refinement Statistics. Values in Parenthesis Are for Highest-Resolution Shell. Data Refined Against a High-Resolution Dataset Collected at the Remote Wavelength and Merged with the “Remote” Dataset (HR-Rem-mrg)
| Form 1 |
||||||
|---|---|---|---|---|---|---|
| Data collection | Remote | HR-Rem-mrg | Peak | Inflection 1 | Inflection 2 | Form 2 |
| Space group | P212121 | P3221 | ||||
| Cell dimensions | ||||||
| a,b,c (Å) | 44.8, 57.3, 113.6 | 79.2, 79.2, 127.7 | ||||
| α,β,γ (°) | 90, 90, 90 | 90, 90, 120 | ||||
| Wavelength (Å) | 0.9793 | 1.0064 | 1.0087 | 1.0084 | 1.5418 | |
| Resolution (Å) | 50−2.2 (2.31−2.2) | 30−1.5 (1.57−1.5) | 50–2.2 (2.31−2.2) | 50−2.2 (2.31−2.2) | 50−2.2 (2.31−2.2) | 68−2.5 (2.65−2.5) |
| Rmerge (%) | 4.2 (6.9) | 10.5 (22.5) | 4.1 (9.7) | 2.9 (4.1) | 3.1 (6.4) | 6.5 (23.2) |
| Average I/σ(I) | 28.7 (15.8) | 11.4 (2) | 20.7 (7.7) | 29.1 (15.5) | 26.5 (13) | 12.6 (4.0) |
| Completeness (%) | 97.2 (97.2) | 93.3 (93.3) | 89.7 (89.7) | 88.9 (88.9) | 89.5 (89.5) | 96.5 (76.2) |
| Redundancy | 6.1 (3.2) | 4.5 (1.9) | 3.4 (1.9) | 3.4 (1.9) | 3.4 (1.9) | 4.3 (4.1) |
| Refinement | ||||||
| Resolution (Å) | 30−1.5 (1.57−1.5) | 68−2.5 (2.65−2.5) | ||||
| No. reflections | 202976 (9403) | 67747 (7252) | ||||
| Unique reflections | 44795 (4823) | 15695 (1764) | ||||
| R/Rfreea (%) | 19.2/22.5 | 20.6/26.4 | ||||
| No. atoms | ||||||
| Protein | 2691 | 2648 | ||||
| Water | 292 | 78 | ||||
| Other | 12 | 24 | ||||
| B-factors (Å2) | ||||||
| Protein | 19.17 | 22.2 | ||||
| Water | 31.52 | 20.6 | ||||
| Other | 38.72 | 52.4 | ||||
| r.m.s. deviations | ||||||
| Bond lengths (Å) | 0.0173 | 0.0201 | ||||
| Bond angles (°) | 1.651 | 1.812 | ||||
| Ramachandran plot (%) | ||||||
| Favored | 98.5 | 95.36 | ||||
| Allowed | 2.5 | 4.33 | ||||
| Disallowed | 0 | 0.31 | ||||
5% of data used for Rfree calculation.
Overall structure of InvA357–685
The overall structure shows a relatively compact globular fold with approximate dimensions of 60 × 50 × 40 Å, that can be divided into four subdomains referred to as InvA357–685SD1–4 [Fig. 1(b)]. InvA357–685SD1 is made of two segments, residues (−4)357–427 and 476–496 (−4 to −1 denote remnant tag residues), and is composed of three helices and four sheets. The central core of the domain is made up of a mixed four stranded beta-sheet in a 1X −2 −1 topology,16 which is packed against three helices in a topological arrangement of β1, α1, α2, β2, β3, β4, α5. The majority of secondary structure elements are at the N-terminus whilst helix α5 is situated in sequence following InvA357–685SD2. Helix α5 kinks sharply ∼90° at the C-terminal ending in a three residue 310-helix that forms part of the interface with InvA357–685SD3. There is a slight variation in the N-terminal region between crystal forms 1 and 2 with the first six residues incorporating the four left over tag residues and residues 357–358 not visible in the form 2 electron density despite being well ordered in the form 1 model.
InvA357–685SD2 (residues 428–475) is the smallest subdomain. It is a mixed α/β subdomain consisting of a four stranded antiparallel β-sheet with −2X 1 2X topology. Helices α3 and α4 pack against the β-sheet to form a wedge shaped fold with the β-hairpin formed by β6 and β7 projecting toward and contacting InvA357–685SD4. InvA357–685SD3 (residues 476–585) is an all helical subdomain composed of six helices (α6-α11) positioned in an anticlockwise arrangement. InvA357–685SD4 (residues 586–685) is composed of three α-helices and six β-strands. Four strands (β9, β12–14) form a parallel β-sheet with 1X 1X −3X topology, which is flanked by helix α14 and the N- and C-terminal ends of helices α12 and α13 on one side and two 310-helices on the other. Strands β10 and β11 comprise an extended β-hairpin protruding from helices α12 and α13 that reaches up to and contacts the N-terminal end of helix α10 from SD3. This motif, in particular residues Arg606 to Thr612 exhibits the most significant degree of structural variability between the two crystal forms with up to a 4 Å shift away from SD3 observed in form 2. This is associated with significantly higher than average backbone temperature factors in both models indicative of a degree of conformational flexibility for this region of the protein.
The overall structure is relatively compact and there are extensive inter-subdomain interactions within the fold. InvA357–685SD1 interacts with each subdomain forming interfaces of 471, 666, and 180 Å2 with subdomains 2, 3, and 4 respectively. In addition to InvA357–685SD1, InvA357–685SD2 also interacts with InvA357–685SD4 with an interface area of 480 Å2. The largest interface is between InvA357–685SD3 and InvA357–685SD4 with an area of 782 Å2.
Structural comparisons
A structural similarity search of the Protein Data Bank with the pairwise structural comparison server DALI17 of the complete InvA C-terminal domain gave no statistically significant global similarities. Searching with the individual subdomains revealed that the overall architecture of InvA357–685SD1 is that of a canonical thioredoxin/glutaredoxin-like fold with the highest Z-score to an annotated thioredoxin of 5.5 (PDB 1V98) with a core RMSD of 2.6 Å. However, the catalytic cysteine residues at the beginning of thioredoxin helix α3 are not conserved and helix α2 of the thioredoxin fold is absent. Of notable interest is a predicted similarity to the E subunit of the peripheral stalk from the archeal A0A1 ATP synthase (Z-score 2.7, core RMSD 2.5 Å, PDB 2DM9) [Fig. 2(a)]. Connectivity is conserved for the core of the fold although helix α5 of InvA357–685SD1, situated in sequence following InvA357–685SD2, superposes with an N-terminal helix from subunit E. F/V/A-type ATPases are related rotary motors that link transmembrane ion transport with ATP metabolism. They are composed of a catalytic soluble component (F/V/A1) and a membrane bound ion channel (F/V/A0) and the peripheral stalk links the two, acting as a stator to prevent unwanted rotation of the catalytic subunits. The E subunit of the A- and V-ATPases has a globular C-terminal domain, similar in structure to InvA357–685SD1, which interacts with the N-terminal domain of the noncatalytic B subunit of the A/V1 component.19 The related catalytic/non-catalytic subunits of the F/V/A-type ATPases (α/β in the F-ATPase and A/B in the V/A-ATPase) are close structural homologues of the T3SS/flagellar ATPase14,15 and other homologues between the T3SS and F/V/A-ATPases have been observed.17 In light of the ability of the C-terminal domain of flagellar FlhA to interact with the F-T3SS ATPase, this observed structural similarity to a protein capable of binding a homologous ATPase may reflect a common mode of interaction for InvA/YscV/FlhA and the ATPase InvC/YscN/FliI although such a hypothesis requires experimental validation.
Figure 2.
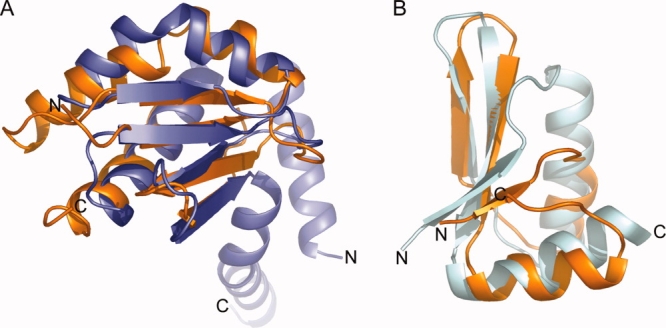
Structural similarity of InvA357–685SD1 and InvA357–685SD2. (a) Structural alignment of InvA357–685SD1 (orange) and subunit E of the A-ATPase peripheral stalk (blue, PDB 2DM9). DALI Z-score 2.7, RMSD 2.5 Å. (b) Structural alignment of InvA357–685SD2 (orange) and a ring building domain from EscJ (cyan, PDB 1YJ7). DALI Z-score 4.0, RMSD 1.8 Å.
The highest scoring annotated DALI hit for InvA357–685SD2 is surprisingly to a domain from EscJ (Z-score 4.0, RMSD 1.8 Å, PDB 1YJ7) [Fig. 2(b)], another component of the EPEC T3SS. EscJ is highly conserved and homo-oligomerises to form the prominent inner membrane ring of the T3SS basal body.20 Despite lack of detectable sequence conservation, the same domain has recently been observed in two further ring forming T3SS basal body components: Salmonella PrgH, which forms a ring structure that encompasses Salmonella EscJ homologue PrgK; and EPEC EscC, a member of the highly conserved secretin family that forms the sole outer membrane ring. In addition, the motif is also found in GspD, a type II secretion system secretin.21 It is proposed that this topologically similar fold provides a flexible ring building motif.22 Although F-T3SS InvA homologue FlhA has been shown to interact with itself5 it has not been shown to form any large stable homomeric complexes in solution as supported by our observations from static light scattering analysis of InvA357–685 at various concentrations (data not shown). The discovery of this motif in an additional member of the T3SS is thus of interest and warrants further investigation into the role of oligomerization in the InvA/YscV/FlhA family with particular focus on the membrane anchoring region and other inner membrane components that may potentiate self-association. Additionally, a similar domain from P. aeruginosa Rhomboid was shown to mediate membrane association.23 Since the C-terminal domain is tethered to the transmembrane N-terminal region it is possible that this subdomain plays some role in peripheral membrane association.
InvA357–685SD3 and SD4 yield multiple significant hits including domains within the ribonucleotide reductase R1 protein (Z-score 5.9) and a tRNA 2-selenouridine synthase (Z-score of 4.5) respectively although the specific function of the similar domains in all cases is not specifically understood.
Materials and Methods
Expression, purification, and crystallization
His6-tagged InvA357–685 was expressed from a pET-28a plasmid in BL21 cells for 4 h at 37°C. The protein was purified using Ni2+ charged fast-flow chelating sepharose (GE Healthcare), dialyzed overnight against 20 mM tris pH 7.5, 50 mM NaCl, 0.5 mM TCEP in the presence of thrombin (HTI) and resolved on a Superdex 200 16/60 (GE Healthcare) equilibrated in the same buffer. Protein was concentrated to 5–10 mg/mL. Crystals were grown at 293 K by sitting-drop vapor diffusion using either 15–20% PEG 500, 0.05 M CaCl2, 0.1 M citrate pH 5.5 (form 1) or 0.2 M LiSO4, 0.1 M CAPS (N-cyclohexyl-3-aminopropanesulfonic acid) pH 10.5, 1.2 M NaH2PO4/0.8 M K2HPO4 (form 2) as reservoir solution.
Data collection and structure determination
Form 1 crystals were cryo-protected by increasing the concentration of PEG 500 to 25% and form 2 crystals were cryo-protected by supplementing the reservoir solution with 20% ethylene glycol. An ethyl mercuric phosphate derivative of form 1 crystals was obtained by soaking in cryo-protectant +2 mM ethyl mercury phosphate for 1 h. A four wavelength MAD experiment24 was carried out at beamline ID08–1 of the Canadian Light Source (Saskatoon, Canada) at 100 K. A high resolution dataset was also collected from the same crystal at 0.9793 Å and merged with the high-energy remote. Native data were collected from form 2 crystals on a rotating anode in-house X-ray generator at 100 K. All data were indexed and integrated using the MOSFLM package25 and scaled using SCALA.26 Substructure determination and phasing was carried out with autoSHARP,27 finding three heavy atoms and yielding excellent phasing statistics and a high quality initial map. Automatic model-building was carried using ARP/wARP.26 Iterative model building and refinement, using the high-resolution dataset, was carried out with COOT28 and REFMAC26 using TLS refinement29 in the later stages with a final R/Rfree of 19.2/22.5%. Data collected from form 2 crystals was phased by molecular replacement with the program PHASER26 using the form 1 structure as a search model and refined with the same protocol to a final R/Rfree of 20.6/26.4%. Data processing and model refinement statistics are summarized in Table I. Structure quality was assessed with MolProbity30 and the final analysis results were included in the file deposited to the Protein Data Bank.
Accession numbers
The structure factors and coordinates have been deposited in the Protein Data Bank under accession codes 2X49 and 2X4A.
Conclusions
In conclusion, we have solved the high resolution crystal structure of the C-terminal cytoplasmic domain of the NF-T3SS export apparatus protein InvA. This is the first structural information of the InvA/YscV/FlhA family and adds to the increasing structural knowledge of the conserved core inner membrane T3SS export apparatus proteins. The structural similarities observed between InvA and the ATPase binding peripheral stalk from the A/V-type ATPases and a proposed ring building motif from other T3SS proteins may be a basis for functional comparison and merit further investigation into the functional interactions of the InvA/YscV/FlhA family.
Acknowledgments
Part of this research was performed at the Canadian Light Source, which was supported by NSERC, NRC, CIHR, and the University of Saskatchewan.
References
- 1.Minamino T, Macnab RM. Interactions among components of the Salmonella flagellar export apparatus and its substrates. Mol Microbiol. 2000;35:1052–1064. doi: 10.1046/j.1365-2958.2000.01771.x. [DOI] [PubMed] [Google Scholar]
- 2.Akeda Y, Galan JE. Chaperone release and unfolding of substrates in type III secretion. Nature. 2005;437:911–915. doi: 10.1038/nature03992. [DOI] [PubMed] [Google Scholar]
- 3.Minamino T, Namba K. Distinct roles of the FliI ATPase and proton motive force in bacterial flagellar protein export. Nature. 2008;451:485–488. doi: 10.1038/nature06449. [DOI] [PubMed] [Google Scholar]
- 4.Paul K, Erhardt M, Hirano T, Blair DF, Hughes KT. Energy source of flagellar type III secretion. Nature. 2008;451:489–492. doi: 10.1038/nature06497. [DOI] [PubMed] [Google Scholar]
- 5.Zhu K, Gonzalez-Pedrajo B, Macnab RM. Interactions among membrane and soluble components of the flagellar export apparatus of Salmonella. Biochemistry. 2002;41:9516–9524. doi: 10.1021/bi0203280. [DOI] [PubMed] [Google Scholar]
- 6.Mcmurry JL, Van Arnam JS, Kihara M, Macnab RM. Analysis of the cytoplasmic domains of Salmonella FlhA and interactions with components of the flagellar export machinery. J Bacteriol. 2004;186:7586–7592. doi: 10.1128/JB.186.22.7586-7592.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saijo-Hamano Y, Minamino T, Macnab RM, Namba K. Structural and functional analysis of the C-terminal cytoplasmic domain of FlhA, an integral membrane component of the type III flagellar protein export apparatus in Salmonella. J Mol Biol. 2004;343:457–466. doi: 10.1016/j.jmb.2004.08.067. [DOI] [PubMed] [Google Scholar]
- 8.Gonzalez-Pedrajo B, Minamino T, Kihara M, Namba K. Interactions between C ring proteins and export apparatus components: a possible mechanism for facilitating type III protein export. Mol Microbiol. 2006;60:984–998. doi: 10.1111/j.1365-2958.2006.05149.x. [DOI] [PubMed] [Google Scholar]
- 9.Delano WL. The PyMOL molecular graphics system. CA: DeLano Scientific LLC; 2008. [Google Scholar]
- 10.Deane JE, Graham SC, Mitchell EP, Flot D, Johnson S, Lea SM. Crystal structure of Spa40, the specificity switch for the Shigella flexneri type III secretion system. Mol Microbiol. 2008;69:267–276. doi: 10.1111/j.1365-2958.2008.06293.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zarivach R, Deng W, Vuckovic M, Felise HB, Nguyen HV, Miller SI, Finlay BB, Strynadka NC. Structural analysis of the essential self-cleaving type III secretion proteins EscU and SpaS. Nature. 2008;453:124–127. doi: 10.1038/nature06832. [DOI] [PubMed] [Google Scholar]
- 12.Lountos GT, Austin BP, Nallamsetty S, Waugh DS. Atomic resolution structure of the cytoplasmic domain of Yersinia pestis YscU, a regulatory switch involved in type III secretion. Protein Sci. 2009;18:467–474. doi: 10.1002/pro.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiesand U, Sorg I, Amstutz M, Wagner S, Van Den Heuvel J, Luhrs T, Cornelis GR, Heinz DW. Structure of the type III secretion recognition protein YscU from Yersinia enterocolitica. J Mol Biol. 2009;385:854–866. doi: 10.1016/j.jmb.2008.10.034. [DOI] [PubMed] [Google Scholar]
- 14.Zarivach R, Vuckovic M, Deng W, Finlay BB, Strynadka NC. Structural analysis of a prototypical ATPase from the type III secretion system. Nat Struct Mol Biol. 2007;14:131–137. doi: 10.1038/nsmb1196. [DOI] [PubMed] [Google Scholar]
- 15.Imada K, Minamino T, Tahara A, Namba K. Structural similarity between the flagellar type III ATPase FliI and F1-ATPase subunits. Proc Natl Acad Sci USA. 2007;104:485–490. doi: 10.1073/pnas.0608090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richardson JS. Beta-sheet topology and the relatedness of proteins. Nature. 1977;268:495–500. doi: 10.1038/268495a0. [DOI] [PubMed] [Google Scholar]
- 17.Pallen MJ, Bailey CM, Beatson SA. Evolutionary links between FliH/YscL-like proteins from bacterial type III secretion systems and second-stalk components of the FoF1 and vacuolar ATPases. Protein Sci. 2006;15:935–941. doi: 10.1110/ps.051958806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Searching protein structure databases with DaliLite v.3. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lokanath NK, Matsuura Y, Kuroishi C, Takahashi N, Kunishima N. Dimeric core structure of modular stator subunit E of archaeal H+ -ATPase. J Mol Biol. 2007;366:933–944. doi: 10.1016/j.jmb.2006.11.088. [DOI] [PubMed] [Google Scholar]
- 20.Yip CK, Kimbrough TG, Felise HB, Vuckovic M, Thomas NA, Pfuetzner RA, Frey EA, Finlay BB, Miller SI, Strynadka NC. Structural characterization of the molecular platform for type III secretion system assembly. Nature. 2005;435:702–707. doi: 10.1038/nature03554. [DOI] [PubMed] [Google Scholar]
- 21.Korotkov KV, Pardon E, Steyaert J, Hol WG. Crystal structure of the N-terminal domain of the secretin GspD from ETEC determined with the assistance of a nanobody. Structure. 2009;17:255–265. doi: 10.1016/j.str.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spreter T, Yip CK, Sanowar S, Andre I, Kimbrough TG, Vuckovic M, Pfuetzner RA, Deng W, Yu AC, Finlay BB, Baker D, Miller SI, Strynadka NC. A conserved structural motif mediates formation of the periplasmic rings in the type III secretion system. Nat Struct Mol Biol. 2009;16:468–476. doi: 10.1038/nsmb.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Del Rio A, Dutta K, Chavez J, Ubarretxena-Belandia I, Ghose R. Solution structure and dynamics of the N-terminal cytosolic domain of rhomboid intramembrane protease from Pseudomonas aeruginosa: insights into a functional role in intramembrane proteolysis. J Mol Biol. 2007;365:109–122. doi: 10.1016/j.jmb.2006.09.047. [DOI] [PubMed] [Google Scholar]
- 24.Ji X, Blaszczyk J, Chen X. The absorption edge of protein-bound mercury and a double-edge strategy for HgMAD data acquisition. Acta Cryst. 2001;57:1003–1007. doi: 10.1107/s0907444901006370. [DOI] [PubMed] [Google Scholar]
- 25.Leslie AGW. Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter Protein Crystallogr. 1992;26 [Google Scholar]
- 26.CCPR. The CCP4 suite: programs for protein crystallography. Acta Cryst. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 27.Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated structure solution with autoSHARP. Methods Mol Biol. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- 28.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 29.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Cryst. 2001;57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 30.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucl Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]