

Abstract
We present a 13C direct detection CACA-TOCSY experiment for samples with alternate 13C–12C labeling. It provides inter-residue correlations between 13Cα resonances of residue i and adjacent Cαs at positions i − 1 and i + 1. Furthermore, longer mixing times yield correlations to Cα nuclei separated by more than one residue. The experiment also provides Cα-to-sidechain correlations, some amino acid type identifications and estimates for ψ dihedral angles. The power of the experiment derives from the alternate 13C–12C labeling with [1,3-13C] glycerol or [2-13C] glycerol, which allows utilizing the small scalar 3JCC couplings that are masked by strong 1JCC couplings in uniformly 13C labeled samples.
Keywords: Alternate 13C labeling, Cα direct detection, TOCSY, Nuclear magnetic resonance (NMR), Resonance assignment, Dihedral angle
Introduction
The motivation for using 13C detection in protein NMR is to take advantage of the slower transverse relaxation rates of 13C magnetization compared to 1H. Carbon detection may extend the limits of NMR in structural and functional analyses of larger, highly dynamic, or paramagnetic proteins. According to this idea, a variety of pulse sequences have recently been developed to perform main-chain resonance assignment and structural analyses of proteins (Serber et al. 2001; Bermel et al. 2003; Arnesano et al. 2005; Lee et al. 2005; Bermel et al. 2006a, b; Takeuchi et al. 2008b; Felli and Brutscher 2009; Hsu et al. 2009; Takeuchi et al. 2010). The majority of these pulse programs use carbonyl carbons (C′) as the detected nuclei. This is mainly due to the simple spin–spin coupling pattern of C′ that has only one strong scalar coupling to the alpha carbon (Cα) (Bermel et al. 2006b). However, in largemolecular-weight systems, particularly at higher magnetic fields, carbonyl carbons suffer from fast transverse relaxation due to their large chemical shift anisotropy (CSA). In contrast, Cα nuclei have a small CSA and consequently slow transverse relaxation if samples are deuterated. Thus, deuterated Cα would be the better nuclei for 13C detection experiment in large and/or fast relaxing systems. Direct detection of 13Cα is not straightforward in uniformly 13C-labeled proteins owing to the presence of strong 13C–13C couplings with 13C′ (∼55 Hz) and 13Cβ (∼35 Hz) that split the signals into multiplets and hence reduce the sensitivity. The spectral complexity can be overcome by computational deconvolution (Shimba et al. 2003) or by spin-state selective schemes such as using IPAP or S3E (Bermel et al. 2007). However, these schemes require more complicated pulse programs and/or processing. Recently, we developed a strategy using 13C enrichment at alternating carbon sites and deuteration at the Cα position to avoid these difficulties (Takeuchi et al. 2008b). This can be achieved by using either [2-13C]- or [1,3-13C] glycerol (or pyruvate) as the carbon source while growing the E. coli bacteria expressing the target protein in D2O/15N containing media (LeMaster and Kushlan 1996; Castellani et al. 2002; Takeuchi et al. 2008b; Guo et al. 2009) (Hereafter the terms 2-13C and 1,3-13C labeling are used for alternate 13C labeling using [2-13C]- or [1,3-13C] glycerol, respectively). This approach offers advantages for the detection of 13Cα NMR signals, while maintaining heteronuclear 13C–15N couplings for establishing complete backbone sequential assignments (Takeuchi et al. 2008b, 2010).
Here we show that alternate 13C–12C labeling enables direct correlations between Cα nuclei in two neighboring amino acids via weak long range 3J coupling (∼2 Hz) (Hennig et al. 2000). These weak couplings are usually masked by the strong 1J carbon–carbon splittings but are readily observed in proteins labeled with the alternate 13C–12C-labeling scheme.
Materials and methods
All chemicals were purchased from Sigma (St. Louis, MO) unless otherwise noted. All stable-isotope-labeled materials were acquired from Cambridge Isotope Laboratories (Cambridge, MA).
Expression and purification of the B domain of protein G (GB1)
The gene for 6His-tagged GB1, consisting of 64 amino acid residues, was cloned into the pET9d vector (Novagen, San Diego, CA) as previously described (Frueh et al. 2005). GB1 was expressed in commercially available BL21 (DE3) E. coli cells (Novagen) at 37°C and protein expression was induced for 6 h at the same temperature. For [2-13C-glycerol] (or [1,3-13C-glycerol]) labeled samples, the cells were cultured in 2H, 15N M9 media containing 8.5 g/L Na2HPO4, 3 g/L KH2PO4, 0.5 g/L NaCl, 2 mM MgCl2, 0.1 mM CaCl2, and 1 g/L of 15NH4Cl in D2O, which was supplemented with 2 g/L [2-13C] (or [1,3-13C]) glycerol and 2 g/L NaH13CO3 (or NaH12CO3). The protein was purified with Ni–NTA affinity chromatography as previously described (Frueh et al. 2005).
NMR experiments
NMR spectra were recorded on a Bruker (Billerica, MA) Avance 500 spectrometer equipped with a triple-resonance carbon-cryogenic probe (TXO) designed for carbon detection experiments. Spectra of the 2-13C labeled GB1 sample (5 mM) were recorded at 15°C in buffer containing 10 mM sodium phosphate (pH 6.8), 100 mM NaCl and 20% w/v deuterated glycerol in D2O. The molecular tumbling of GB1 under those conditions corresponds to a 90 kDa protein at 25°C (Takeuchi et al. 2008a). For the 1,3-13C labeled GB1 sample (2.5 mM), spectra were recorded at 25°C in buffer containing 10 mM sodium phosphate (pH 6.8), 100 mM NaCl in D2O, without deuterated glycerol. 2D CACA-TOCSY experiments were recorded with a spectral width of 6,887 Hz (direct) × 6,910 Hz (indirect), centered at 43 ppm. CA(N)CA experiments were recorded with a spectral width of 3,531 Hz (direct) × 3,517 Hz (indirect) centered at 55 ppm. 512 and 128 complexed data points were recorded for direct and indirect directions, respectively, in both experiments. Eight scans were applied for each increment. The wider spectrum width for the 2D CACA-TOCSY was chosen to avoid folding of side chain resonances. The digital resolution in the direct and indirect dimensions after Fourier transformation was 13.5 and 54.0 Hz for the 2D CACA-TOCSY and 6.9 and 27.5 Hz for CA(N)CA. The recycling delay was optimized to be 1.25 × T1 (longitudinal relaxation time) = 4.5 s. All spectra were analyzed with the program Sparky (Goddard and Kneller 2004).
Results and discussion
We show that direct sequential connectivities between Cα spins via coherence transfer can be established by a CACA-TOCSY experiment. Figure 1 depicts the pulse scheme optimized for deuterated and alternately 13C–12C-labeled proteins. Although not necessary for this experiment, our sample was 15N labeled to compare the sensitivity of the CACA-TOCSY experiment with the previously reported CA(N)CA experiment, as we will discuss below. In short, a spin lock, which follows excitation and chemical shift evolution of Cα, correlates with the adjacent via 3J couplings between Cα spins. A related pulse scheme has been proposed previously for uniformly 13C labeled proteins and was used for obtaining side-chain assignments (Bermel et al. 2005). However, the authors also predicted that a modified version that uses Cα-selective pulses throughout, including those for the spin-lock mixing periods, could provide also sequential connectivities. To eliminate carbon–carbon couplings, the experiment also employs a double IPAP module, which adds to the length of the pulse sequence, however. Due to the selective Cα pulses and their non-ideal inversion properties, the signals from Gly, Ser, and high-field Cα resonances would become significantly weaker. Furthermore, a weak and less homogeneous B1 field produced by a spin mixing with longer selective pulses would significantly decrease the efficiency of TOCSY transfers. In addition, Cα selective pulses do not eliminate evolution under 1J(CαCβ) during t1, which broadens the lines and may even split each signal into a doublet at high resolution. Probably because of these reasons no experimental CACA-TOCSY spectrum has been reported in a literature so far showing sequential correlations. The 13Cα-detected CACA-TOCSY experiment presented here is simple and sensitive. It is designed for proteins expressed to have an alternate 13C–12C labeling pattern using procedures originally described in (LeMaster and Kushlan 1996).
Fig. 1.
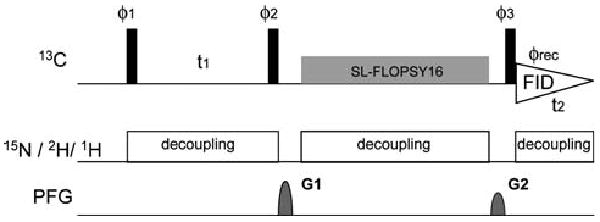
Pulse scheme of the CACA-TOCSY experiment optimized for uniformly 2H15N- and alternate 13C–12C -labeled samples. Black bars indicate non-selective π/2 pulses. FLOPSY-16 was applied for spin locking (3.5 kHz). The phase cycle employed was ϕ1 = (x, −x), ϕ2 = (x, x, −x, −x), ϕ3 = (x, x, x, x, −x, −x, −x, −x), and ϕrec = (x, −x, −x, x, −x, x, x, −x). Phase sensitive spectra in the indirect dimension are obtained by incrementing the phases ϕ1 in a States-TPPI manner (Marion et al. 1989). The recycling delay is optimized based on the estimated longitudinal relaxation time (4.5 s was used in here). The two sine-shaped pulsed field gradients were applied along the z-axis for 1.0 ms with maximum intensities of G1 = 40 G/cm and G2 = 35 G/cm. Broad-band decoupling was achieved by using WALTZ16 (Shaka et al. 1983) for proton and deuterium (3.1 and 1 kHz, respectively) and GARP (Shaka et al. 1985) for nitrogen (1.5 kHz)
Experiments were recorded on a 5 mM sample of the uniformly 2H15N and alternately 13C–12C labeled B1 domain of protein G (GB1) in D2O. Here, 2-13C glycerol was used as carbon source to maximize the 13Cα labeling rate (Takeuchi et al. 2008b). To simulate the tumbling of a 90 kDa protein, 20% glycerol was added, and the experiment was recorded at 285 K (for a calibration of the correlation time, see Takeuchi et al. 2008b). All experiments were recorded in D2O to minimize the long-range protoncarbon dipole interaction. Under these conditions, average Cα transverse and longitudinal relaxation times were determined with spin echo and inversion recovery experiments and found to be 90 ms and 3.6 s, respectively. Usually, an effective relaxation rate 1/Teff = Wt/T2 + (1 − Wt)/T1 is used to estimate the signal decay during the mixing scheme (Felli et al. 2009). The transverse weight Wt depends on the spin-lock scheme, and one wants to achieve a low value of Wt to minimize the decay of coherence during the mixing sequence. Since Wt for the FLOPSY-16 pulse sequence is ∼0.65 (Felli et al. 2009), the estimated effective relaxation time at this condition is ∼140 ms. The effective relaxation rate during the spin lock can be decreased by using a MOCCA-XY16 mixing sequence which has a smaller Wt value (<0.2) than FLOPSY-16 as shown in a recent publication (Felli et al. 2009). This might be particularly important for large molecular weight systems in order to take advantage of the long T1 of deuterated 13Cα. However, despite the apparent advantages, the MOCCA-XY16 mixing scheme has the disadvantage that it covers a much narrower bandwidth compared to FLOPSY-16. Thus, the MOCCA mixing scheme may not be an ideal choice, especially for high magnetic fields. Furthermore, as discussed below, the correlations between 13Cα and side-chain 13C spins would be difficult to observe with the MOCCA-XY16 mixing scheme.
To shorten recycling delays and thus enhancing sensitivity by acquiring more scans one can safely use paramagnetic enhancement reagents, such as Gd(DTPA-BMA) or Ni(DO2A) (Eletsky et al. 2003; Cai et al. 2006; Takeuchi et al. 2010). Fortunately, Teff is rather insensitive to shortening of the longitudinal relaxation time T1 in particular for FLOPSY-16, which has a larger Wt value but also in the MOCCA-XY16 mixing sequence (Eletsky et al. 2003). Addition of 3 mM Gd(DTPA-BMA) to the “90 kDa” GB1 sample decreased T1 values from 3.6 to 0.9 s as reported previously (Takeuchi et al. 2010) while the T2 values are only decreased from 90 to 80 ms. Thus it becomes possible to acquire scans four times faster, with only a 10% decrease in Teff when using FLOPSY-16 (from 140 to 120 ms). When using the MOCCA-XY16 mixing sequence the value of Teff is estimated to decrease about 25% from 400 to 300 ms. Thus, when a moderately narrow mixing band width is acceptable, such as for just alpha carbon correlations, the MOCCA-XY16 sequence may have advantages over FLOPSY-16. Development of more powerful mixing sequences is still desirable.
Figure 2a shows 2D CACA-TOCSY spectra for mixing times ranging from 13 to 514 ms. The measuring time of each spectrum was 5.5 h. At mixing times of 13 and 26 ms, only diagonal signals and a few cross-peaks are observed. Except for several weak Cα–Cα signals seen in the 26-ms mixing-time spectrum, strong signals corresponding to 1J carbon correlation from natural abundance 13C in the 20% glycerol in the NMR sample solution (indicated by asterisks in the 13-ms mixing time spectrum). The correlations of the protein become obvious in spectra with mixing times ≥66 ms. At 132 ms, all expected inter-residue signals are observed, except for three crosspeaks that overlap with diagonal peaks (Ala23-Ala24, Glu27-Lys28, and Asn35-Adp36) and the peaks of the three Leu residues. The Cα carbons of Leu are not 13C-labeled with the 2-13C labeling scheme. However, these correlations can be readily observed with the 1,3-13C labeling (Fig. S1). An alternative would be the addition of 2H, 15N, 13Cα-labeled Leu to the 2-13C labeling culture media, which would increase the cost of labeling, but would allow obtaining all assignments with a single sample.
Fig. 2.
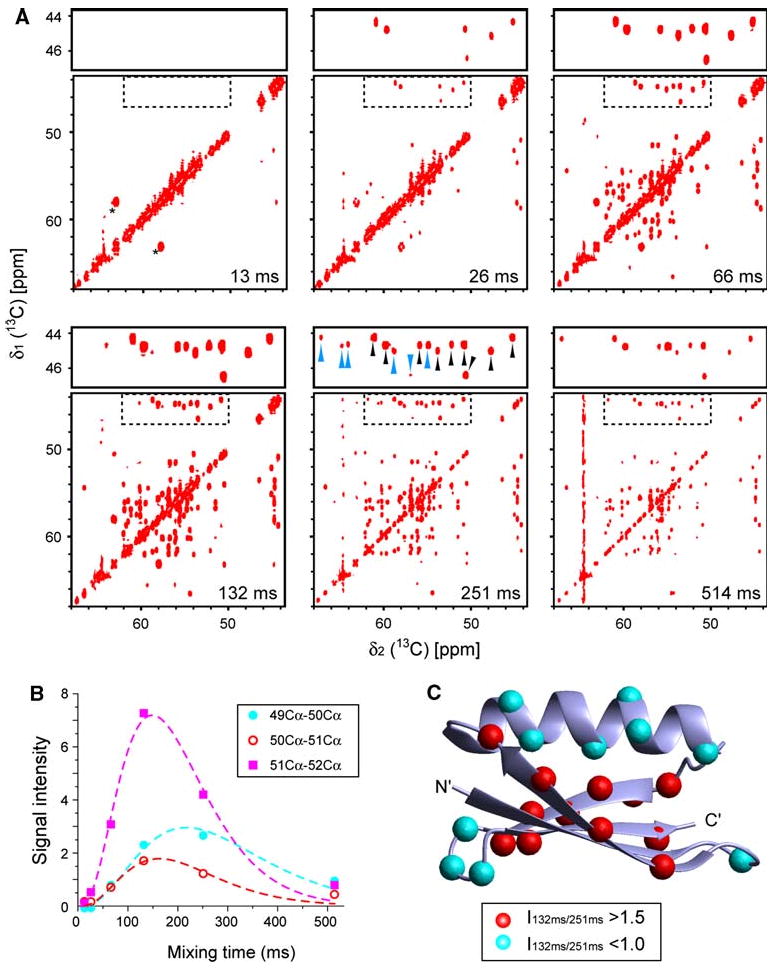
Mixing efficiency of the CACA-TOCSY experiment with [U-2H, 15N + 2-13C]-GB1 (5 mM) recorded in 20% glycerol at 285 K, which corresponds to a 90 kDa protein in aqueous solution at room temperature. a The CACA-TOCSY spectra at various mixing times ranging from 13 to 514 ms. As easily analyzable areas, the regions containing correlations to glycine residues (dotted square) are enlarged on top of each spectrum. Black and cyan arrow heads in the enlarged section at 251-ms mixing time indicate sequential and long range (i:i−2 or i:i+2) correlations between Cα nuclei, respectively. The measuring time for each spectrum was 5.5 h. The black asterisks in the 13 ms mixing time spectrum denote signals arising from natural abundance 13C-nuclei of glycerol. 2D CACA-TOCSY experiments were recorded with a spectral width of 6,887 Hz (direct) × 6,910 Hz (indirect), on a Bruker (Billerica, MA) Avance 500 spectrometer equipped with a triple-resonance carbon-cryogenic probe (TXO) designed for carbon detection experiments. 512 and 128 complexed data points were recorded for direct and indirect directions, respectively. Eight scans were applied for each increment. The digital resolution in the direct and indirect dimensions after Fourier transformation was 13.5 and 54.0 Hz for the 2D CACA-TOCSY. b Examples of build-up curves for three sequential correlations (residues 49–50, 50–51, and 51–52). Curves are drawn to approximately fit the TOCSY time course. c Location of residues that have fast and slow build up rates, respectively. On the three-dimensional structure of GB1, correlations with ratios of intensities between 132 and 251 ms, I132/I256 ≥ 1.5 or ≤ 1.0 are indicated by the red and cyan spheres in positions, respectively
Interestingly, at a mixing time of 251 ms, “supra-sequential” correlations are observed for 23 out of 56 possible residue pairs (41%). This represents a unique feature of the CACA-TOCSY spectrum. It is in principle possible to observe correlations farther than and indeed one weak correlation between Trp43 and Asp46 was observed in the 2D CACA-TOCSY spectrum at a mixing time of 251 ms.
Most of the cross peaks showed a maximum intensity at 132-ms mixing time while some of the correlations give strongest intensity at 251 ms (Fig. 2b). We realized that the maxima of the time courses of correlation signals depend on the secondary structure. We use the ratios of the peak heights of correlations at mixing times of 132 and 251 ms, I132/I251, to quantify this dependence. Residues in β-sheets were found to have I132/I251 values >1.5, whereas those located in α-helical or in loop regions had I132/I251 values <1.0 (Fig. 2c). This observation is consistent with a previous report showing that the coupling constants depend on the ψi angle, albeit not by a Karplus-type relation (Hennig et al. 2000). The mean coupling constants reported for β-sheet secondary structure were 1.7 ± 0.1 Hz, while they were substantially smaller for α-helical secondary structure (Hennig et al. 2000; Peti et al. 2000). To verify these reported coupling constant values, we recorded 13C-detected DQF-COSY spectra with the same protein sample but at higher temperature (308 K). For residues in β-sheets, we observed a ∼3 Hz separation between the most upfield and downfield components of the unresolved doublet of doublet (data not shown), which corresponds to the sum of and couplings and thus confirming the reported values. However, the coupling constants for the α-helical region were too small to be observed in the DQF-COSY experiment and we estimate their value to be less than 1 Hz. Thus, the CACA-TOCSY experiment (or DQF-COSY) with a 13C–12C alternate sample provides qualitative information on the ψ torsion angle by distinguishing larger for β-sheet secondary structures from smaller values for helical or loop conformations.
Since the COCO-TOCSY experiment provides information on the ϕ dihedral angle by a Karplus dependence (Balayssac et al. 2006), the complementary use of the CACA- with COCO-TOCSY experiments would particularly be interesting to obtain structural information of perdeuterated proteins. In this context, it is also worth mentioning that most of the couplings are smaller than 1.5 Hz (Hu and Bax 1996; Balayssac et al. 2006). Thus, at least for β-sheet regions, the CACA-TOCSY experiment is more sensitive than COCO-TOCSY due to the larger coupling (1.5–2 Hz) and slower relaxation properties of Cα atoms compared to C′.
It is of interest to compare the sensitivity of the 2D CACA-TOCSY experiment with the previously reported CA(N)CA experiment (Takeuchi et al. 2010). Figure 3 shows such a comparison of the 2D CACA-TOCSY (132-ms mixing time) and 2D CA(N)CA experiments (Takeuchi et al. 2010) recorded under the same conditions and measuring times. The signal intensities of most of the correlations are substantially higher in the CACA-TOCSY experiment (see slices in Fig. 3a, b). On average, the 2D CACA-TOCSY was found to have a S/N ratio 3.7 times higher than the one for the 2D CA(N)CA experiment. Only eight correlations had smaller S/N ratios than in the 2D CA(N)CA experiment (red bar in Fig. 3b). These eight correlations involve the 4 Val residues, which are labeled in an exceptional way under 13C–12C alternate labeling. For each Val residue in position i, the and correlations have weak intensity. Unlike all other residues (Fig. 3c), Val residues, as well as Ile, are 13C labeled simultaneously in Cα and Cβ positions with 2-13C glycerol. Therefore the strong 1J carbon–carbon coupling causes detrimental coherence “leaking”, which prevents the weak correlations to be observed. However, this effect can be used to identify valine and isoleucine residues as discussed below. A tailored mixing sequence designed to avoid touching the Cβ resonances of Val and Ile would prevent the “leakage” of coherence.
Fig. 3.
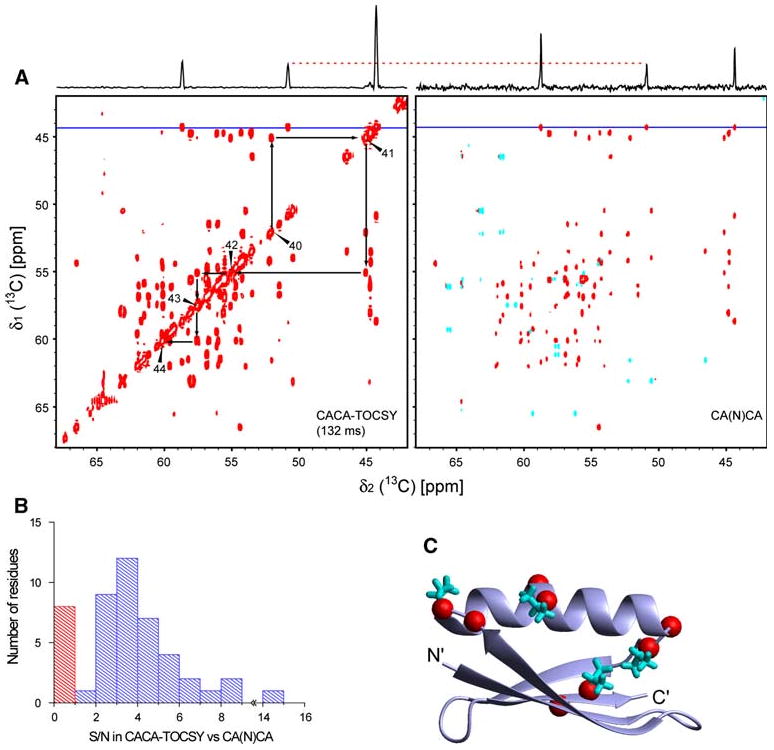
Comparison between the CACA-TOCSY and CANCA experiments. a The CACA-TOCSY (132 ms) and CANCA spectra were recorded at the same condition as in Fig. 2. The measuring time for each experiment was 5.5 h. Slices at the position of the horizontal blue lines (corresponding to Gly9) are shown on top of each spectrum. The vertical scales of the slices are normalized by the peak intensity of the middle resonances originating from Cα of residue Asn8. Black arrows in the CACA-TOCSY indicate the assignment walk connecting the Cα of residues 40–44. Starting from diagonal at , one can find the Cα frequency of the preceding or succeeding residue ω ( or ). b Histogram of the ratios of the signal-to-noise values (S/N) in the CACA-TOCSY over the S/N in CANCA. The average of the S/N values for and correlations is used. Bars representing ratios larger and smaller than 1 are colored blue and red, respectively. c Mapping of correlations that have smaller S/N value in the CACA-TOCSY. On the three-dimensional structure of GB1, red spheres positioned at the positions indicate correlations with S/N(CACA-TOCSY)/S/N(CA(N)CA) < 1.0. The side chains of Val residues are shown in cyan stick representations
The CACA-TOCSY experiment with alternate labeling also correlates Cα to specific side chain carbons in certain types of amino acid residues. As it is summarized in Fig. 4 for observed and expected Cα to side chain carbon correlations, the information can be quite useful for identifying the amino acid type in the same experiment that also provides the sequential assignments. As shown in Fig. 5, intra residue Cα–Cδ correlations via 3J(Cα–Cδ) coupling were observed for all Lys residues in the spectra with 132 and 251-ms mixing time (black arrowheads). More interestingly, two sequential correlations between Lys Cδ and the Cα of the neighboring residues via intra-residual 3J(Cα–Cδ) and sequential 3J(Cα–Cα) couplings were observed (Fig. 5, cyan arrowheads). Those Cα–Cδ correlations are also expected for arginine residues (no Arg is present in GB1), as Arg has the same labeling pattern as Lys under 2-13C labeling (13Cα–12Cβ–12Cγ–13Cδ). Thus, this unique information is helpful in specifying amino acid types as well as in providing additional sequential cross peaks. Such cross peaks can be particularly valuable, if the sequential connections cannot be resolved due to the overlapping in Cα signals. The peaks from Lys and Arg can be unambiguously distinguished because of their substantially different Cδ chemical shifts. In addition, since proton (or deuterium) in the δ sites of these residues originates from the solvent used for protein expression (H2O or D2O); the Cδ positions are highly deuterated in our experimental condition. Thus, those correlations are observable in proteins with a high molecular weight. While Glx (Glu and Gln), His, and Trp are also labeled with the same pattern, observing those correlations is not straightforward considering the chemical shift separation between Cα and Cδ nuclei in those residues (Average Cδ chemical shifts in BMRB are 181.5 ppm for Glu, 179.5 ppm for Gln, 119.8 ppm for His, and 126.4 (Cδ1) and 127.5 (Cδ2) ppm for Trp). Leu and Ile sidechain would also have the same 13C-labeling pattern with 1,3-13C labeling. Cα–Cδ correlations in Leu are rather easy to detect (Figure S2), providing additional amino acid specific information. The Cα–Cδ correlation of the Ile residue (Ile6) in GB1 was not observed in this study. This might be because the coupling constant happens to be small or the spin mixing was not efficient due to the rather large chemical shift difference between Cα and Cδ (Cα: 61.6 ppm and Cδ: 13.5 ppm, in average), in this residue. One can easily detect one bond Cα–Cβ correlation for Ile in 2-13C labeling with a short mixing time (13 ms, Fig. S3). With 2-13C labeling, Val residues are also simultaneously 13C labeled at Cα and Cβ positions as discussed above. However, Val and Ile Cα–Cβ correlation can be easily distinguished from each other by their Cβ chemical shifts (Fig. S3). Pro residues also exhibit Cα–Cγ correlations via 3J(13Cα–15N–12Cδ–13Cγ) couplings with 1,3-13C labeling. However, 2H and 13C labeled media have to be used to achieve complete deuteration of the Cγ and Cδ positions in 1,3-13C labeling.
Fig. 4.
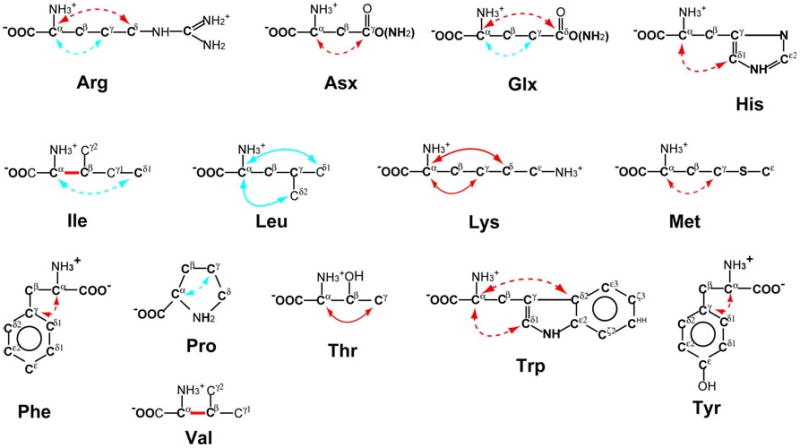
Summary of the amino acid specific correlations between Cα and side chain carbon in CC-TOCSY experiment with 13C–12C alternate labeling strategies. Red and cyan arrows indicate 2J and 3J carbon–carbon couplings that are expected to be observed with 2-13C and 1,3-13C-labeling, respectively. Red bonds indicate 1J carbon–carbon coupling observed with 2-13C labeling. Note that most amino acids can be 13C labeled in a couple of different patterns as it is exemplified in Arg (see text). The correlations observed in this study are shown in solid lines, whereas, the expected but not observed correlations are shown by broken lines. The correlations with broken lines are not observed due to the absence of the residues in the GB1 protein or the inefficient TOCSY transfer
Fig. 5.
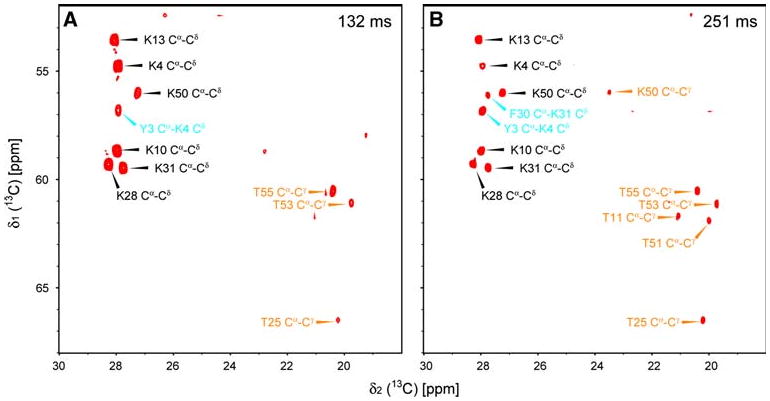
Cα to sidechain carbon correlations in the CACA-TOCSY experiment. The spectra were recorded with [U-2H, 15N + 2-13C]-GB1 (5 mM) under the same conditions as those in Fig. 2, with mixing times of 132 ms (a) and 251 ms (b). The measuring time for each experiment was 5.5 h. Black and cyan arrowheads in the spectra indicate intra-residual and sequential Cα–Cδ correlations, respectively. Orange arrowheads are for intra-residual Cα–Cγ correlations
In addition to correlations via 3J and 1J couplings mentioned above, intra-residual Cα–Cγ crosspeaks via 2J(Cα–Cγ) coupling were also observed in one out of six Lys and five out of ten Thr residues (Fig. 5, orange arrow heads). Because the coupling constants for these 2J C–C couplings are smaller than those for 3J couplings, those correlations have a maximum at 256 ms mixing time and are relatively weak in intensity. Those cross peaks would be easier to observe in smaller molecular weight systems. With 2-13C labeling schemes, one can also identify Cα–Cγ correlations in Met, Asx (Asp and Asn), Tyr and Phe, which have the same labeling pattern as Thr (13Cα–12Cδ–13Cγ). However, observing Cα–Cγ correlations in Asx, Phe, and Tyr are not straightforward unless broadband spin-locking schemes are used (Average chemical shifts of Asp, Asn, Phe, and Tyr Cδ in the BMRB are 179.2, 176.8, 137.3, 129.1 ppm, respectively). Note that 2J Cα–Cγ correlations can additionally be observed for Arg and Glx with the 1,3-13C labeled schemes (see Fig. S3 for Glx correlations). The amino-acid selective information mentioned above can be used complementary with amino-acid spin type information, such as obtained from the CC-TOCSY-DIPAP experiment using a uniformly labeled sample (Bermel et al. 2005).
Conclusion
In summary, we presented the CACA-TOCSY, a novel 13C direct detection experiment that provides sequential and supra-sequential correlations among Cα resonances via 3J(Cα–Cα) couplings. The experiment becomes possible with the alternate 13C–12C labeling strategy, which makes weak scalar couplings to be the dominant routes for coherence transfer pathways, and except for two residue types, there is no need for eliminating large one-bond carbon–carbon couplings during detection. The CACA-TOCSY with alternate 13C labeling has improved sensitivity compared to the established coherence transfer pathways that rely on heteronuclear scalar coupling, such as CA(N)CA experiment. Furthermore, with a longer spin-mixing time, the experiment provides unique supra-sequential correlations between Cα atoms separated by ≥2 residues. In addition, since couplings correlate with the ψi protein backbone dihedral angles, CACA-TOCSY build-up curves reveal information about protein secondary structure. In the same experiment as used for sequential assignments, the C–C TOCSY mixing scheme with the alternate 13C–12C labeling strategy also provides Cα to Cβ, Cγ, and Cδ correlations for certain amino acids depending on side chain 13C labeling pattern. These correlations are useful for unambiguous determination of amino acid types. Val (Cβ), Ile (Cβ), Met (Cγ), Thr (Cγ), Arg (Cδ), and Lys (Cδ) can be identified in 2-C13 labeled samples and Arg (Cγ), Glx (Cγ), Pro (Cγ), and Leu (Cδ) in 1,3-13C labeled samples (Fig. 4). However, a deuterated 13C source might be needed for 1,3-13C labeling to detect those couplings in large molecular weight proteins, so that side-chain carbons of interest are deuterated. Lys and Arg in 2-13C labeling and Leu in 1,3-13C labeling are of particular interest providing also sequential correlations. These sequential correlations would be useful to solve problems with the degeneracy of overlapped Cα signals. The experimental scheme also has a potential to observe correlation between Cα and sidechain carbonyl carbons as well as with aromatic carbons. These correlations are difficult to observe because of the large chemical shift difference between the spins of interests. Thus, implementation of broadband mixing schemes would be particularly interesting and are under development. While the 2D spectra shown here provide quite narrow lines and good dispersion, one could further improve the spectral resolution and sensitivity by applying non-uniform sampling and processing schemes, which will be especially helpful for peaks near the diagonal (Hyberts et al. 2010). In addition, a 3D version of this experiment, which avoids signal overlap in larger proteins, is currently under development. Although the sensitivity of the experiment was largely improved by using the homonuclear TOCSY transfer step rather than C–N heteronuclear coherences, further improvement in sensitivity might be needed in the routine use of this experiment for very large single polypeptide proteins. In this regard, an experiment utilizing the larger 1H polarization as a magnetization source but not for the detection can be considered (Bermel et al. 2009a, b). For systems with less severe relaxation losses, proton-excited/detected 3D experiments that contain CACA-TOCSY transfers can also provide unique sequential information. Experiments of this type have been shown already for a series of 3D pulse sequences that use C′C′-TOCSY transfers. These include 3D (H)N(CO-TOCSY)NH, 3D (H)CA(CO-TOCSY)NH, and 3D (H)CBCA (CO-TOCSY)NH but used uniformly 13C-labeled samples (Liu et al. 2000). Proton detected experiments that use the CACA-TOSY module might provide the method of choice given the increased polarization of proton nuclei.
Supplementary Material
Acknowledgments
Dr. Tsyr-Yan (Dharma) Yu, Dr. Arthanari Haribabu, and Dr. Alexander Koglin are acknowledged for stimulating discussions. This work was supported by the NIH (grants AI37581, GM47467 and EB 002026).
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s10858-010-9410-3) contains supplementary material, which is available to authorized users.
References
- Arnesano F, Banci L, Piccioli M. NMR structures of paramagnetic metalloproteins. Q Rev Biophys. 2005;38:167–219. doi: 10.1017/S0033583506004161. [DOI] [PubMed] [Google Scholar]
- Balayssac S, Jimenez B, Piccioli M. 13C direct detected COCO-TOCSY: a tool for sequence specific assignment and structure determination in protonless NMR experiments. J Magn Reson. 2006;182:325–329. doi: 10.1016/j.jmr.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Bermel W, Bertini I, Felli IC, Kummerle R, Pierattelli R. 13C Direct detection experiments on the paramagnetic oxidized monomeric copper zinc superoxide dismutase. J Am Chem Soc. 2003;125:16423–16429. doi: 10.1021/ja037676p. [DOI] [PubMed] [Google Scholar]
- Bermel W, Bertini I, Duma L, Felli IC, Emsley L, Pierattelli R, Vasos PR. Complete assignment of heteronuclear protein resonances by protonless NMR spectroscopy. Angew Chem Int Ed Engl. 2005;44:3089–3092. doi: 10.1002/anie.200461794. [DOI] [PubMed] [Google Scholar]
- Bermel W, Bertini I, Felli IC, Lee YM, Luchinat C, Pierattelli R. Protonless NMR experiments for sequence-specific assignment of backbone nuclei in unfolded proteins. J Am Chem Soc. 2006a;128:3918–3919. doi: 10.1021/ja0582206. [DOI] [PubMed] [Google Scholar]
- Bermel W, Bertini I, Felli IC, Piccioli M, Pierattelli R. 13C-detected protonless NMR spectroscopy of proteins in solution. Prog Nucl Magn Res Spec. 2006b;48:25–45. [Google Scholar]
- Bermel W, Bertini I, Felli IC, Matzapetakis M, Pierattelli R, Theil EC, Turano P. A method for Cα direct-detection in protonless NMR. J Magn Reson. 2007;188:301–310. doi: 10.1016/j.jmr.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Bermel W, Bertini I, Csizmok V, Felli IC, Pierattelli R, Tompa P. H-start for exclusively heteronuclear NMR spectroscopy: the case of intrinsically disordered proteins. J Magn Reson. 2009a;198:275–281. doi: 10.1016/j.jmr.2009.02.012. [DOI] [PubMed] [Google Scholar]
- Bermel W, Bertini I, Felli IC, Pierattelli R. Speeding up 13C direct detection biomolecular NMR spectroscopy. J Am Chem Soc. 2009b;131:15339–15345. doi: 10.1021/ja9058525. [DOI] [PubMed] [Google Scholar]
- Cai S, Seu C, Kovacs Z, Sherry AD, Chen Y. Sensitivity enhancement of multidimensional NMR experiments by paramagnetic relaxation effects. J Am Chem Soc. 2006;128:13474–13478. doi: 10.1021/ja0634526. [DOI] [PubMed] [Google Scholar]
- Castellani F, van Rossum B, Diehl A, Schubert M, Rehbein K, Oschkinat H. Structure of a protein determined by solid-state magic-angle-spinning NMR spectroscopy. Nature. 2002;420:98–102. doi: 10.1038/nature01070. [DOI] [PubMed] [Google Scholar]
- Eletsky A, Moreira O, Kovacs H, Pervushin K. A novel strategy for the assignment of side-chain resonances in completely deuterated large proteins using 13C spectroscopy. J Biomol NMR. 2003;26:167–179. doi: 10.1023/a:1023572320699. [DOI] [PubMed] [Google Scholar]
- Felli I, Brutscher B. Recent advances in solution NMR: fast methods and heteronuclear direct detection. ChemPhysChem. 2009;10:1356–1368. doi: 10.1002/cphc.200900133. [DOI] [PubMed] [Google Scholar]
- Felli I, Pierattelli R, Glaser S, Luy B. Relaxation-optimised Hartmann–Hahn transfer using a specifically Tailored MOCCA-XY16 mixing sequence for carbonyl–carbonyl correlation spectroscopy in 13C direct detection NMR experiments. J Biomol NMR. 2009;43:187–196. doi: 10.1007/s10858-009-9302-6. [DOI] [PubMed] [Google Scholar]
- Frueh DP, Arthanari H, Wagner G. Unambiguous assignment of NMR protein backbone signals with a time-shared triple-resonance experiment. J Biomol NMR. 2005;33:187–196. doi: 10.1007/s10858-005-3204-z. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Kneller DG. SPARKY 3. University of California; San Francisco: 2004. [Google Scholar]
- Guo C, Geng C, Tugarinov V. Selective backbone labeling of proteins using 1, 2-13C2-pyruvate as carbon source. J Biomol NMR. 2009;44:167–173. doi: 10.1007/s10858-009-9326-y. [DOI] [PubMed] [Google Scholar]
- Hennig M, Bermel W, Schwalbe H, Griesinger C. Determination of ψ torsion angle restraints from 3J(Cα, Cα) and 3J(Cα, HN) coupling constants in proteins. J Am Chem Soc. 2000;122:6268–6277. [Google Scholar]
- Hsu S-TD, Bertoncini CW, Dobson CM. Use of protonless NMR spectroscopy to alleviate the loss of information resulting from exchange-broadening. J Am Chem Soc. 2009;131:7222–7223. doi: 10.1021/ja902307q. [DOI] [PubMed] [Google Scholar]
- Hu JS, Bax A. Measurement of three-bond 13C–13C J couplings between carbonyl and carbonyl/carboxyl carbons in isotopically enriched proteins. J Am Chem Soc. 1996;118:8170–8171. [Google Scholar]
- Hyberts SG, Takeuchi K, Wagner G. Poisson-gap sampling and forward maximum entropy reconstruction for enhancing the resolution and sensitivity of protein NMR Data. J Am Chem Soc. 2010;132:2145–2147. doi: 10.1021/ja908004w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Vögeli B, Pervushin K. Detection of C′, Cα correlations in proteins using a new time- and sensitivity-optimal experiment. J Biomol NMR. 2005;31:273–278. doi: 10.1007/s10858-005-2361-4. [DOI] [PubMed] [Google Scholar]
- LeMaster DM, Kushlan DM. Dynamical mapping of E. coli thioredoxin via 13C NMR relaxation analysis. J Am Chem Soc. 1996;118:9255–9264. [Google Scholar]
- Liu A, Riek R, Wider G, von Schroetter C, Zahn R, Wuthrich K. NMR experiments for resonance assignments of 13C, 15N doubly-labeled flexible polypeptides: application to the human prion protein hPrP(23–230) J Biomol NMR. 2000;16:127–138. doi: 10.1023/a:1008305022907. [DOI] [PubMed] [Google Scholar]
- Marion D, Ikura M, Tschudin R, Bax A. Rapid recording of 2D NMR spectra without phase cycling. Application to the study of hydrogen exchange in proteins. J Magn Reson. 1989;85:393–399. [Google Scholar]
- Peti W, Hennig M, Smith LJ, Schwalbe H. NMR spectroscopic investigation of ψ torsion angle distribution in unfolded ubiquitin from analysis of 3J(Cα,Cα) coupling constants and crosscorrelated relaxation rates. J Am Chem Soc. 2000;122:12017–12018. [Google Scholar]
- Serber Z, Richter C, Dotsch V. Carbon-detected NMR experiments to investigate structure and dynamics of biological macromolecules. Chembiochem. 2001;2:247–251. doi: 10.1002/1439-7633(20010401)2:4<247::AID-CBIC247>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Shaka AJ, Keeler J, Frenkiel T, Freeman R. An improved sequence for broadband decoupling: WALTZ-16. J Magn Reson. 1983;52:335–338. [Google Scholar]
- Shaka AJ, Barker PB, Freeman R. Computer-optimized decoupling scheme for wideband applications and low-level operation. J Magn Reson. 1985;64:547–552. [Google Scholar]
- Shimba N, Stern AS, Craik CS, Hoch JC, Dotsch V. Elimination of 13Calpha splitting in protein NMR spectra by deconvolution with maximum entropy reconstruction. J Am Chem Soc. 2003;125:2382–2383. doi: 10.1021/ja027973e. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Sun ZY, Wagner G. Alternate 13C–12C labeling for complete mainchain resonance assignments using C alpha direct-detection with applicability toward fast relaxing protein systems. J Am Chem Soc. 2008a;130:17210–17211. doi: 10.1021/ja806956p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Sun ZY, Wagner G. Alternate 13C–12C labeling for complete mainchain resonance assignments using Cα direct-detection with applicability toward fast relaxing protein systems. J Am Chem Soc. 2008b;130:17210–17211. doi: 10.1021/ja806956p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K, Frueh DP, Hyberts SG, Sun ZJ, Wagner G. Highresolution 3D CANCA NMR experiments for complete mainchain assignments using Cα direct-detection. J Am Chem Soc. 2010;132:2945–2951. doi: 10.1021/ja907717b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.