

Abstract
We used data from studies of copy-number variants (CNVs), single-gene associations, growth-signaling pathways, and intermediate phenotypes associated with brain growth to evaluate four alternative hypotheses for the genomic and developmental relationships between autism and schizophrenia: (i) autism subsumed in schizophrenia, (ii) independence, (iii) diametric, and (iv) partial overlap. Data from CNVs provides statistical support for the hypothesis that autism and schizophrenia are associated with reciprocal variants, such that at four loci, deletions predispose to one disorder, whereas duplications predispose to the other. Data from single-gene studies are inconsistent with a hypothesis based on independence, in that autism and schizophrenia share associated genes more often than expected by chance. However, differentiation between the partial overlap and diametric hypotheses using these data is precluded by limited overlap in the specific genetic markers analyzed in both autism and schizophrenia. Evidence from the effects of risk variants on growth-signaling pathways shows that autism-spectrum conditions tend to be associated with up-regulation of pathways due to loss of function mutations in negative regulators, whereas schizophrenia is associated with reduced pathway activation. Finally, data from studies of head and brain size phenotypes indicate that autism is commonly associated with developmentally-enhanced brain growth, whereas schizophrenia is characterized, on average, by reduced brain growth. These convergent lines of evidence appear most compatible with the hypothesis that autism and schizophrenia represent diametric conditions with regard to their genomic underpinnings, neurodevelopmental bases, and phenotypic manifestations as reflecting under-development versus dysregulated over-development of the human social brain.
Keywords: genetics, evolution, psychiatry
The Swiss psychiatrist Eugen Bleuler coined the terms “schizophrenia”, for the splitting of psychic functions, and “autism”, for withdrawal from external reality in patients with schizophrenia, almost exactly a century ago (1). Ever since 1943, when Leo Kanner (2) coopted autism to refer to a new condition involving “disturbance of affective contact” manifested in children, the relationship between schizophrenia and Kanner’s autism has remained unclear (3). Kanner originally conceived autism as an early, distinct subtype of schizophrenia (model 1A) (Fig. 1A), a view he later renounced in favor of a model, which was also supported by Rutter (4) with the conditions as distinct, separate, and unrelated (model 1B) (Fig. 1B). Under each of these two hypotheses, autism and schizophrenia may each grade more or less smoothly, and independently, into so-called normality. Schizophrenia and autism have also been considered as diametric, or opposite sets of conditions (model 1C) (Fig. 1C) along a spectrum of social-brain phenotypes from hypodevelopment in autism, to normality, to hyperdevelopment in schizophrenia (5). By a fourth model, autism overlaps broadly yet partially with schizophrenia, sharing some risk factors and phenotypes but not others (model 1D) (Fig. 1D). This latter model has been motivated by recent genetic evidence of shared loci and pathways, mediating both autism and schizophrenia (6–8), and by work describing social deficits as central to both Kanner’s autism and the “autistic” symptoms of Blueler’s schizophrenia (9,10).
Fig. 1.

Alternative models for the genomic and etiological relationships of autism with schizophrenia and bipolar disorder. (A) Subsumed. (B) Separate. (C) Diametric. (D) Overlapping.
Differentiating alternative models for the relationships between major human psychiatric conditions has important implications for diagnoses, pharmacological and psychological treatment, and strategies for dissection of the etiology of these disorders at all levels from genes to cognition. In a series of recent papers, Neil Craddock and others (11–13) have demonstrated how genetic data can be deployed to evaluate alternative hypotheses for the relationship of schizophrenia with bipolar disorder, originally described as a dichotomy in work that followed from the pioneering studies of Emil Kraepelin, but now increasingly viewed in terms of intergradation and some form of partial overlap, based on the presence of shared genetic risk factors mediating shared phenotypes (Fig. 1).
In this article, we evaluate alternative hypotheses for the relationship of autistic spectrum conditions with schizophrenia spectrum conditions, by using recent genomic and genetic data to test the predictions that differentiate between models. Thus, under model 1A, alleles or haplotypes affecting autism risk should represent some subset of a larger pool of schizophrenia risk alleles or haplotypes; under model 1B, the two sets of conditions should exhibit independent risk factors; under model 1C, the conditions should be mediated by alternative risk factors with diametric effects on development; and under model 1D, autism and schizophrenia are expected to overlap for some set of genetic or genomic factors underlying liability to shared psychiatric phenotypes, as do bipolar disorder and schizophrenia. Model 1A and model 1D also predict that overlapping risk factors should influence developmental and phenotypic similarities between autism and schizophrenia. By contrast, model 1C predicts that these two sets of conditions should exhibit phenotypes that deviate in opposite directions from normality, at least for traits (such as aspects of growth or synaptic function) that are underlain by loci subject to variation or perturbation that can cause deviations from typical neurodevelopment or function in opposite directions.
Results
Copy Number Data.
Rare copy-number variants (CNVs) at seven loci, 1q21.1, 15q13.3, 16p11.2, 16p13.1, 17p12, 22q11.21, and 22q13.3 (Tables S1 and S2), have been independently ascertained and associated with autism and schizophrenia in a sufficient number of microarray-based comparative genomic hybridization (aCGH) and SNP-based studies to allow statistical analysis of the frequencies of deletions versus duplications in these two conditions (Table 1, Tables S3–S9). For five of the loci (1q21.1, 16p11.2, 16p13.1, 22q11.21, and 22q13.3), specific risk variants have been statistically supported for both autism and schizophrenia using case-control comparisons, which allows direct evaluation of the alternative hypotheses in Fig. 1. One locus (16p13.1) supports a model of overlap, and four loci support the reciprocal model, such that deletions are associated with increased risk of autism and duplications with increased risk of schizophrenia (16p11.2, 22q13.3), or deletions are associated with increased risk of schizophrenia and duplications with increased risk of autism (1q21.1, 22q11.21). For 1q21.1 and 22q11.21, contingency table analyses also indicate highly-significant differences in the frequencies of deletions compared with duplications for the two disorders, such that schizophrenia is differentially associated with deletions and autism with duplications. By contrast, for 16p11.2 and 22q13.3 such analyses show that autism is differentially associated with deletions and schizophrenia with duplications.
Table 1.
Contingency table analyses of the relative frequencies of copy-number deletions and duplications associated with autism or schizophrenia
| CNV locus | Condition | Deletion cases (refs.) | Duplication cases (refs.) | P-value (Fisher’s exact test) |
| 1q21.1 | Autism | 2 (14, 15) | 10 (14–17) | 0.001 |
| Schizophrenia | 15 (18–21) | 4 (18, 22) | ||
| 15q13.3 | Autism | 3 (19, 23) | 2 (24) | 0.849 |
| Schizophrenia | 10 (18, 19) | 4 (18) | ||
| 16p11.2 | Autism | 14 (16, 25–30) | 5 (16, 28, 30) | 0.00013 |
| Schizophrenia | 5 (16, 18, 19, 31) | 24 (18, 20, 31) | ||
| 16p13.1 | Autism | 0 | 3 (32) | 0.434 |
| Schizophrenia | 8 (18, 21, 33) | 23 (18, 22, 33) | ||
| 17p12 | Autism | 4 (14, 16, 26) | 1 (14) | 0.385 |
| Schizophrenia | 8 (18, 19, 22) | 0 | ||
| 22q11.21 | Autism | 1 (14) | 8 (14, 26, 28, 30) | 0.000049 |
| Schizophrenia | 16 (18, 21, 22, 34) | 1 (21) | ||
| 22q13.3 | Autism | 5 (25, 28, 30) | 0 | 0.0079 |
| Schizophrenia | 0 | 4 (18) |
Genetic Association Data.
Of 45 genes evaluated for association with both autism and schizophrenia and with replicated positive associations, 20 genes exhibit one or more positive associations with both conditions, compared with 2 genes positive for autism but not schizophrenia and 11 genes positive for schizophrenia but not autism; 12 genes showed negative results for both conditions (Table 2). Under a hypothesis in which associations with schizophrenia and autism are distributed independently of each other (corresponding to one interpretation of the “separate” model) (Fig. 1B), 20 or more genes associated with both conditions would be observed with a probability of 0.002 (Table S10). We interpret these results as inconsistent with a separate and independent relationship of autism and schizophrenia. The existence of genes associated with autism but not schizophrenia is inconsistent with a strict interpretation of Kanner’s original “subsumed” model (Fig. 1A) in which autism represents a subtype of schizophrenia; however, the presence of only two genes in this category mitigates against robust rejection of this model, and the relative lack of genetic-association studies of autism compared with schizophrenia may also partially explain this result.
Table 2.
Patterns in overlap between autism-associated genes and alleles and schizophrenia-associated genes and alleles
| Autism and schizophrenia | ||||||
| Shared alleles | Different alleles | Different markers | Complex overlap | Autism, not schiz. | Schiz., not autism | Not autism, not schiz. |
| DAO† | AHI1† | BDNF | CNTNAP2† | NRCAM† | CTLA4 | CYP21A2 |
| DISC1† | APOE | DRD3† | COMT† | SLC25A12 | DAOA | DBH |
| GRIK2 | DRD1 | EGF | NRXN1† | DRD2 | DDC | |
| GSTM1† | FOXP2† | NTNG1† | SLC6A4† | DRD4 | GABRA5 | |
| MTHFR† | HLA-DRB1 | RELN† | HTR2A | GABRG2 | ||
| SHANK3† | HTR7 | GABRP | ||||
| NOTCH4 | GSTP1 | |||||
| NRG1 | PENK | |||||
| SLC6A3 | RYR3 | |||||
| TH | SYNGAP1 | |||||
| TPH1 | TYR | |||||
| YWHAB | ||||||
Bold genes are included in the list of top 30 SZgene candidates (http://www.schizophreniaforum.org/res/sczgene/default.asp), as determined by effect size.
†At least one of the studies of autism showing positive associations used diagnostic criteria of “autism spectrum,” “autistic spectrum,” PDD-NOS, or “broad spectrum” autism, as compared to just “autism.”
Models 1C (diametric) and 1D (overlapping) both predict broad overlap in risk genes between autism and schizophrenia, and do not necessarily predict an absence or paucity of genes affecting one condition but not the other. In theory, these models can be differentiated by using data on specific risk alleles for specific loci (such as single-nucleotide polymorphisms, haplotypes, or genotypes), which should be partially shared under the overlapping model but different under the diametric model. For the genes DAO, DISC1, GRIK2, GSTM1, and MTHFR, the same allele, genotype, or haplotype was associated with both autism and schizophrenia, and for the genes AHI1, APOE, DRD1, FOXP2, HLA-DRB1, and SHANK3, alternative alleles, genotypes, or haplotypes at the same loci appear to mediate risk of these two conditions (SI Text). For the other genes that have been associated with both conditions, heterogeneity in the genetic markers used, heterogeneity among results from multiple studies of the same genes, and the general lack of functional information preclude interpretation in terms of shared or alternative risk factors.
Discussion
Delineation and classification of psychiatric conditions evolves through an iterative process, driven by societal pressures (35) and advances in our understanding of symptoms and etiology (36). The last several years have been characterized by remarkable advances in our understanding of the genetic and genomic underpinnings of autism, schizophrenia, and bipolar disorder, and evaluation of explicit alternative hypotheses (37) has led to progress well beyond a simple, century-old Kraepelian dichotomy of bipolar disorder distinguished from schizophrenia (12). In this article, we have evaluated alternative hypotheses for the relationship between schizophrenia and autism, using evidence from CNVs and genetic-association studies.
Models of autism as a subset of schizophrenia (Fig. 1A), and autism and schizophrenia as independent or separate (model 1B), can be rejected with some degree of confidence, but models involving diametric etiology (model 1C) or partial overlap (model 1D) cannot be clearly rejected. Taken together, most of the data and analyses described here appear to support the hypothesis of autism and schizophrenia as diametric conditions, based primarily on the findings that reciprocal variants at 1q21.1, 16p11.2, 22q11.21, and 22q13.3 represent statistically-supported, highly-penetrant risk factors for the two conditions (Table 1), and that for a number of genes, alternative alleles or haplotypes appear to mediate risk of autism versus schizophrenia.
Additional lines of evidence supporting the diametric hypothesis, from previous studies of autism and schizophrenia, include:
Data showing notable rarity of familial coaggregation of autism with schizophrenia (38), in contrast, for example, to strong patterns of co-occurance within pedigrees of schizophrenia, schizoaffective disorder, and bipolar disorder (39).
Psychiatric contrasts of Smith-Magenis syndrome with Potocki-Lupski syndrome (due to the reciprocal duplication at the Smith-Magenis locus), Williams syndrome with cases of Williams-syndrome region duplication, and Klinefelter syndrome with Turner syndrome, each of which tends to involve psychotic-affective spectrum phenotypes in the former syndrome, and autistic spectrum conditions in the latter (5, 40).
Effects of autism and schizophrenia risk alleles on common growth-signaling pathways, such that autism has been associated with loss of function in genes, such as FMR1, NF1, PTEN, TSC1, and TSC2 that act as negative regulators of the PI3K, Akt, mTOR, or other growth-signaling pathways (41–45), whereas schizophrenia tends to be associated with reduced function or activity of genes that up-regulate the PI3K, Akt, and other growth-related pathways (46–49).
Increased average head size, childhood brain volume, or cortical thickness in individuals with: (i) idiopathic autism (50–53), (ii) the autism-associated duplications at 1q21.1 (17) and 16p13.1 (32) and the autism-associated deletions at 16p11.2 (31), and (iii) autism due to loss of function (or haploinsufficiency) of FMR1 (54), NF1 (55), PTEN (56) and RNF135 (57). By contrast, reduced average values for brain size and cortical thickness, due to some combination of reduced growth and accelerated gray matter loss, have been demonstrated with notable consistency across studies of schizophrenia (58–62), and such reduced head or brain size has also been associated with the schizophrenia-linked CNVs at 1q21.1 and 22q11.21 (17, 63, 64), and with deletions of 16p13.1 (65).
Despite a convergence of evidence consistent with a model of autism and schizophrenia as diametric conditions, the presence of genetically-based risk factors common to autism and schizophrenia, including deletions, duplications, or specific alleles shared between the conditions (Tables 1 and 2), deletions within the NRXN1 gene (66), down-regulated RELN signaling (67), reduced GAD1 expression (68), high levels of oxidative stress mediated in part by the GSTM1 deletion allele (69, 70), and altered folate metabolism mediated by the TT genotype of the MTHFR C677T locus (71) support a model of partially-overlapping etiology or indicate that some genetic variants may increase liability to both conditions in otherwise highly-vulnerable individuals.
Epidemiological reports of comorbidity between autism and schizophrenia (72, 73) may be generated by several processes: (i) true etiological overlap, (ii) diagnostic perspectives, common at least through the 1980s, that conflate autism with “childhood schizophrenia”, and (iii) false-positive diagnoses of children with premorbidity to schizophrenia as subject to autistic spectrum conditions, especially Pervasive Developmental Disorder Not Otherwise Specified (PDD-NOS) (74–77). Similarly, evidence for risk loci, rather than specific risk alleles or haplotypes, differentially shared between autism and schizophrenia (6, 7, 78), supports exclusion of model 1B (Fig. 1) but cannot differentiate between models 1A, 1C, and 1D without data on association of specific alleles with these conditions. Further analyses of alternative models for the relationship of autism with schizophrenia may usefully test for genetic associations of specific alleles, haplotypes, or CNVs at relatively well-established schizophrenia risk loci with autism, using autistic patient populations strictly diagnosed to exclude children with conditions such as PDD-NOS or “autism spectrum”, some of whom may actually be premorbid for schizophrenia.
Interpretation of autistic-spectrum conditions and schizophrenia-spectrum conditions as diametric for psychological and psychiatric traits is predicated on a hypothesized axis of neurodevelopment and cognitive-affective functioning that reflects the degree to which the human social brain may be under-developed versus over-developed to diverse forms of dysfunction (5). Under-development of social phenotypes such as theory of mind, language, sense of self in relation to others, and reciprocal social interaction represent well-recognized manifestations of autism (79–81), which can in some models be linked with neurological phenotypes such as imbalance toward excitatory glutamatergic cortical neurotransmission (82–84) or increased local versus global processing of information (85, 86). By contrast, such psychotic traits as auditory hallucination and thought disorder, paranoia, megalomania, and ascription of causal purpose to inanimate objects may be interpretable in terms of dysregulated hyperdevelopment of language, theory of mind and sense of self, all traits that are highly derived and elaborated in the human lineage (5).
Differentiating between alternative models for the relationship of autism with schizophrenia has far-reaching implications for diagnosis, treatment, and causal analyses of both sets of conditions. Current diagnostic categorization places the most common childhood condition likely to involve premorbidity to schizophrenia, PDD-NOS, formally within the autism spectrum; this system thus implictly presumes either a general lack of premorbidity to schizophrenia sufficiently severe to result in autism spectrum diagnoses, or that autism and schizophrenia overlap in etiology and symptoms. The tendency for males to exhibit worse premorbidity to schizophrenia than females (87, 88), and for earlier-onset schizophrenia to exhibit a higher male bias and a stronger tendency to be mediated by CNVs rather than other factors (89, 90) suggests a notable risk for false-positive diagnoses of autistic spectrum conditions (75–77, 91–93). Apparent direct evidence of such risk comes from tendencies to diagnose autism spectrum conditions in children with deletions at 15q11.2, 15q13.3, and 22q11.21, and duplications of 16p11.2, CNVs for which high risk of schizophrenia has been established from studies of adults (16, 23, 31, 94–97). To the degree that autism and schizophrenia exhibit diametric genetically-based risk factors, inclusion of children premorbid for schizophrenia-spectrum conditions in studies of the genetic bases of autism will substantially dilute and confound the detection of significant results. Until the relationship between autism and schizophrenia is better understood, such risks could be minimized through explicit differential diagnosis of autism versus multiple complex developmental disorder (which is indicative of schizophrenia premorbidity, see ref. 98), and by subsetting genetic analysis of autism by infantile autism versus more broadly-conceived autistic spectrum categories. Finally, to the extent that autism and schizophrenia involve diametric alterations to aspects of neurological function such as hyperglutamatergic, over-excitatory cortical signaling in autism (82, 99) compared with schizophrenia symptoms mediated by hypoglutamatergic cortical states (100, 101), conceptual frameworks for developing pharmaceutical treatment of one set of conditions may prove useful for understanding the other. Fragile X syndrome provides an apparent example, as this autistic condition can be rescued in mice using antagonists of mGlur5 signaling (102), whereas agonists of this receptor represent among the more promising treatments for schizophrenia (103).
The Kraepelian dichotomy between schizophrenia and bipolar disorder has usefully guided psychiatric research programs for many years, but may now impede progress in the development of genet-ically-based models for the causes of psychotic and affective phenotypes in schizophrenia, bipolar disorder, and major depression (11, 12). Kanner’s genius was to recognize a new syndrome among the apparent chaos of human childhood psychopathology, but his adoption of the term autism, already established by Bleuler in the context of social withdrawal in schizophrenia, has led to >65 years of conflating the two sets of conditions, the autistic spectrum and the schizophrenia spectrum, that share social deficits as central phenotypes, but whose causes may differ substantially. To the extent that the autism spectrum and the schizophrenia spectrum represent diametric disorders of the social brain, as suggested by some of the analyses described here, a predictive framework based in evolutionary theory can be developed to guide research into the etiologies of both sets of conditions.
Materials and Methods
Copy Number.
CNVs for analysis were determined exclusively from publicly-available data in the aCGH and SNP-based studies listed in Table S2. The seven loci analyzed here, 1q21.1, 15q13.3, 16p11.2, 16p13.1, 17p12, 22q11.21, and 22q13.3 were selected for study strictly on the basis of two criteria: (i) statistical support from at least one study, or from analyses conducted here, for the loci as harboring risk variants for schizophrenia and/or autism enriched over controls, and (ii) the presence of at least six cases of autism (per se) or schizophrenia combined, with at least one case of autism and one case of schizophrenia associated with CNVs at the locus. Overlap in cases among studies, mainly involving AGRE, Stefansson et al. (19) and ISC (18) samples, was accounted for in ascertainment of cases for inclusion. Data from ref. 18 include information available at http://pngu.mgh.harvard.edu/isc/, and data from ref. 19 include only the authors’ phase 1 sample, for which complete data are available for deletions and duplications in regions other than 1q21.1, 15q11.2, and 15q13.3 (19).
Deletions at 15q11.2 that include four genes, TUBGCP5, CYFIP1, NIPA2, and NIPA1, are a statistically-documented risk factor for schizophrenia (8, 19), but deletions or duplications of this specific locus have not been reported in studies of autism. Duplications of a much larger region from 15q11.2 to 15q13 (and sometimes including these four genes) are a well-documented risk factor for autism, with seven such duplications reported in the autism studies considered here (16, 25, 26, 28). These data and work implicating overexpression of CYFIP1 in autism due to duplications of 15q11.2-q13 and Fragile X syndrome (104) suggest that CYFIP1, which interacts with the protein product (FMRP) of the FMR1 gene, may mediate associations of this locus to autism and schizophrenia. However, some cases of autism associated with duplications of 15q11.2-q13 do not include the CYFIP1 gene, which is inconsistent with any simple causative role for this gene.
Genes affected by CNVs that appear to be associated with both autism and schizophrenia, but have not been reported in a sufficient number of cases for inclusion here, include APBA2 [2 duplications in schizophrenia (8, 21), one duplication in autism (26)], CDH8 [4 deletions in autism (14), 1 duplication in schizophrenia (18)], and ZNF804A [3 duplications in autism (14), 1 deletion in schizophrenia (18)]. Deletions involving PARK2 have been reported in association with autism (30), and two deletions and three duplications involving this gene have been reported in cases of schizophrenia (18), but the CNVs exhibit complex patterns of partial overlap that are difficult to interpret without further information.
Genetic Association.
Genes in Table 2 were ascertained from the Szgene and AutDB databases and PubMed searches using the terms “autism or schizophrenia” and “gene or genetic”. Genes in AutDB in the “functional” category, and in the “syndromic” category, where associations with autism per se have not been reported (e.g., ALDH5A1), were not included. Positive results for schizophrenia also include genes that are positively-associated from meta-analyses in Szgene. PubMed searches are to date as of June 15, 2009. To minimize the possible effects of false-positive results, only genes that exhibited replicated positive associations in one or both conditions or positive associations in meta-analysis for schizophrenia were included in the sets of genes showing positive associations in one or both conditions.
Under a null hypothesis, associations with schizophrenia and autism are distributed among genes independently. Let s be the number of genes associated with schizophrenia, a be the number of genes associated with autism, and n be the total number of genes. The total number of possible combinations of genes with the specified number of associations is given by:
where
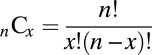 |
Let w be the number of genes associated with both schizophrenia and autism, x the number associated only with autism, y the number associated only with schizophrenia, and z the number associated with neither condition. The total number of possible combinations of genes with the specified sets of associations is given by:
Because the latter term is equal to one, the probability of obtaining some configuration of associations under the null hypothesis is given by:
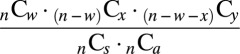 |
The possible combinations of associations and their probabilities are provided in Table S10.
Acknowledgments
We thank C. Badcock, J. Friedman, D. Geschwind, R. Glessner, H. Hakonarson, R. Holt, C. Marshall, S. Scherer, J. Sebat, D. St. Clair, H. Stefansson, J. Stone, R. Ullmann, and members of the Simon Fraser University Fab-lab for helpful comments, discussion, or provision of information to us. We thank R. Nesse and S. Stearns for inviting B.C. to the Sackler Colloquium, and NSERC for financial support.
Footnotes
This paper results from the Arthur M. Sackler Colloquium of the National Academy of Sciences, “Evolution in Health and Medicine” held April 2–3, 2009, at the National Academy of Sciences in Washington, DC. The complete program and audio files of most presentations are available on the NAS web site at www.nasonline.org/Sackler_Evolution_Health_Medicine.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. S.C.S. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/cgi/content/full/0906080106/DCSupplemental.
References
- 1.Bleuler E. In: Dementia praecox or the group of schizophrenias. Zinkin J, translator. New York: International Universities Press; 1911. 1950. (German) [Google Scholar]
- 2.Kanner L. Autistic disturbances of affective contact. Nervous Child. 1943;2:217–250. [PubMed] [Google Scholar]
- 3.Petty LK, Ornitz EM, Michelman JD, Zimmerman EG. Autistic children who become schizophrenic. Arch Gen Psychiatry. 1984;41:129–135. doi: 10.1001/archpsyc.1984.01790130023003. [DOI] [PubMed] [Google Scholar]
- 4.Rutter M. Childhood schizophrenia reconsidered. Journal of Autism and Childhood Schizophrenia. 1972;2:315–337. doi: 10.1007/BF01537622. [DOI] [PubMed] [Google Scholar]
- 5.Crespi B, Badcock C. Psychosis and autism as diametrical disorders of the social brain. Behav Brain Sci. 2008;31:241–261. doi: 10.1017/S0140525X08004214. and discussion (2008) 31:261–320. [DOI] [PubMed] [Google Scholar]
- 6.Burbach JP, van der Zwaag B. Contact in the genetics of autism and schizophre-nia. Trends Neurosci. 2009;32:69–72. doi: 10.1016/j.tins.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Iossifov I, Zheng T, Baron M, Gilliam TC, Rzhetsky A. Genetic-linkage mapping of complex hereditary disorders to a whole-genome molecular-interaction network. Genome Res. 2008;18:1150–1162. doi: 10.1101/gr.075622.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kirov G, et al. Comparative genome hybridization suggests a role for NRXN1 and APBA2 in schizophrenia. Hum Mol Genet. 2008;17:458–465. doi: 10.1093/hmg/ddm323. [DOI] [PubMed] [Google Scholar]
- 9.Frith CD, Frith U. Elective affinities in schizophrenia and childhood autism. In: Bebbington P, editor. Social Psychiatry: Theory, Methodology and Practice. New Brunswick, NJ: Transactions Press; 1991. pp. 65–88. [Google Scholar]
- 10.Tordjman S. Reunifying autism disorder and early-onset schizophrenia in terms of social communication disorders. Behav Brain Sci. 2008;31:278–281. [Google Scholar]
- 11.Craddock N, Owen MJ. The beginning of the end for the Kraepelinian dichot-omy. Br J Psychiatry. 2005;186:364–366. doi: 10.1192/bjp.186.5.364. [DOI] [PubMed] [Google Scholar]
- 12.Craddock N, O’Donovan MC, Owen MJ. Psychosis genetics: Modeling the rela-tionship between schizophrenia, bipolar disorder, and mixed (or “schizoaffective”) psychoses. Schizophr Bull. 2009;35:482–490. doi: 10.1093/schbul/sbp020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moskvina V, et al. Gene-wide analyses of genome-wide association data sets: Evidence for multiple common risk alleles for schizophrenia and bipolar disorder and for overlap in genetic risk. Mol Psychiatr. 2009;14:252–260. doi: 10.1038/mp.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Autism Genome Project Consortium, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mefford HC, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss LA, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 17.Brunetti-Pierri N, et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 2008;40:1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stefansson H, et al. Large recurrent microdeletions associated with schizophre-nia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walsh T, et al. Rare structural variants disrupt multiple genes in neurodevelop-mental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 21.Need AC, et al. A genome-wide investigation of SNPs and CNVs in schizophrenia. PLoS Genet. 2009;5:e1000373. doi: 10.1371/journal.pgen.1000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kirov G, et al. Support for the involvement of large copy number variants in the pathogenesis of schizophrenia. Hum Mol Genet. 2009;18:1497–1503. doi: 10.1093/hmg/ddp043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ben-Shachar S, et al. Microdeletion 15q13.3: A locus with incomplete pen-etrance for autism, mental retardation, and psychiatric disorders. J Med Genet. 2009;46:382–388. doi: 10.1136/jmg.2008.064378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller DT, et al. Microdeletion/duplication at 15q13.2q13.3 among individuals with features of autism and other neuropsychiatric disorders. J Med Genet. 2009;46:242–248. doi: 10.1136/jmg.2008.059907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Christian SL, et al. Novel submicroscopic chromosomal abnormalities detected in autism spectrum disorder. Biol Psychiatry. 2008;63:1111–1117. doi: 10.1016/j.biopsych.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kumar RA, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- 28.Marshall CR, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bijlsma EK, et al. Extending the phenotype of recurrent rearrangements of 16p11.2: Deletions in mentally retarded patients without autism and in normal indi-viduals. European Journal of Medical Genetics. 2009;52:77–87. doi: 10.1016/j.ejmg.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 30.Glessner JT, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCarthy S, et al. Microduplications of 16p11.2 are associated with schizophre-nia. Nat Genet. 2009 doi: 10.1038/ng.474. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ullmann R, et al. Array CGH identifies reciprocal 16p13.1 duplications and deletions that predispose to autism and/or mental retardation. Hum Mutat. 2007;28:674–682. doi: 10.1002/humu.20546. [DOI] [PubMed] [Google Scholar]
- 33.Ingason A, et al. Copy number variations of chromosome 16p13.1 region associated with schizophrenia. Mol Psychiatry. 2009 doi: 10.1038/mp.2009.101. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu B, et al. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet. 2008;4:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 35.Mayes R, Horwitz AV. DSM-III and the revolution in the classification of mental illness. Journal of the History of the Behavioral Sciences. 2003;41:249–267. doi: 10.1002/jhbs.20103. [DOI] [PubMed] [Google Scholar]
- 36.Kendler KS. An historical framework for psychiatric nosology. Psychol Med. 2009 doi: 10.1017/S0033291709005753. 10.1017/S0033291709005753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cannon TD. What is the role of theories in the study of schizophrenia? Schizophr Bull. 2009;35:563–567. doi: 10.1093/schbul/sbp008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rutter M. Concepts of autism: A review of research. J Child Psychol Psyc. 1968;9:1–25. doi: 10.1111/j.1469-7610.1968.tb02204.x. [DOI] [PubMed] [Google Scholar]
- 39.Craddock N, O’Donovan MC, Owen MJ. The genetics of schizophrenia and bipolar disorder: Dissecting psychosis. J Med Genet. 2005;42:193–204. doi: 10.1136/jmg.2005.030718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crespi B, Summers K, Dorus S. Genomic sister-disorders of neurodevelopment: An evolutionary approach. Evolutionary Applications. 2009;2:81–100. doi: 10.1111/j.1752-4571.2008.00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Belmonte MK, Bourgeron T. Fragile X syndrome and autism at the intersection of genetic and neural networks. Nat Neurosci. 2006;9:1221–1225. doi: 10.1038/nn1765. [DOI] [PubMed] [Google Scholar]
- 42.Kwon CH, et al. Pten regulates neuronal arborization and social interaction in mice. Neuron. 2006;50:377–388. doi: 10.1016/j.neuron.2006.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoeffer CA, et al. Removal of FKBP12 enhances mTOR-Raptor interactions, LTP, memory, and perseverative/repetitive behavior. Neuron. 2008;60:832–845. doi: 10.1016/j.neuron.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kelleher RJ, 3rd, Bear MF. The autistic neuron: Troubled translation? Cell. 2008;135:401–437. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 45.Cuscó I, et al. Autism-specific copy number variants further implicate the phosphatidylinositol signaling pathway and the glutamatergic synapse in the etiology of the disorder. Hum Mol Genet. 2009;18:1795–1804. doi: 10.1093/hmg/ddp092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emamian ES, Hall D, Birnbaum MJ, Karayiorgou M, Gogos JA. Convergent evidence for impaired AKT1-GSK3beta signaling in schizophrenia. Nat Genet. 2004;36:131–137. doi: 10.1038/ng1296. [DOI] [PubMed] [Google Scholar]
- 47.Stopkova P, et al. Identification of PIK3C3 promoter variant associated with bipolar disorder and schizophrenia. Biol Psychiatry. 2004;55:981–988. doi: 10.1016/j.biopsych.2004.01.014. [DOI] [PubMed] [Google Scholar]
- 48.Kalkman HO. The role of the phosphatidylinositide 3-kinase-protein kinase B pathway in schizophrenia. Pharmacol Ther. 2006;110:117–134. doi: 10.1016/j.pharmthera.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 49.Krivosheya D, et al. ErbB4-neuregulin signaling modulates synapse develop-ment and dendritic arborization through distinct mechanisms. J Biol Chem. 2008;283:32944–32956. doi: 10.1074/jbc.M800073200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hardan AY, Muddasani S, Vemulapalli M, Keshavan MS, Minshew NJ. An MRI study of increased cortical thickness in autism. Am J Psychiatry. 2006;163:1290–1292. doi: 10.1176/appi.ajp.163.7.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bethea TC, Sikich L. Early pharmacological treatment of autism: A rationale for developmental treatment. Biol Psychiatry. 2007;61:521–537. doi: 10.1016/j.biopsych.2006.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Elder LM, Dawson G, Toth K, Fein D, Munson J. Head circumference as an early predictor of autism symptoms in younger siblings of children with autism spectrum disorder. J Autism Dev Disord. 2008;38:1104–1111. doi: 10.1007/s10803-007-0495-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stanfield AC, et al. Towards a neuroanatomy of autism: A systematic review and meta-analysis of structural magnetic resonance imaging studies. Eur Psychiat. 2008;23:289–299. doi: 10.1016/j.eurpsy.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 54.Chiu S, et al. Early acceleration of head circumference in children with fragile x syndrome and autism. J Dev Behav Pediatr. 2007;28:31–35. doi: 10.1097/01.DBP.0000257518.60083.2d. [DOI] [PubMed] [Google Scholar]
- 55.Szudek J, Evans DG, Friedman JM. Patterns of associations of clinical features in neurofibromatosis 1 (NF1) Hum Genet. 2003;112:289–297. doi: 10.1007/s00439-002-0871-7. [DOI] [PubMed] [Google Scholar]
- 56.Butler MG, et al. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mu-tations. J Med Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Douglas J, et al. Mutations in RNF135, a gene within the NF1 microdeletion region, cause phenotypic abnormalities including overgrowth. Nat Genet. 2007;39:963–965. doi: 10.1038/ng2083. [DOI] [PubMed] [Google Scholar]
- 58.Goghari VM, Rehm K, Carter CS, MacDonald AW., 3rd Regionally specific cortical thinning and gray matter abnormalities in the healthy relatives of schizophrenia patients. Cereb Cortex. 2007;17:415–424. doi: 10.1093/cercor/bhj158. [DOI] [PubMed] [Google Scholar]
- 59.Gur RE, Keshavan MS, Lawrie SM. Deconstructing psychosis with human brain imaging. Schizophr Bull. 2007;33:921–931. doi: 10.1093/schbul/sbm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bose SK, et al. The effect of ageing on grey and white matter reductions in schizophrenia. Schizophr Res. 2009;112:7–13. doi: 10.1016/j.schres.2009.04.023. [DOI] [PubMed] [Google Scholar]
- 61.Goldman AL, et al. Widespread reductions of cortical thickness in schizophrenia and spectrum disorders and evidence of heritability. Arch Gen Psychiatry. 2009;66:467–477. doi: 10.1001/archgenpsychiatry.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Haukvik UK, et al. Cerebral cortical thickness and a history of obstetric complications in schizophrenia. J Psychiat Res. 2009;526 doi: 10.1016/j.jpsychires.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 63.Eliez S, Schmitt JE, White CD, Reiss AL. Children and adolescents with velocar-diofacial syndrome: A volumetric MRI study. Am J Psychiatry. 2000;157:409–415. doi: 10.1176/appi.ajp.157.3.409. [DOI] [PubMed] [Google Scholar]
- 64.Chow EW, Zipursky RB, Mikulis DJ, Bassett AS. Structural brain abnormalities in patients with schizophrenia and 22q11 deletion syndrome. Biol Psychiatry. 2002;51:208–215. doi: 10.1016/s0006-3223(01)01246-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hannes FD, et al. Recurrent reciprocal deletions and duplications of 16p13.11: The deletion is a risk factor for MR/MCA while the duplication may be a rare benign variant. J Med Genet. 2008;46:223–232. doi: 10.1136/jmg.2007.055202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rujescu D, et al. Disruption of the neurexin 1 gene is associated with schizo-phrenia. Hum Mol Genet. 2009;18:988–996. doi: 10.1093/hmg/ddn351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Persico AM, Levitt P, Pimenta AF. Polymorphic GGC repeat differentially regu-lates human reelin gene expression levels. J Neural Transm. 2006;113:1373–1382. doi: 10.1007/s00702-006-0441-6. [DOI] [PubMed] [Google Scholar]
- 68.Akbarian S, Huang HS. Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res Rev. 2006;52:293–304. doi: 10.1016/j.brainresrev.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 69.Harada S, Tachikawa H, Kawanishi Y. Glutathione S-transferase M1 gene dele-tion may be associated with susceptibility to certain forms of schizophrenia. Biochem Biophys Res Commun. 2001;281:267–271. doi: 10.1006/bbrc.2001.4347. [DOI] [PubMed] [Google Scholar]
- 70.Buyske S, et al. Analysis of case-parent trios at a locus with a deletion allele: Association of GSTM1 with autism. BMC Genetics. 2006;7:8. doi: 10.1186/1471-2156-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zogel C, et al. Identification of cis- and trans-acting factors possibly modifying the risk of epimutations on chromosome 15. Eur J Hum Genet. 2006;14:752–758. doi: 10.1038/sj.ejhg.5201602. [DOI] [PubMed] [Google Scholar]
- 72.Mouridsen SE, Rich B, Isager T. Psychiatric disorders in adults diagnosed as children with atypical autism. A case control study. J Neural Transm. 2008;115:135–138. doi: 10.1007/s00702-007-0798-1. [DOI] [PubMed] [Google Scholar]
- 73.Rzhetsky A, Wajngurt D, Park N, Zheng T. Probing genetic overlap among complex human phenotypes. Proc Natl Acad Sci USA. 2007;104:11694–11699. doi: 10.1073/pnas.0704820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sporn AL, et al. Pervasive developmental disorder and childhood-onset schizo-phrenia: Comorbid disorder or a phenotypic variant of a very early onset illness? Biol Psychiatry. 2004;55:989–994. doi: 10.1016/j.biopsych.2004.01.019. [DOI] [PubMed] [Google Scholar]
- 75.Feinstein C, Singh S. Social phenotypes in neurogenetic syndromes. Child Adol Psych Cl. 2007;16:631–647. doi: 10.1016/j.chc.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 76.Reaven JA, Hepburn SL, Ross RG. Use of the ADOS and ADI-R in children with psychosis: Importance of clinical judgment. Clinical Child Psychiatry and Psychology. 2008;13:81–94. doi: 10.1177/1359104507086343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Starling J, Dossetor D. Pervasive developmental disorders and psychosis. Current Psychiatry Reports. 2009;11:190–196. doi: 10.1007/s11920-009-0030-0. [DOI] [PubMed] [Google Scholar]
- 78.Konneker T, et al. A searchable database of genetic evidence for psychiatric disorders. Am J Med Genet Part B. 2008;147B:671–675. doi: 10.1002/ajmg.b.30802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hill EL, Frith U. Understanding autism: Insights from mind and brain. Philos Trans R Soc London Ser B. 2003;358:281–289. doi: 10.1098/rstb.2002.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Williams JH. Self-other relations in social development and autism: Multiple roles for mirror neurons and other brain bases. Autism Research. 2008;1:73–90. doi: 10.1002/aur.15. [DOI] [PubMed] [Google Scholar]
- 81.Baron-Cohen S. Autism: The empathizing-systemizing (E-S) theory. Ann NY Acad Sci. 2009;1156:68–80. doi: 10.1111/j.1749-6632.2009.04467.x. [DOI] [PubMed] [Google Scholar]
- 82.Rubenstein JL, Merzenich MM. Model of autism: Increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fatemi SH. The hyperglutamatergic hypothesis of autism. Prog Neuropsychop-harmacol Biol Psychiatry. 2007;32:911. doi: 10.1016/j.pnpbp.2007.11.004. author reply (2007) 32:912–913. [DOI] [PubMed] [Google Scholar]
- 84.Markram H, Rinaldi T, Markram K. The intense world syndrome - an alternative hypothesis for autism. Front Neurosci. 2007;1:77–96. doi: 10.3389/neuro.01.1.1.006.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Happé F, Frith U. The weak coherence account: Detail-focused cognitive style in autism spectrum disorders. J Autism Dev Disord. 2006;36:5–25. doi: 10.1007/s10803-005-0039-0. [DOI] [PubMed] [Google Scholar]
- 86.Lewis JD, Elman JL. Growth-related neural reorganization and the autism phenotype: A test of the hypothesis that altered brain growth leads to altered connectivity. Dev Sci. 2008;11:135–155. doi: 10.1111/j.1467-7687.2007.00634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sobin C, et al. Early, non-psychotic deviant behavior in schizophrenia: A possible endophenotypic marker for genetic studies. Psychiatry Res. 2001;101:101–113. doi: 10.1016/s0165-1781(00)00246-8. [DOI] [PubMed] [Google Scholar]
- 88.Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia, “just the facts” 4. Clinical features and conceptualization. Schizophr Res. 2009;110:1–23. doi: 10.1016/j.schres.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 89.Remschmidt HE, Schulz E, Martin M, Warnke A, Trott GE. Childhood-onset schizophrenia: History of the concept and recent studies. Schizophr Bull. 1994;20:727–745. doi: 10.1093/schbul/20.4.727. [DOI] [PubMed] [Google Scholar]
- 90.Rapoport J, Chavez A, Greenstein D, Addington A, Gogtay N. Autism spectrum disorders and childhood-onset schizophrenia: Clinical and biological contributions to a relation revisited. J Am Acad Child Adolesc Psychiatry. 2009;48:10–18. doi: 10.1097/CHI.0b013e31818b1c63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Eliez S. Autism in children with 22q11.2 deletion syndrome. J Am Acad Child Adolesc Psychiatry. 2007;46:433–434. doi: 10.1097/CHI.0b013e31802f5490. author reply (2007) 46:434. [DOI] [PubMed] [Google Scholar]
- 92.Solomon M, Ozonoff S, Carter C, Caplan R. Formal thought disorder and the autism spectrum: Relationship with symptoms, executive control, and anxiety. J Autism Dev Disord. 2008;38:1474–1484. doi: 10.1007/s10803-007-0526-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sugihara G, Tsuchiya KJ, Takei N. Distinguishing broad autism phenotype from schizophrenia-spectrum disorders. J Autism Dev Disord. 2008;38:1998–1999. doi: 10.1007/s10803-008-0638-7. author reply (2008) 38:2000–2001. [DOI] [PubMed] [Google Scholar]
- 94.Antshel KM, et al. Autistic spectrum disorders in velo-cardio facial syndrome (22q11.2 deletion) J Autism Dev Disord. 2007;37:1776–1786. doi: 10.1007/s10803-006-0308-6. [DOI] [PubMed] [Google Scholar]
- 95.Niklasson L, Rasmussen P, Oskarsdóttir S, Gillberg C. Autism, ADHD, mental retardation and behavior problems in 100 individuals with 22q11 deletion syndrome. Res Dev Disabil. 2008;30:763–773. doi: 10.1016/j.ridd.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 96.Doornbos M, et al. Nine patients with a microdeletion 15q11.2 between breakpoints 1 and 2 of the Prader-Willi critical region, possibly associated with behav-ioural disturbances. Eur J Med Genet. 2009;52:108–115. doi: 10.1016/j.ejmg.2009.03.010. [DOI] [PubMed] [Google Scholar]
- 97.Jolin EM, Weller RA, Weller EB. Psychosis in children with velocardiofacial syndrome (22q11.2 deletion syndrome) Curr Psychiatry Rep. 2009;11:99–105. doi: 10.1007/s11920-009-0016-y. [DOI] [PubMed] [Google Scholar]
- 98.Sprong M, et al. Pathways to psychosis: A comparison of the pervasive devel-opmental disorder subtype Multiple Complex Developmental Disorder and the “At Risk Mental State”. Schizophr Res. 2008;99:38–44. doi: 10.1016/j.schres.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 99.Spence SJ, Schneider MT. The role of epilepsy and epileptiform EEGs in autism spectrum disorders. Pediatr Res. 2009;65:599–606. doi: 10.1203/01.pdr.0000352115.41382.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Coyle JT. The GABA-glutamate connection in schizophrenia: Which is the prox-imate cause? Biochem Pharmacol. 2004;68:1507–1514. doi: 10.1016/j.bcp.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 101.Kehrer C, Maziashvili N, Dugladze T, Gloveli T. Altered excitatory-inhibitory balance in the NMDA-hypofunction model of schizophrenia. Front Mol Neurosci. 2008;1:6. doi: 10.3389/neuro.02.006.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dölen G, Bear MF. Role for metabotropic glutamate receptor 5 (mGluR5) in the pathogenesis of fragile X syndrome. J Physiol. 2008;586:1503–1508. doi: 10.1113/jphysiol.2008.150722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Conn PJ, Lindsley CW, Jones CK. Activation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends Pharmacol Sci. 2009;30:25–31. doi: 10.1016/j.tips.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nishimura Y, et al. Genome-wide expression profiling of lymphoblastoid cell lines distinguishes different forms of autism and reveals shared pathways. Hum Mol Genet. 2007;16:1682–1698. doi: 10.1093/hmg/ddm116. [DOI] [PubMed] [Google Scholar]